

Abstract
The genetic basis underlying the majority of hereditary pancreatic adenocarcinoma (PC) is unknown. Since DNA repair genes are widely implicated in gastrointestinal malignancies, including PC, we hypothesized that there are novel DNA repair PC susceptibility genes. As germline DNA repair gene mutations may lead to PC subtypes with selective therapeutic responses, we also hypothesized that there is an overall survival (OS) difference in mutation carriers versus non-carriers. We therefore interrogated the germline exomes of 109 high-risk PC cases for rare protein-truncating variants (PTVs) in 513 putative DNA repair genes. We identified PTVs in 41 novel genes among 36 kindred. Additional genetic evidence for causality was obtained for 17 genes, with FAN1, NEK1 and RHNO1 emerging as the strongest candidates. An OS difference was observed for carriers versus non-carriers of PTVs with early stage (≤ IIB) disease. This adverse survival trend in carriers with early stage disease was also observed in an independent series of 130 PC cases. We identified candidate DNA repair PC susceptibility genes and suggest that carriers of a germline PTV in a DNA repair gene with early stage disease have worse survival.
Keywords: Pancreatic adenocarcinoma, Familial pancreatic cancer, Exome sequencing, DNA repair genes
1. Introduction
Pancreatic ductal adenocarcinoma (PC) has the worst prognosis of any solid tumor type, which is largely attributable to late diagnosis.1, 2 Since genetic predisposition is thought to underlie 10% of PC, early detection programs for individuals at increased risk may improve clinical outcomes. A role for screening programs is supported by estimates that PC develops over a decade following the initiating somatic mutation, providing significant lead-time for screening.3 Unfortunately, screening strategies based on family history alone have been largely ineffective.4, 5 An understanding of the full spectrum of causative germline mutations will help identify individuals at highest risk and allow for more specific screening programs.
The evidence for hereditary PC is based on familial clustering suggestive of Mendelian inheritance, as well as the occurrence of PC within the tumor spectrums of characterized genetic syndromes.6 Family history is an important risk factor for PC, with a 2.3- to 32-fold increased risk depending on the number and relatedness of affected relatives in a family.7 Hereditary PC occurring either alone or as part of a tumor spectrum in families is partially attributable to rare, loss-of-function mutations, usually protein-truncating variants (PTVs) in the BRCA2, BRCA1, PALB2, ATM, CDKN2A, PRSS1, SPINK1 and mismatch repair (MLH2, MSH2, MSH6 and PMS2) genes.8 However, these genes account for less than 10% of hereditary PC, and one of the most important questions in the field remains the identification of the genetic causes where known genes are not implicated.
Segregation analyses suggest autosomal dominant inheritance of a rare allele(s) with variable penetrance to explain the missing heritability of PC.9 However, traditional linkage and genome-wide association studies have been largely unsuccessful in identifying novel medium or high penetrant PC susceptibility loci.8, 10 This is likely owing to unavailability of DNA from multiple affected family members due to the rapid progression of PC, locus heterogeneity, and variable penetrance of disease-causing alleles.8 The unbiased nature of next generation sequencing (NGS) overcomes many of these limitations, making this a promising approach for discovery of novel PC susceptibility genes, as evidenced by the recent identification of PALB2 and ATM using this approach.11, 12
Searching among thousands of genetic variants identified by NGS for the causative mutation is analogous to identifying the proverbial “needle in a haystack”. An a priori candidate gene approach is one method of overcoming this challenge and has been successful in identifying novel cancer susceptibility genes.13-15 Since DNA repair genes are widely implicated in gastrointestinal malignancies,16 and account for the majority of hereditary PC attributable to known PC predisposition genes (BRCA1, BRCA2, ATM, PALB2, mismatch repair genes),6, 7 we hypothesized that additional DNA repair genes are involved in hereditary PC. Therefore, we employed a DNA repair candidate gene approach to interrogate whole exome sequencing (WES) data for novel susceptibility genes. In addition, since there is a growing body of literature suggesting that PC associated with germline mutations in homology-directed DNA repair (HDR) genes (i.e., BRCA1, BRCA2, PALB2) have distinct genomic signatures, therapeutic responses and possibly clinical outcomes,17-19 we questioned whether there is an overall survival (OS) difference in carriers versus non-carriers of germline mutations in putative DNA repair genes.
2. Materials and Methods
2.1 Participants
PC cases enrolled in the Ontario20 or Quebec21 Pancreas Cancer Studies (OPCS, QPCS) were selected for WES of lymphocyte or white blood cell (surrogate germline) DNA. This series of cases (discovery set) included 8 young onset cases (diagnosed at 50 years of age or less) and 101 cases from 85 families with two or more PC-affected relatives (Table S1). These cases were not known to carry causal mutations in known PC susceptibility genes (i.e., BRCA2, BRCA1, PALB2, ATM, CDKN2A, PRSS1, SPINK1 and mismatch repair genes). Of the familial cases, WES data were generated from 70 cases for which DNA was available from a single affected family member and in 15 families where DNA was available from multiple PC-affected family members. We also generated WES data from matched tumor DNA that was available for cases 32B and 72. Fresh-frozen tumor samples for these cases were macrodissected to enrich for higher tumor cellularity prior to extracting DNA for WES. For case 58B, we used existing tumor whole genome sequencing (WGS) data.22 Available tissues from relatives of patients included in the discovery set were used for segregation and loss of heterozygosity (LOH) studies. These individuals were also enrolled in the OPCS or QPCS.
The validation set was composed of 130 PC cases with existing WGS data.22 All of these cases were explored surgically for resectability. This independent series of PC cases did not carry predisposing germline mutations in known genes and were unselected for increased hereditary PC risk.
The study was approved by the McGill University and Mount Sinai Hospital ethics review boards and written informed consent was obtained for all participants.
2.2 Whole exome sequencing
Library capture, variant calling and filtering (Figure 1) methods are described in Supplemental Materials and Methods.23-31 We evaluated for rare PTVs in genes implicated in DNA repair (n=513; Table S2) and in recognized PC susceptibility genes that are not implicated in DNA repair (i.e., CDKN2A, PRSS1, and SPINK1). Table S2 lists the 513 recognized and putative DNA repair genes evaluated, which includes genes defined as DNA repair genes in the Gene Ontology project (via AmiGO browser),32 genes included in the REPAIRtoire database33 and other genes identified through PUBMED literature search. Primers used to validate variants by Sanger sequencing are listed in Table S3.
Figure 1.
