

Abstract
Background
Chronic kidney disease (CKD) is a leading cause of end stage renal disease (ESRD) and cardiovascular morbidity and mortality worldwide, resulting in a growing social and economic burden. The prevalence and burden of CKD is anticipated to further increase over the next decades as a result of aging.
Main body of abstract
In the pathogenesis of CKD, irrespective of the etiology, resident fibroblasts are key players and have been demonstrated to play crucial roles for disease initiation and progression. In response to injury, resident fibroblasts transdifferentiate into myofibroblasts that express alpha smooth muscle actin (αSMA) and have an increased capacity to produce large amounts of extracellular matrix (ECM) proteins, leading to renal fibrosis. In addition to this fundamental role of fibroblasts as drivers for renal fibrosis, growing amounts of evidence have shown that resident fibroblasts are also actively involved in initiating and promoting inflammation during kidney injury. During the myofibroblastic transition described above, resident fibroblasts activate NF-κB signaling and produce pro-inflammatory cytokines and chemokines, promoting inflammation. Furthermore, under aging milieu, resident fibroblasts transdifferentiate into several distinct phenotypic fibroblasts, including CXCL13/CCL19-producing fibroblasts, retinoic acid-producing fibroblasts, and follicular dendritic cells, in response to injury and orchestrate tertiary lymphoid tissue (TLT) formation, which results in uncontrolled aberrant inflammation and retards tissue repair. Anti-inflammatory agents can improve myofibroblastic transdifferentiation and abolish TLT formation, suggesting that targeting these inflammatory fibroblasts can potentially ameliorate kidney disease.
Short conclusion
Beyond its conventional role as an executor of fibrosis, resident fibroblasts display more pro-inflammatory phenotypes and contribute actively to driving inflammation during kidney injury.
Keywords: Fibroblast, Inflammation, Myofibroblast, Chronic kidney disease, Erythropoietin, Heterogeneity, Tertiary lymphoid tissue, CXCL13
Background
Fibroblasts reside in virtually all tissues in our body and provide three-dimensional architecture and mechanical strength to the tissues. Emerging evidence suggests that they also have tissue-specific physiologic functions and participate actively in pathogenesis during injury. In the kidney, resident fibroblasts produce erythropoietin (EPO) in response to hypoxic insults to maintain homeostasis under physiologic condition, whereas, under pathologic conditions, resident fibroblasts transdifferentiate into myofibroblasts, which execute renal fibrosis by producing large amounts of extracellular matrix proteins, at the cost of EPO production [1, 2]. Recently, the role and phenotype of resident fibroblasts in the kidney during injury have been demonstrated to be more diverse and crucial for disease initiation and progression. Under the aging milieu, for example, resident fibroblasts further gain a variety of distinct phenotypes in response to injury and orchestrate tertiary lymphoid tissue formation, which results in uncontrolled inflammation and retards tissue repair [3]. In this review, we provide the current state of knowledge of the renal fibroblasts as a driver of fibrosis and inflammation, and consider a novel therapeutic strategy to treat patients with kidney disease.
Resident fibroblasts as sentinels in the kidney
The kidney plays a central role in body fluid homeostasis and metabolic waste elimination. Each human kidney is composed of about 1 million nephrons, which are functional units of the kidney that comprise the glomerulus and the tubules. The glomerulus is a capillary loop that is specialized for plasma filtration. The glomeruli receive blood supply from the renal artery, and the glomerular filtrate subsequently travels through renal tubules, where metabolic exchange, and reabsorption and secretion occur. Resident fibroblasts are spindle-shaped mesenchymal cells that reside in the renal interstitium [4], which is the extracellular compartment between tubules and peritubular capillaries [5]. Several pathological studies have shown that the magnitude of renal impairment correlates better with interstitial changes than the glomerular changes in most forms of chronic kidney disease (CKD), suggesting that renal function is critically dependent on the environment in this compartment.