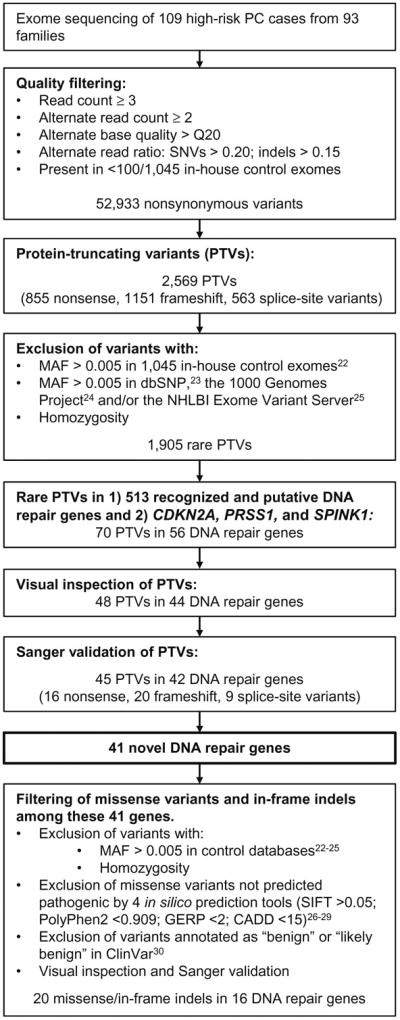
Schematic of the exome sequencing data analysis. Variants remaining after each filtering step are indicated. SNV, single nucleotide variant; indel, insertion/deletion; PTV, protein-truncating variant; MAF, minor allele frequency.
2.3 Segregation
Segregation of variants with PC was assessed in kindreds where WES data were available from multiple PC-affected family members. In cases where archived formalin-fixed, paraffin-embedded (FFPE) non-tumor tissue samples were available from relatives affected with PC, genomic DNA was extracted and tested for segregation by Sanger sequencing (see Table S4 for primers). In kindreds where samples were unavailable from PC-affected family members, DNA available from unaffected family members was used to infer segregation.
2.4 Loss of heterozygosity
In cases where tumor WES (52B, 72) or WGS22 (58B) data were available, variants were assessed for LOH or somatic inactivation of the second allele. In cases where archived FFPE tumor blocks were available, LOH was assessed by Sanger sequencing (see Table S4 for primers). Regions of tumor cellularity >50% were macrodissected prior to DNA extraction. LOH was analyzed by visually comparing allelic ratios of tumor and respective normal tissue DNA.
2.5 Overall survival
Univariate and multivariate Cox proportional hazard models were used to identify variables impacting survival. OS, defined as the time from diagnosis to death by any cause, was chosen as the primary end-point since this closely reflects cancer-related death in PC. Censoring events were created for patients alive at the time of last follow-up or patients lost to follow-up. Date of diagnosis was defined as date of first documentation of radiologic evidence or biopsy confirmation of PC. Date of surgery was used in cases where date of diagnosis was unknown. We included only the affected probands from kindreds in which multiple PC-affected relatives were sequenced. Covariates included age at diagnosis, gender, stage [early stage (≤ IIB) versus advanced stage (≥ III)] and DNA repair PTV carrier status (carrier versus non-carrier). Chemotherapy status was unavailable for 14 patients and was therefore excluded as a covariate. The results are reported as hazard ratios (HRs) with 95% confidence intervals (CIs). Kaplan-Meier survival curves were generated for carriers versus non-carriers of DNA repair gene PTVs and compared using the log-rank test. All statistical analyses were performed using SPSS version 22.0 (SPSS Inc., Chicago, IL, USA). Statistical significance was defined as P≤0.05.
Existing WGS data from an independent series of 130 PC cases22 were used to validate the OS correlations observed in the discovery set. Variant calling and filtering steps for these data are described in the Supplemental Materials and Methods. The OS analyses were carried out as described for the discovery set, with date of surgery used as a surrogate for date of diagnosis for all cases. Family history status [sporadic (n=122) versus familial (n=18)] was included as a covariate.
3. Results
3.1 Whole exome sequencing
WES data were generated for 109 high-risk PC cases from 93 families, as well as matched tumor DNA from two patients. The mean read depth obtained for target regions was 61.8±39.8 and the average percentage of Consensus Coding Sequence (CCDS)34 bases covered by at least 5, 10 and 20 reads were 94.0, 89.4 and 77.5, respectively. As expected, coverage was superior in the newer generation capture kits. The average percentage of CCDS bases covered by at least 5 reads were 92.2, 96.7 and 97.5 for the Illumina TruSeq Exome Enrichment Kit (n=69), Agilent SureSelect Human All Exon V4 (n=14) and Roche NimbleGen SeqCap EZ kit v3.0 (n=26) kits, respectively. The mean read depth obtained for the tumor exomes (n=2) was 130.8±3.3, and the average percentage of CCDS bases covered by at least 5, 10 and 20 reads were 97.1, 96.1 and 94.5, respectively.
3.2 Identification of DNA repair gene variants
Variant filtering was applied to the germline WES dataset (n=109) according to the algorithm outlined in Figure 1. Following quality filtering, a total of 52,933 nonsynonymous variants remained. Of these, 2,569 variants were PTVs. Next, variants were excluded if homozgyous or if present at a MAF >0.005 in unaffected in-house control exomes (n=1045), the 1000 Genomes Project or the NHLBI Exome Variant Server, leaving 1,905 rare PTVs. Of these, variants in recognized and putative DNA repair genes (n=513, Table S2) were selected for further evaluation. We also evaluated for PTVs in known PC susceptibility genes that are not implicated in DNA repair (i.e., CDKN2A, PRSS1, and SPINK1). A total of 70 variants in 56 DNA repair genes were identified. Following visual inspection, 48 variants (68.6%) in 44 genes remained. Sanger sequencing confirmed 45 PTVs in 42 DNA repair genes, resulting in a validation frequency of 93.8% Of the confirmed PTVs, 16 were nonsense, 20 were frameshift indels, and 9 were splice-site variants (Table S5).
Forty-one PC cases in 37 (39.8%) kindreds had one or more PTVs in a DNA repair gene (Table S5). Notably, we identified one previously unrecognized PTV in a known PC susceptibility gene [BRCA2:c.4691dupC (p.Thr1566Aspfs*9)]. Of the remaining 41 novel genes identified in 36 (38.7%) kindreds, four genes (FANCL, MC1R, NEK1 and RHNO1) had PTVs in multiple kindreds. Seven individuals were carriers of two PTVs, one individual was a carrier of 3 PTVs, while 2 kindreds had different affected family members carrying different PTVs (Table S6).
To further prioritize candidate genes, the WES data were also searched for nonsynonymous (missense and in-frame indel) variants in the 41 putative DNA repair genes that were confirmed by Sanger sequencing (Figure 1). The same quality and control filtering were utilized as for PTVs. Excluding variants annotated as “benign” in ClinVar31 and considering only missense variants predicted to be pathogenic by 4 in silico prediction tools, 18 missense variants and 2 in-frame indels in 16 DNA repair genes were identified (Table S7). All variants were confirmed by Sanger sequencing. Twenty-two PC cases in 19 (20.4%) kindreds had one or more missense variant or in-frame indel (Table S7). Three PC cases were carriers of multiple nonsynonymous variants and five cases were carriers of both a PTV and one or more nonsynonymous variant (Table S6). Five genes (DCLRE1A, FAN1, POLQ, TEX15, TONSL) had a missense variant or in-frame indel in multiple kindreds (Table S7).
3.3 Segregation and loss of heterozygosity analyses
For all validated variants, segregation with PC was assessed in families where either WES sequencing data were available from affected family members or DNA was available from affected or unaffected relatives (Tables S5 and S7). Fourteen genes demonstrated segregation of variants with PC in two or more affected family members, including AATF, BLM, CEP164, CHD1L, FAN1, FANCG, MC1R, NEIL1, NEK1, NEK11, RHNO1, SPP1, TONSL, and WRN. Notably, the following variants were found to co-segregate in 3 affected family members: NEK11:c.455+1G>A, SPP1:c.94-1G>A and FAN1:c.149T>G (p.Met50Arg). Five genes had variants segregating in 2 families: AATF, CHD1L, FAN1, NEK1 and RHNO1.
LOH was assessed in all cases where tumor WES data were available or in cases where archived FFPE tumor blocks were available. In total, LOH was assessed for 27 variants in 29 tumors, with loss of the wild-type allele observed for three variants [MGMT:c.593G>A (p.Trp198*), RHNO1:c.250C>T (p.Arg84*), WDR48:c.1278_1279del (p.Gly427Aspfs*8)], heterozygosity retained for 22 variants in 24 tumors and loss of the alternate allele observed for two variants [MLH3:c.1856A>T (p.Lys619Ile) and PMS1:c.1826G>A (p.Trp609*)]. Additionally, no second somatic mutation was observed in the tumor WES data for cases 52B and 72 carrying variants MC1R:c.456C>A (p.Tyr152*) and NINL:c.4142_4143del (p.Ser1381Cysfs*17), respectively, or in the tumor WGS data for case 58B carrying the FAN1:c.149T>G (p.Met50Arg) variant.
3.4 Top candidate genes
Among the 41 putative DNA repair genes identified with at least 1 PTV among high-risk PC cases, 17 genes have stronger genetic evidence supporting their roles as candidate novel PC predisposition genes (Table 1). This includes genes with more than 1 kindred with a PTV in that gene, genes with segregation of a predicted-pathogenic variant in at least one kindred and/or genes with at least one predicted-pathogenic variant and LOH of the corresponding wild-type allele. Of particular note are FAN1, NEK1 and RHNO1, which have variants present in 3 kindred and co-segregation of a variant with PC in at least 2 kindred. Figures 2, 3 and 4 show the pedigrees for families carrying variants in FAN1, NEK1 and RHNO1, respectively.
Table I.
Top candidate PC susceptibility genes.
| Gene | Chr. Pos. | Variant (HGVS nomenclature) | Samples | > 1 PTV | Segregation | LOH |
|---|---|---|---|---|---|---|
| AATF | chr17:35307578 | c.158_159dup (p.Gly54Trpfs*157) | 76A, 76B | Y | ||
| AATF | chr17:35311130 | c.755A>G (p.Asn252Ser) | 32 | Y | ||
| BLM | chr15:91292792 | c.298_299del (p.Gln100Glufs*42) | 78B | Y | ||
| CEP164 | chr11:117282575 | c.4228C>T (p.Gln1410*) | 16 | |||
| CEP164 | chr11:117244534 | c.1220C>T (p.Ser407Phe) | 53A, 53B | Y | ||
| CHD1L | chr1:146742591 | c.1086-2A>G | 90 | Y | ||
| CHD1L | chr1:146756048 | c.1730G>A (p.Gly373Asp) | 25 | Y | ||
| FAN1 | chr15:31214513 | c.2128C>T (p.Arg710*) | 42 | |||
| FAN1 | chr15:31197015 | c.149T>G (p.Met50Arg) | 58A, 58B, 34 | Y×2 | ||
| FANCG | chr9:35074472 | c.1652_1655del (p.Tyr551Phefs*7) | 50 | Y | ||
| FANCL | chr2:58386928 | c.1096_1099dup (p.Thr367Asnfs*13) | 47, 55B | Y | ||
| MC1R | chr16:89985733 | c.67C>T (p.Gln23*) | 14 | Y | ||
| MC1R | chr16:89985750 | c.86dup (p.Asn29Lysfs*14) | 69 | Y | ||
| MC1R | chr16:89986122 | c.456C>A (p.Tyr152*) | 52B | Y | Y | |
| MC1R | chr16:89986522 | c.862_864del (p.Ile288del) | 89 | Y | ||
| MGMT | chr10:131565137 | c.593G>A (p.Trp198*) | 63A | Y | ||
| NEIL1 | chr15:75641315 | c.330_331insAGGC (p.Ala111Argfs*46) | 43 | Y | ||
| NEK1 | chr4:170428209 | c.1687_1688del (p.Ala563Tyrfs*36) | 17, 78 | Y | Y | |
| NEK1 | chr4:170398474 | c.2235T>G (p.Asn648Lys) | 89 | Y | Y | |
| NEK11 | chr3:130828766 | c.455+1G>A | 68C, 68B | Y | ||
| RHNO1 | chr12:2997158 | c.250C>T (p.Arg84*) | 43 | Y | Y | |
| RHNO1 | chr12:2997245 | c.337C>T (p.Arg113*) | 18 | Y | Y | |
| RHNO1 | chr12:2994578 | c.45_46delinsAG (p.Leu16Val) | 70A, 70B | Y | Y | |
| SPP1 | chr4:88901197 | c.94-1G>A | 78A, 78B | Y | ||
| TONSL | chr8:145668147 | c.490del (p.Leu164Serfs*72) | 2 | |||
| TONSL | chr8:145660507 | c.2899C>T (p.Arg967Cys) | 3A | |||
| TONSL | chr8:145662005 | c.1950C>G (p.Asp650Glu) | 86 | Y | ||
| WDR48 | chr3:39125749 | c.1278_1279del (p.Gly427Aspfs*8) | 72 | Y | ||
| WRN | chr8:30999118 | c.3138+2T>G | 51 | |||
| WRN | chr8:31012237 | c.3785C>G (p.Thr1262Arg) | 44 | Y |
Chr. Pos. chromosomal position; >1 PTV, more than one kindred with a PTV in the corresponding gene; segregation. segregation of the variant in 2 or more PC-affected family members within a kindred; LOH, loss of heterozygosity (loss of the wild-type allele); Y, yes; ×2, occurs in 2 kindreds.