The kidney interstitium contains two cellular components: resident fibroblasts and resident renal mononuclear phagocytes (rMoPh) [6, 7]. Although these two types of cells reside in virtually all tissues, they are versatile cell types with strong organ-specific modifications. The kidney is continually exposed to various kinds of endogenous and exogenous substances, which must be monitored and possibly eliminated, and most of the reabsorbed substances have to traverse the interstitium before entering the capillaries. Fibroblasts and rMoPh are strategically positioned at the interstitium to sense these circulating substances and environmental changes [5]. Indeed, with the progression of kidney disease, many kinds of uremic toxins have accumulated in the body and have various effects on these renal cells. For instance, indoxyl sulfate (IS), a typical uremic toxin derived from indole, suppresses EPO production in resident fibroblasts in the kidney [8], which may partly explain the relative deficiency of EPO production in CKD patients described in the next chapter.
The readiness to respond to diverse environmental cues has been well described for rMoPh, and these cells have been considered to be sentinels in the kidney [6]. However, it has recently been demonstrated that fibroblasts also express most immune receptors, including pattern recognition receptors such as Toll-like receptors (TLRs), and that they are also highly sensitive to local tissue injury. Leaf et al. demonstrated that, although various types of cell are likely to become activated through TLRs, fibroblasts respond to damage-associated molecular patterns (DAMPs) more sensitively than other cell types including epithelial cells, endothelial cells, and even monocyte-derived macrophage in the context of sterile inflammation, and they produce higher amount of pro-inflammatory cytokine, participating actively in the initiation of renal inflammation [9]. Macrophages, on the other hand, have a higher sensitivity to pathogen ligands, suggesting that these two cell types may collaborate together and serve as a sophisticated network that senses both intrinsic and extrinsic substances under physiologic and pathologic conditions.
Pericytes, which are defined as mesenchymal cells wrapping around the microvessels, also reside in interstitium and are positive for CD73 and PDGFRβ, both of which are also utilized as markers for resident fibroblasts [10]. Overlapping definitions of resident fibroblasts and pericytes have generated confusion and controversy, although it is becoming increasingly clear that they are overlapping populations in the kidney [11]. Recent studies of the lineage relationships demonstrated that almost all fibroblasts in the renal cortex and outer medulla, including EPO-producing cells, are derived from cells that are lineage-labeled with myelin protein zero (P0)-Cre [12], which labels migrating neural crest cells and neural crest-derived Schwann cells [13], whereas almost all pericytes are derived from Foxd1-Cre lineage-labeled stromal cells [14]. P0-Cre lineage-labeled cells transiently express FoxD1 during development, whereas FoxD1 is expressed in the migrating neural crest, indicating that these two populations are overlapping [11].
Role of resident fibroblasts during kidney injury
Fibrosis is a common pathologic feature in CKD patients, and myofibroblasts are major drivers of fibrosis. Myofibroblasts are not present under physiologic conditions, but emerge de novo in injured tissues. The origin of myofibroblasts has been controversial for a long time, and various precursor cells of myofibroblasts in fibrotic kidneys have been reported [1]. Over the last 5 years, comprehensive cell fate mapping experiments using various Cre mouse lines have been conducted by several groups and the origin of myofibroblasts has been reconsidered. We also demonstrated that P0-Cre lineage-labeled resident fibroblasts transdifferentiate αSMA-positive myofibroblasts in response to kidney injury [12]. Together with the results from other groups, it is currently believed that resident fibroblasts or pericytes seem to be the most important precursor of myofibroblasts, which is consistent with recent studies in liver [15], lung [16, 17], and skin fibrosis [18, 19], all of which concluded that myofibroblasts are derived from resident fibroblasts. Interestingly, Kramman et al. recently identified the myofibroblast progenitor, which represents a small fraction of renal pericytes in the healthy kidney, and they were lineage-labeled with Gli1 [20]. Gli1-positive pericytes fulfill the criteria of mesenchymal stem cells, having tri-lineage differentiation potential and colony-forming capability in vitro. The blood vessel wall was shown to be a niche for mesenchymal stem cells in multiple human organs including the skeletal muscle, pancreas, adipose tissue, placenta, and kidney [21, 22].
In CKD patients, fibrosis progresses for decades. This clinical course suggests that epigenetic changes, which can persist long after the removal of initial trigger, have been involved and played an important role in this process. A recent genome-wide methylation scan of fibroblasts in the kidney identified epigenetic silencing of RASAL1, a suppressor of the Ras oncoprotein, as the cause of spontaneous proliferation of fibroblasts [23], providing a new molecular explanation for a sustained activation of fibroblasts in the injured kidneys.