Figure 2.

Pedigrees of the families with FAN1 variants. Carrier status is depicted for all the cases in which germline DNA was available and tested. +/- indicates heterozygous carrier status. +/+ indicates wild-type. Probands are indicated with an arrow. Individuals shaded in black are affected with PC, while individuals shaded in grey are affected with a tumor other than PC. The ages of living family members and the ages of death (d.) for deceased individuals are indicated in years. Tumor types and ages at diagnoses are indicated in years. Other illnesses with ages in years at diagnosis (if known) are shown. NHL, non-Hodgkin's lymphoma; CLL, Chronic lymphocytic leukemia. Maternal and paternal ancestries are indicated.
Figure 3.

Pedigrees of the families with NEK1 variants. Carrier status is depicted for all the cases in which germline DNA was available and tested. +/- indicates heterozygous carrier status. +/+ indicates wild-type. Probands are indicated with an arrow. Individuals shaded in black are affected with PC, while individuals shaded in grey are affected with a tumor other than PC. The ages of living family members and the ages of death (d.) for deceased individuals are indicated in years. Tumor types and ages at diagnoses are indicated in years. Other illnesses with ages in years at diagnosis (if known) are shown. BCC, basal cell carcinoma. Maternal and paternal ancestries are indicated.
Figure 4.

Pedigrees of the families with RHNO1 variants. Carrier status is depicted for all the cases in which germline DNA was available and tested. +/- indicates heterozygous carrier status. +/+ indicates wild-type. Probands are indicated with an arrow. Individuals shaded in black are affected with PC, while individuals shaded in grey are affected with a tumor other than PC. The ages of living family members and the ages of death (d.) for deceased individuals are indicated in years. Tumor types and ages at diagnoses are indicated in years. NM, non-melanoma. Maternal and paternal ancestries are indicated.
3.5 Overall survival - Discovery set
The results of the univariate and multivariate Cox models of variables implicated in OS in the discovery set are shown in Table S8. Seventy-five (82.4%) cases were deceased. Considering all stages combined (n=91), significant associations were found for stage (early versus advanced) in both univariate and multivariate analyses (HR 4.3, 95% CI 2.5-7.0; P<0.001 and HR 5.9, 95% CI 3.4-10.4, P<0.001, respectively), and age at diagnosis in multivariate analysis (HR 1.03, 95% CI 1.01-1.05; P=0.003). Subset analyses were carried out for patients who presented with early (≤ IIB, n=50) and advanced (≥ III, n=41) stages. Interestingly, carriers of DNA repair gene PTVs with early stage (≤ IIB) had worse OS by univariate and multivariate analyses (HR 2.5, 95% CI 1.2-5.0; P=0.010 and HR 2.4, 95% CI 1.2-4.9, P=0.015, respectively). However, for patients with advanced stage (≥ III), carrier status did not correlate with OS. Figure 5 (panels A to C) show the Kaplan-Meier survival curves for carriers of DNA repair gene PTVs versus non-carriers.
Figure 5.

Discovery set Kaplan-Meier survival curves for carriers versus non-carriers of DNA repair gene PTVs for all stages (A), early stage (B) and advanced stage (C) cases. Log-rank p-values are indicated.
3.6 Overall survival - Validation set
The WGS data of the 130 PC cases in the validation set were assessed for rare germline PTVs in the 41 candidate DNA repair genes identified in the discovery set. We found 10 PTVs in 8 genes [AATF, BLM, CHD1L, DCLRE1A (2), NEK1, POLL, POLQ, TEX15 (2)] (Table S9). Ninety-nine (76.2%) cases were deceased. The results of the univariate and multivariate Cox regression analyses are shown in Table S10. Considering all stages combined (n=130), stage was a significant factor by univariate and multivariate analyses (HR 3.58, 95% CI 1.7-7.5; P=0.001 and HR 4.2, 95% CI 1.9-9.1, P<0.001, respectively), and a significant survival disadvantage for carriers of DNA repair gene PTVs versus non-carriers was observed by multivariate analysis (HR 2.8, 95% CI 1.2-6.3; P=0.017). Considering early stage alone (n=122), carrier status remained a significant variable in both univariate and multivariate analyses (HR 2.6, 95% CI 1.3-5.5; P=0.011 and HR 3.1, 95% CI 1.4-6.7; P=0.006 in univariate and multivariate analyses, respectively). The validation case series was enriched for early stage cases, with only 8 patients presenting with advanced stage disease and none carrying PTVs. The Kaplan-Meier survival curves for carriers of DNA repair gene PTVs versus non-carriers for all stages and early stage, respectively, are shown in Figure 6 (panels A and B).
Figure 6.
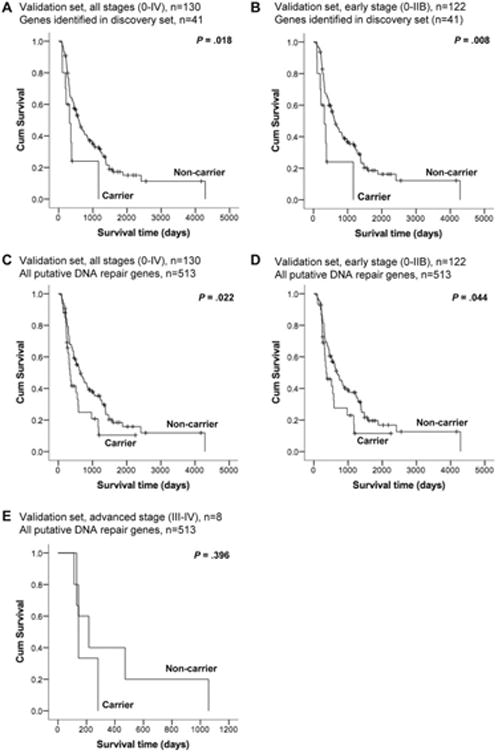
Validation set Kaplan-Meier survival curves for carriers versus non-carriers of PTVs in the genes identified in the discovery set (n=41) for all stages (A) and early stage (B), as well as for all 513 putative DNA repair genes (n=513) for all stages (C), early stage (D) and advanced stage (E) cases. Log-rank p-values are indicated.