In parallel with renal fibrosis, as the severity of kidney disease progresses, renal anemia increases in prevalence. Renal anemia is driven predominantly by a relative deficiency in the production of EPO, a principal regulatory hormone of red blood cell production [2], which is produced by renal resident fibroblasts in response to hypoxia [24]. We have previously demonstrated that, during kidney injury, EPO-producing cells transdifferentiate into myofibroblasts, same as other resident fibroblasts in the kidney, at the cost of EPO production [12]. EPO production is mainly regulated by hypoxia-inducible factors (HIFs) in healthy kidney. However, in injured kidneys, despite their hypoxic conditions, most of the HIF target gene expression is insufficient to counteract hypoxia [25]. Notably, we also showed that these transdifferentiated myofibroblasts regain their EPO production ability after the induction of severe anemia or the administration of neuroprotective agents such as neurotrophin and selective estrogen receptor modulator (SERM) [12]. These results indicate that resident fibroblasts possess functional plasticity and myofibroblasts still have the potential to produce EPO in response to hypoxic insults, which is consistent with epidemiological evidence indicating the presence of hypoxia-driven EPO regulation even in the patients with ESRD who require hemodialysis [26].
What triggers this phenotypic change in fibroblast in the kidney? In the previous study, we have demonstrated that proximal tubule injury alone can drive this phenotypic change and lead to renal fibrosis and deficiency in EPO production [27]. This phenomenon has been supported by the evidences from other groups, which demonstrate that TGF-β derived from injured tubules promotes the transdifferentiation from fibroblast into myofibroblast [28]. In addition to this, the pathways regulating this phenotypic change, including PDGFR pathway and hedgehog pathway, have been already identified and considered as targets of novel therapeutic approaches [11]. Interestingly, during this phenotypic transition, fibroblasts also become both extracellular matrix (ECM)-producing cells and inflammatory effector cells [29, 30]. In response to injury, renal fibroblasts have been shown to activate NF-κB signaling, which leads to the production of pro-inflammatory cytokines and chemokines. The pro-inflammatory cytokines such as IL-1 and TNFα activate GATA-2 and NF-κB, both of which inhibit EPO transcription by binding the EPO promoter [29, 31], leading to relative EPO deficiency in CKD patients. Together with the findings that anti-inflammatory agents such as dexamethasone can restore the myofibroblast phenotype [12, 29], the inflammatory phenotypes of fibroblasts can be a promising therapeutic target and require more characterization in future studies.
In addition, various types of cells in the kidney, including fibroblasts, produce prostaglandins (PGs), which have also been recognized as a mediator of inflammatory responses [32]. Growing amounts of evidence have shown that PGs are involved in tissue fibrosis and inflammation. PGE2 is the most abundant PG in the kidney, and it plays a suppressive role in renal fibrosis via its receptor EP4 [33], although specific EP4 depletion in podocytes, which are a critical component of the filtration barrier in the glomerulus, results in milder glomerular injury [34]. These potential confounding features suggest that the PG cellular source and function are highly variable depending on the conditions and the cell type. PG signaling is considered to be a promising therapeutic target because PGs have been shown to amplify cytokine signaling and induce chemokine expression in other organs [32].
Heterogeneity of resident fibroblasts involved in tertiary lymphoid tissue formation in aged injured kidney
Several phenotypically novel heterogeneous fibroblasts in the injured kidney have been identified recently, and they were characterized in both the rodent and human kidneys, which are involved in tertiary lymphoid tissue (TLT) formation [3]. TLTs are inducible ectopic lymphoid tissues that are composed of a hematopoietic compartment, which comprises mostly T cells and B cells, and stromal components, which include fibroblasts in particular (Fig. 1) [35]. TLTs can propagate local antigen-specific immune responses within non-lymphoid tissues, although their roles are context dependent and can be either beneficial or detrimental [36]. In chronic inflammatory disorders, for instance, TLTs are generally considered to be perpetuators of aberrant immune responses and detrimental to the host [37], whereas, during infections, TLTs generate robust immune responses to pathogens and play protective roles for the host [38]. Besides anatomical and functional similarities, TLT and secondary lymphoid organs, such as lymph nodes, both depend on related mechanisms and molecules for their development [39, 40]. In lymph nodes, homeostatic chemokines, including CXCL13, CCL19, and CCL21, play essential roles in their development, maturation, and homeostasis [39, 40]. The homeostatic chemokine is a powerful driving force for recruiting lymphocytes and is also sufficient to drive TLTs in non-lymphoid tissue, as transgenic expression of homeostatic chemokines in non-lymphoid organs induce the development of functional TLTs [41, 42].