We also evaluated the 130 cases in the validation set for rare PTVs in all 513 putative DNA repair genes (Table S2) and repeated the OS analysis. We observed 39 PTVs in 34 genes in 33 cases (25.4% Table S9). The results of the Cox regression analyses are shown in Table S11. For all stages combined (n=130), stage remained a significant factor (HR 3.8, 95% CI 1.8-8.3, P=0.001 in multivariate analysis). Considering all stages, a survival disadvantage for carriers of DNA repair gene PTVs versus non-carriers was observed in both univariate and multivariate analyses (HR 1.7, 95% CI 1.1-2.7; P=0.024 and HR 1.7, 95% CI 1.1-2.7, P=0.022, respectively). For early stage alone (n=122), carrier status retained association with worse OS in univariate and multivariate analyses (HR 1.6, 95% CI 1.0-2.7; P=0.046 and HR 1.6, 95% CI 1.0-2.7; P=0.051, respectively). The Kaplan-Meier survival curves for carriers of DNA repair gene PTVs versus non-carriers for all stages, early stage and advanced stage are shown in Figure 6 (panels C to E).
4. Discussion
We report a large-scale NGS study aimed at identifying novel genetic causes of hereditary PC in which 109 high-risk PC cases from 93 families underwent WES. Using a filter-based candidate gene approach focused on DNA repair genes, we identified PTVs in 41 putative DNA repair genes among 36 (38.7%) kindreds. We also found a rare PTV in BRCA2, a known PC susceptibility gene, demonstrating the ability of the approach to identify causal variants. The WES data were also evaluated for mutations in known PC predisposition genes not implicated in DNA repair (i.e., CDKN2A, SPINK1 and PRSS1). Since WES is unable to detect large genomic deletions and rearrangements, the possibility of such variants in known and candidate PC predisposition genes cannot be excluded and represents a limitation of our study. The 41 candidate genes were further characterized for predicted pathogenic nonsynonymous variants, segregation of putative pathogenic variants with disease in families, and LOH of the wild-type allele in tumors.
Some of the challenges in identifying causal genes in hereditary PC are the occurrence of phenocopies, genetic heterogeneity, and variable penetrance of disease-causing alleles.8 Although young age of onset is a risk factor for hereditary cancer, the majority of hereditary PC cases have the same age of onset as sporadic cases.35 As such, genetic studies of hereditary PC are often confounded by phenocopies. This notion is highlighted by our previous report showing lack of segregation of the PALB2:c.3256C>T (p.Arg1086*) and ATM:c.1931C>A (p.Ser644*) PTVs with PC-affected relatives.36 Consequently, in the present study, we used segregation status of variants to prioritize but not exclude candidate genes.
Although LOH was viewed favourably for causation, its absence did not exclude candidate genes since there are other mechanisms of somatic loss of the wild-type allele, as well as the possibility that haploinsufficiency is sufficient for tumorigenesis. Consistent with this possibility, the BRCA2:c.4691dupC (p.Thr1566Aspfs*9) variant identified in the present study did not exhibit LOH, suggesting other mechanisms of wild-type allele silencing in the tumor.
Another challenge in identifying causative genes in hereditary PC is the presence of multiple predicted-pathogenic variants in a single individual. As shown in Table S6, we observed cases with PTVs or predicted-pathogenic missense variants in multiple putative DNA repair genes. Double heterozygosity of pathogenic variants in multiple cancer predisposition genes has been previously reported in breast and ovarian cancers and likely reflects the variable penetrance of disease-causing alleles, where only one germline variant is needed to drive tumorigenesis.37, 38 The variable penetrance of PC predisposition genes identified to date suggests that the presence of double heterozygotes of disease-causing variants in our high-risk case series is plausible. In addition, the possibility that mutations in two genes results in predisposition “synergy” in the form of “additive” haploinsufficiency is an interesting hypothesis.
Based on the evidence obtained from the genetic investigations, we prioritized our list of 41 candidate PC susceptibility genes. Top candidate genes (n=17, Table 1) were considered those with more than 1 kindred with a PTV in that gene, genes with segregation of a predicted-pathogenic variant (PTV or nonsynonymous variant) in at least one kindred, and/or genes with LOH associated with at least one predicted-pathogenic variant. The strongest candidate PC predisposition genes, in view of case frequency, segregation and somatic silencing, are FAN1, NEK1 and RHNO1. Each of these genes had variants present in 3 out of 94 high-risk kindreds (3.2%), with at least partial co-segregation of the variants with PC in two or more kindreds (Figures 2, 3 and 4).
FAN1 (FANCD2/FANCI-associated nuclease 1) is required for the repair of interstrand cross-links.39, 40 Interestingly, FAN1 has recently been reported as a putative colon cancer susceptibility gene.41 Here, we identified a PTV in FAN1 (p.Arg710*) in Family 42, as well as a missense variant (p.Met50Arg) present in 2 kindreds. While there were no samples available for segregation or LOH analyses of the p.Arg710* variant, the p.Met50Arg variant demonstrated complete co-segregation with PC in tested family members (Figure 2). Notably, the p.Met50Arg variant occurs at a highly conserved amino acid residue within the RAD18-like ubiquitin-binding (UBZ) domain, which is essential for FAN1 localization to sites of DNA damage.39 The absence of LOH in two tumors from carriers of the p.Met50Arg variant and lack of evidence of a second hit in one of these cases (58B) for which WGS was available, is consistent with the results reported by Seguí et al,41 where somatic inactivation of the wild-type allele was not observed in any of the colon cancer cases with germline FAN1 mutations.
NEK1 [NIMA (Never In Mitosis Gene A)-Related Kinase 1] is the second strongest candidate gene. Its protein product is a dual serine-threonine and tyrosine kinase required for efficient DNA damage checkpoint activation and for maintaining chromosome stability.42 Moreover, there is evidence to suggest that NEK1 functions as a tumor suppressor.42 In the present study, 2 of 93 high-risk PC families were found to carry a novel NEK1:p.Ala563Tyrfs*36 variant. This variant has not been previously reported in the public control databases,25, 26, 43 nor was observed in 1,045 in-house control exomes. One family is of Greek origin and the other of English and Scottish descent. Therefore, this recurrent variant is unlikely to represent cryptic relatedness or an ethnic-specific variant but, perhaps, a mutation “hot spot” in NEK1 (deletion occurs within a triple “AG” repeat). Segregation of this variant was observed in one bilineal family (78), and only partial segregation was observed in the second family (17) (Figure 3). Notably, the affected relative found to be wild-type for the NEK1 variant in Family 17 was the eldest diagnosed in the family (75 years of age) and may represent a phenocopy. A third family was found to carry the predicted-pathogenic p.Asn648Lys variant with segregation of the variant observed in the 2 PC-affected siblings. As well, an additional PTV in NEK1 (c.868+1G>C) was identified in the case series used for the OS validation studies, providing further support for NEK1 as a candidate PC susceptibility gene.
Interestingly, NEK1 maps to chromosomal region 4q33, within a previously reported PC susceptibility locus (4q32-34) identified by linkage analysis of a kindred with 9 PC-affected family members and additional relatives with precancerous pancreatic lesions.44 Sequencing of candidate genes in the region in affected family members led to the identification of a variant in PALLD (P239S) which is considered to be the causative mutation in this family.45 Although PALLD may be causative in this kindred, subsequent studies have not supported PALLD as a common PC susceptibility gene.46-48 Interestingly, NEK1 was among the candidate genes sequenced in the aforementioned study and even though sequencing failed to identify a NEK1 variant, assays for large genomic structural changes at this locus were not performed. Thus, the possibility that NEK1 underlies PC predisposition in this kindred cannot be fully excluded.
The third top ranking candidate gene is RHNO1 (Rad9-Hus1-Rad1 Interacting Nuclear Orphan 1), which has an important role in DNA damage response signaling.49 Two PC kindreds were found to carry different PTVs in RHNO1. The p.Arg84* variant was not found to segregate with PC in the second affected family member tested (Figure 4), but LOH of the wild-type allele was observed in the proband tumor. The p.Arg113* variant was found to segregate with both PC-affected family members. A third family was found to carry the p.Leu16Val predicted-pathogenic missense variant, with co-segregation of the variant among PC-affected third degree relatives. This variant has also been observed in a thyroid cancer sample reported in the COSMIC database (COSM4146987). It is noteworthy that a recent study did not observe a statistically significant difference in the frequency of inactivating RHNO1 mutations (including the p.Arg84* and p.Leu16Val variants identified in the present study) among Finnish breast cancer families versus population controls.50 However, this finding does not exclude a possible role for RHNO1 in PC predisposition.
Additional genes identified in the present study have been implicated in other hereditary cancer syndromes and might also have a role in PC susceptibility. In particular, BLM has been implicated in hereditary breast cancer and BARD1 has been implicated in hereditary breast and ovarian cancers.14, 51 The Fanconi Anemia genes, FANCG and FANCL, are also noteworthy since HDR genes are of particular interest in PC.18-21 A nonsynonymous variant previously associated with Fanconi Anemia in FANCG has been previously described in a cell line derived from an early onset PC and demonstrated LOH.52 However only intronic variants were observed in a follow up study of 38 familial PC kindreds.53 Another gene of interest is POLQ, which has recently been shown to have a key role in the microhomology-mediated end joining of double stranded DNA breaks, and has been suggested as a potential target for synthetic lethality in HDR-deficient tumors.54, 55 One PTV and three predicted-pathogenic missense variants were identified in five cases from the discovery set, however the variants did not segregate with PC in the four families that were tested. An additional PTV was identified in the validation set.
Since PC is likely a heterogeneous disease,19 it is possible that causative germline mutations among genes with similar cellular roles (i.e., DNA repair) may give rise to unique PC clinical subtypes. Therefore, we evaluated whether carriers versus non-carriers of PTV germline variants in a putative DNA repair gene have different clinical outcomes. We used OS as a marker of clinical outcome since these clinical data were available and OS closely reflects cancer deaths due to the lethality of PC. Only PTVs were considered in these analyses since missense variants were primarily evaluated as a means of prioritizing candidate genes in the discovery set as pathogenicity cannot be concluded with the same degree of confidence in the absence of functional assays, despite our strict in silico selection criteria. In the discovery set, we found a significant adverse difference in OS among carriers versus non-carriers of DNA repair gene PTVs in cases with early (≤ IIB), but not advanced (≥ III), stage disease.
This adverse OS for carriers versus non-carriers was validated in an independent series of 130 PC cases.22 We first confirmed a survival disadvantage for carriers of PTVs of the 41 genes identified in the discovery set. Next, we searched for PTVs in the full panel of 513 putative DNA repair genes, since additional genes not identified in the discovery set may be contributory. The adverse OS trend with carrier status persisted when we examined all 513 genes. The carrier status association with survival that was observed for all stages in the validation, but not in the discovery, set likely reflects the sampling bias of the validation set (only 8 patients with advanced stage). Most notably, a survival disadvantage was observed in both the discovery and validation sets for early, but not advanced, stage disease. Since early stage patients are expected to have better outcomes, the adverse survival observation for these cases is intriguing and cannot be explained by an earlier stage selection bias.
Although the OS correlation with carrier status needs to be validated in a larger case series of prospectively collected cases, our observations points to a hypothesis that PC patients with inherent DNA repair deficiencies, perhaps even haploinsufficient, may have a distinct clinical outcome. This concept is not without precedent. PC tumors from patients with germline mutations in BRCA1, BRCA2 and PALB2 show unique treatment responses to DNA-damaging agents (e.g., platinums and PARP inhibitors).17-19 As well, Waddell et al.19 have recently described an “unstable” genomic subtype of PC defined by a large number of structural variation events that reflect underlying defects in DNA maintenance. This unstable PC subtype was associated with inactivation of DNA repair genes (BRCA1, BRCA2 and PALB2).
Interestingly, only half of tumors within this group were accounted for by germline or somatic mutations in these three genes, suggesting that additional DNA repair genes may be important. Our findings that PTV carrier status is associated with an adverse clinical outcome may reflect a more aggressive PC subtype and/or a PC subtype with unique therapeutic sensitivities, akin to mismatch repair gene mutation carriers in colon cancer.56 Since gemcitabine was predominantly used to treat both early and advanced PC stages in the era in which these cases were collected, our observations may be reflecting poor efficacy, or perhaps even a deleterious effect, of gemcitabine in cases with germline DNA repair mutations.