Fig. 1.
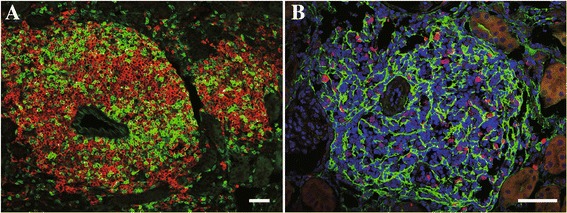
Tertiary lymphoid tissues in aged injured mouse kidney. Tertiary lymphoid tissues are mainly composed of T cells and B cells, some of which are proliferating. p75NTR-positive fibroblasts extend their projections and form a structural backbone within TLTs. a green: CD3ε, red: B220. b green: p75NTR, red: Ki67. Scale bar (a, b) 50 μm
Recent studies have increasingly highlighted the potential roles for TLTs in regulating local immune responses in various pathological conditions. We showed that aged mice, but not young mice, developed multiple TLTs in the kidney after acute kidney injury (AKI) (Fig. 2). This unique response program in aged injured kidneys might explain why aged kidneys fail to repair themselves after kidney injury and progress to ESRD [43], since aberrant chronic inflammation hinders normal tissue repair and results in worse remodeling and dysfunction [44, 45]. Administration of anti-CD4 monoclonal antibody and dexamethasone abolished TLT formation and improved renal outcomes. Thus, the molecular mechanisms that govern the development and maintenance of TLT identity are of great interest, having implications for the prevention of TLT formation and the subsequent aberrant inflammation [46].
Fig. 2.
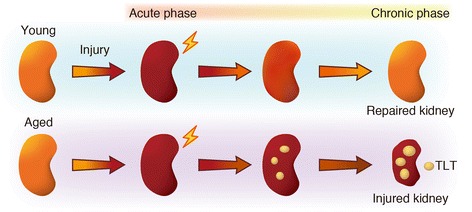
The distinct injury response in young and aged mice. Aged mice, but not young mice, developed multiple tertiary lymphoid tissues (TLTs) in the kidney after acute kidney injury (AKI). TLTs sustain and amplify inflammation and retard regeneration, resulting a poor renal outcome in aged mice
In aged injured kidneys, resident fibroblasts transdifferentiate into myofibroblasts and also into several distinct phenotypic fibroblasts, which are involved in TLT formation (Fig. 3). After kidney injury, some resident fibroblasts acquire the ability to produce retinoic acid, which induces the neural crest marker p75NTR. Some p75NTR-positive fibroblasts in aged injured kidneys produce CXCL13 and CCL19, resulting in TLT formation [3]. Additionally, in the later phase of TLT formation, some of the p75NTR-positive fibroblasts appear to lose their p75NTR expression and mature into follicular dendritic cells (FDCs). FDCs express high amounts of CD21, complement receptors-2, and CXCL13, resulting in forming B cell areas and supporting germinal center response [47]. Lineage tracing demonstrated that P0-Cre lineage-labeled resident fibroblasts diversified into fibroblasts with these several distinct phenotypes essential for TLT formation (Fig. 3). These findings in renal TLT are consistent with the results of lineage-tracing studies in stromal cells in secondary lymphoid organs [48, 49], indicating that FDCs in the spleen are lineage-labeled with PDGFRβ-Cre, whereas FDCs in lymph nodes are lineage-labeled with Wnt1-Cre, which is another Cre line which labels neural crest-derived cells. Collectively, our results confirm and extend the concept that resident fibroblasts in the kidney exhibit striking plasticity and functional diversity depending on their residing microenvironments. One important remaining question to be addressed is “why the renal environment is prone to TLT formation with aging.” One possibility is the contribution of hematopoietic cell aging, especially CD4 positive T cells, because depletion of CD4 positive cells abolished TLTs [3]. Because of thymic involution, T cells undergo a global phenotype shift from naïve to memory T cells with aging, and unique age-dependent memory CD4 positive T cell subpopulation has been identified [50]. Another possibility is the contribution of aging in stromal cells, especially resident fibroblasts in the kidney. Further studies will be required to determine which cell aging is critical for the TLT formation.