In summary, we have undertaken the first detailed characterization of germline variants in putative DNA repair genes, using NGS, in a large series of selected cases with increased risk of genetic PC predisposition. Our findings suggest that several novel DNA repair genes may have a role in hereditary PC. The heterogeneity of PC susceptibility (i.e., 12 susceptibility genes described to date) and the failure of previous linkage studies to identify major causal loci, suggest that the remaining familial aggregation of PC may be due to several genes, with each gene accounting for only a small fraction of PC susceptibility. Although our study does not provide confirmatory evidence for the candidate genes described, we have prioritized these genes based on available genetic data and propose FAN1, NEK1 and RHNO1 as the strongest candidates, providing an opportunity for further validation using additional kindreds with PC. The observed survival trend suggests that patients with mutations in DNA repair genes may have more aggressive disease or tumor subtype(s) requiring tailored treatment approaches and warrants validation in a larger series of prospectively collected cases. Such advances will help with the molecular cataloguing of PC as well as the development of gene-based early detection strategies and targeted therapies.
Supplementary Material
Highlights.
The genetic susceptibility of hereditary PC is highly heterogeneous
Rare germline protein-truncating DNA repair gene variants are common in PC
FAN1, NEK1 and RHNO1 are candidate PC susceptibility genes
Carriers of DNA repair gene mutations with early stage PC have worse survival
Acknowledgments
This work was funded by the Cancer Research Society and the National Pancreatic Cancer Canada Foundation (to G.Z.), the Research Institute McGill University Health Centre (to G.Z.), the Department of Oncology of the McGill University Health Centre (to G.Z.), the Goodman Cancer Research Centre and the McGill University Innovation Centre (to G.Z.), the National Pancreatic Cancer Canada Foundation (to S.G.), the W. Garfield Weston Foundation (to S.G.), Ontario Institute for Cancer Research (to S.G., J.Mc., L.S.) and the National Cancer Institute/National Institutes of Health (R01CA97075, to S.G., G.P.). A.L.S. is supported by a Cedars Cancer Institute Fellowship. G.Z. is a clinical research scholar of the Fonds de recherche du Québec – Santé. We would like to thank the hepato-pancreato-biliary and pathology services of the McGill University Health Centre and the University Health Network for their support with patient enrollment and biospecimen collection. We would also like to acknowledge the staff of the Biospecimen Repository of Mount Sinai Hospital for their technical support.
Footnotes
Conflict of Interest Disclosures: The authors declare no conflict of interest.
Author Contributions: Study concept, design and supervision: A.L.S., S.G., G.Z.
Acquisition of data and biospecimens: A.L.S., N.A., A.C., R.G., I.S., C.B., A.B., A.H., T.W., S.H., T.Mc.,
S.C., G.M.P., A.O., E.S., J.Mc., L.D.S., W.D.F., J.M., S.G., G.Z.
Bioinformatics support: A.L.S., N.A., A.C., M.C-S-Y., J.M.
Analysis and interpretation of data: A.L.S, N.A., A.C., W.D.F, J.M., S.G., G.Z
Manuscript writing: A.L.S., S.G., G.Z.
Final approval of manuscript: All authors
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Canadian Cancer Society's Advisory Committee on Cancer Statistics. (2015) Canadian Cancer Statistics 2015. Toronto, ON: Canadian Cancer Society; 2015. [Google Scholar]
- 2.Howlader N, Noone A, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2011. Bethesda, MD: National Institutes of Health, National Cancer Institute; 2013. [cited November 2013]. Current version available at: http://seer.cancer.gov/csr/1975_2011. [Google Scholar]
- 3.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Sukhni W, Borgida A, Rothenmund H, et al. Screening for pancreatic cancer in a high-risk cohort: an eight-year experience. Journal of Gastrointestinal Surgery. 2012;16:771–783. doi: 10.1007/s11605-011-1781-6. [DOI] [PubMed] [Google Scholar]
- 5.Langer P, Kann PH, Fendrich V, et al. Five years of prospective screening of high-risk individuals from families with familial pancreatic cancer. Gut. 2009;58:1410–1418. doi: 10.1136/gut.2008.171611. [DOI] [PubMed] [Google Scholar]
- 6.Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer--current knowledge. Nature Reviews Gastroenterology & Hepatology. 2012;9:445–453. doi: 10.1038/nrgastro.2012.111. [DOI] [PubMed] [Google Scholar]
- 7.Hruban RH, Canto MI, Goggins M, et al. Update on familial pancreatic cancer. 2010;44:293–311. doi: 10.1016/j.yasu.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nat Rev Cancer. 2012;13:66–74. doi: 10.1038/nrc3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein AP, Beaty TH, Bailey-Wilson JE, et al. Evidence for a major gene influencing risk of pancreatic cancer. Genetic epidemiology. 2002;23:133–149. doi: 10.1002/gepi.1102. [DOI] [PubMed] [Google Scholar]
- 10.Childs EJ, Mocci E, Campa D, et al. Common variation at 2p13.3, 3q29, 7p13 and 17q25.1 associated with susceptibility to pancreatic cancer. Nature Genetics. 2015;47:911–916. doi: 10.1038/ng.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217–217. doi: 10.1126/science.1171202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. 2012;2:41–46. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stadler ZK, Schrader KA, Vijai J, et al. Cancer genomics and inherited risk. Journal of Clinical Oncology. 2014;32:687–698. doi: 10.1200/JCO.2013.49.7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson ER, Doyle MA, Ryland GL, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genetics. 2012;8:e1002894. doi: 10.1371/journal.pgen.1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kiiski JI, Pelttari LM, Khan S, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:15172–15177. doi: 10.1073/pnas.1407909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubinstein WS, Weissman SM. Managing hereditary gastrointestinal cancer syndromes: the partnership between genetic counselors and gastroenterologists. Nature clinical practice Gastroenterology & hepatology. 2008;5:569–582. doi: 10.1038/ncpgasthep1235. [DOI] [PubMed] [Google Scholar]
- 17.Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. The Oncologist. 2011;16:1397–1402. doi: 10.1634/theoncologist.2011-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andrei AZ, Hall A, Smith AL, et al. Increased in vitro and in vivo sensitivity of BRCA2-associated pancreatic cancer to the poly(ADP-ribose) polymerase-1/2 inhibitor BMN 673. Cancer Letters. 2015;364:8–16. doi: 10.1016/j.canlet.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Waddell N, Pajic M, Patch AM, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borgida AE, Ashamalla S, Al-Sukhni W, et al. Management of pancreatic adenocarcinoma in Ontario, Canada: a population-based study using novel case ascertainment. Canadian Journal of Surgery. 2011;54:54–60. doi: 10.1503/cjs.026409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith AL, Bascuñana C, Hall A, et al. Establishing a clinic-based pancreatic cancer and periampullary tumour research registry in Quebec. Current Oncology. 2015;22:113–10. doi: 10.3747/co.22.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stein LD, Denroche RE, Timms L, et al. Driver Pathways and Mutational Processes in Pancreatic Ductal Adenocarcinoma. Submitted to Nat Commun. 2015 [Google Scholar]
- 23.Witkowski L, Carrot-Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nature Genetics. 2014;46:438–443. doi: 10.1038/ng.2931. [DOI] [PubMed] [Google Scholar]
- 24.Database of Single Nucleotide Polymorphisms (dbSNP) Bethesda (MD): National Center for Biotechnology Information NLoM; dbSNP Build ID: 135. [Google Scholar]
- 25.Consortium GP. Abecasis GR, Altshuler D, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Exome Variant Server NGESPE. Seattle, WA: Jan, 2015. http://evs.gs.washington.edu/EVS/ [Google Scholar]
- 27.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 28.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nature Publishing Group. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Research. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research. 2014;42:D980–5. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carbon S, Ireland A, Mungall CJ, et al. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;25:288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milanowska K, Krwawicz J, Papaj G, et al. REPAIRtoire--a database of DNA repair pathways. Nucleic Acids Research. 2011;39:D788–92. doi: 10.1093/nar/gkq1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pruitt KD, Harrow J, Harte RA, et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Research. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brune KA, Lau B, Palmisano E, et al. Importance of age of onset in pancreatic cancer kindreds. Journal of the National Cancer Institute. 2010;102:119–126. doi: 10.1093/jnci/djp466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grant RC, Al-Sukhni W, Borgida AE, et al. Exome sequencing identifies nonsegregating nonsense ATM and PALB2 variants in familial pancreatic cancer. Human genomics. 2013;7:11. doi: 10.1186/1479-7364-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell DW, Erban J, Sgroi DC, et al. Selective loss of heterozygosity in multiple breast cancers from a carrier of mutations in both BRCA1 and BRCA2. Cancer Research. 2002;62:2741–2743. [PubMed] [Google Scholar]
- 38.Sokolenko AP, Bogdanova N, Kluzniak W, et al. Double heterozygotes among breast cancer patients analyzed for BRCA1, CHEK2, ATM, NBN/NBS1, and BLM germ-line mutations. Breast Cancer Research and Treatment. 2014;145:553–562. doi: 10.1007/s10549-014-2971-1. [DOI] [PubMed] [Google Scholar]
- 39.MacKay C, Déclais AC, Lundin C, et al. Identification of KIAA1018/FAN1, a DNA Repair Nuclease Recruited to DNA Damage by Monoubiquitinated FANCD2. Cell. 2010;142:65–76. doi: 10.1016/j.cell.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trujillo JP, Mina LB, Pujol R, et al. On the role of FAN1 in Fanconi anemia. Blood. 2012;120:86–89. doi: 10.1182/blood-2012-04-420604. [DOI] [PubMed] [Google Scholar]
- 41.Seguí N, Mina LB, Lazaro C, et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology. 2015;149:563–566. doi: 10.1053/j.gastro.2015.05.056. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Chen CF, Chiang HC, et al. Mutation of NIMA-related kinase 1 (NEK1) leads to chromosome instability. Molecular cancer. 2011;10:5. doi: 10.1186/1476-4598-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Exome Aggregation Consortium (ExAC) C, MA. 2015 Jan; URL: http://exac.broadinstitute.org.
- 44.Eberle MA, Pfützer R, Pogue-Geile KL, et al. A new susceptibility locus for autosomal dominant pancreatic cancer maps to chromosome 4q32-34. 2002;70:1044–1048. doi: 10.1086/339692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pogue-Geile KL, Chen R, Bronner MP, et al. Palladin mutation causes familial pancreatic cancer and suggests a new cancer mechanism. PLoS Medicine. 2006;3:e516. doi: 10.1371/journal.pmed.0030516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salaria SN, Illei P, Sharma R, et al. Palladin is overexpressed in the non-neoplastic stroma of infiltrating ductal adenocarcinomas of the pancreas, but is only rarely overexpressed in neoplastic cells. 2007;6:324–328. doi: 10.4161/cbt.6.3.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zogopoulos G, Rothenmund H, Eppel A, et al. The P239S palladin variant does not account for a significant fraction of hereditary or early onset pancreas cancer. Human Genetics. 2007;121:635–637. doi: 10.1007/s00439-007-0361-z. [DOI] [PubMed] [Google Scholar]
- 48.Klein AP, Borges M, Griffith M, et al. Absence of deleterious palladin mutations in patients with familial pancreatic cancer. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2009;18:1328–1330. doi: 10.1158/1055-9965.EPI-09-0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cotta-Ramusino C, McDonald ER, Hurov K, et al. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science. 2011;332:1313–1317. doi: 10.1126/science.1203430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heikkinen T, Khan S, Huovari E, et al. Evaluation of the RHINO gene for breast cancer predisposition in Finnish breast cancer families. Breast Cancer Res Treat. 2014;144:437–441. doi: 10.1007/s10549-014-2884-z. [DOI] [PubMed] [Google Scholar]
- 51.Ratajska M, Antoszewska E, Piskorz A, et al. Cancer predisposing BARD 1 mutations in breast-ovarian cancer families. Breast Cancer Research and Treatment. 2011;131:89–97. doi: 10.1007/s10549-011-1403-8. [DOI] [PubMed] [Google Scholar]
- 52.van der Heijden MS, Yeo CJ, Hruban RH, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Research. 2003;63:2585–2588. [PubMed] [Google Scholar]
- 53.Rogers CD, van der Heij den MS, Brune K, et al. The genetics of FANCC and FANCG in familial pancreatic cancer. 2004;3:167–169. doi: 10.4161/cbt.3.2.609. [DOI] [PubMed] [Google Scholar]
- 54.Ceccaldi R, Liu JC, Amunugama R, et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015;518:258–262. doi: 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mateos-Gomez PA, Gong F, Nair N, et al. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nature. 2015;518:254–257. doi: 10.1038/nature14157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. New England Journal of Medicine. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.