Fig. 3.
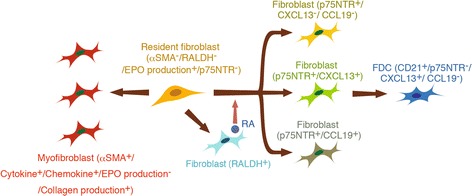
Fibroblasts have two jobs during CKD: fibrosis and inflammation. Resident fibroblasts critically contribute to fibrosis and the persistence of inflammation in the injured kidney [3]. Upon injury, resident fibroblasts transdifferentiate into myofibroblasts, which produce a large amount of ECM protein and pro-inflammatory cytokines/chemokines, at the cost of EPO production. Moreover, in the aging milieu, resident fibroblasts also transdifferentiate into several distinct phenotypic fibroblasts, which orchestrate TLT formation. In response to injury, resident fibroblasts differentiate into RALDH-positive fibroblasts, which induce transdifferentiation of other fibroblasts into p75NTR-positive fibroblasts with three phenotypes, which include CXCL13- and CCL19-producing fibroblasts. In the later phase of TLT formation, some of these p75NTR-positive fibroblasts lose this expression and mature into CD21/CXCL13-positive FDCs
The developmental mechanism of age-dependent TLTs in the kidney and inducible bronchus-associated lymphoid tissue (iBALT) in the lung is similar in that both TLTs are driven by CXCL13 and CCL19 [3, 51]. Although lymphoid tissue is normally absent in the lung, iBALT develops following to various kinds of infection and inflammatory diseases [52]. iBALT has separated T and B cell areas, some of which contain FDCs and germinal centers, and generate immune responses. Although various types of cells, such as monocyte-lineage cells and fibroblasts, have been reported as to be a source of CXCL13 in the lung, bone marrow chimera experiments have demonstrated that the majority of CXCL13 producing cells are non-hematopoietic cells in iBALT [53]. Rangel et al. have showed that wild type mice reconstituted with CXCL13-deficient bone marrow exhibited nearly identical lung expression of CXCL13 compared with wild type controls [53]. Furthermore, the same group has also demonstrated that interleukin-17 produced by CD4-positive T cells triggers the expression of CXCL13 and CCL19, but not CCL21, in pulmonary fibroblasts, which results in iBALT formation [51]. Altogether, these results suggested that resident fibroblasts have the potential to become homeostatic chemokine-producing cells in various organs. Though monocyte lineage cells have also been reported as CXCL13-producing cells in murine lupus models in the kidney [54, 55], the expression of CXCL13 in fibroblasts was not examined in these studies, and the relative contribution of hematopoietic cells and non-hematopoietic cells to overall CXCL13 expression in the kidney has yet to be determined in this model. Further studies are required to determine the main cellular source of renal CXCL13 in this model.
It is difficult to determine whether TLT is beneficial, harmful, or neutral for the host. This is partly because it is technically challenging to deplete TLTs specifically at any time without affecting the immune system systemically. Another way to determine whether TLTs play pathogenic roles is to determine whether TLTs produce autoantibodies. Given that TLTs lead to the production of tissue-specific autoantibodies, targeting TLT formation could be beneficial. Indeed, this idea has been already tested. Lehmann-Horn et al. demonstrated that in experimental autoimmune encephalomyelitis (EAE), autoantibodies with modified affinity for myelin self-antigens are generated within TLT in the meninges of central nerve system [56], suggesting the pathological roles of TLT in this context. In addition to the conventional roles of TLT as an amplifier of inflammation, recent studies have shown that, in some pathological conditions, TLTs can function as niches for tumor progenitor cells [57] and pathogenic memory T cells [58], which might represent new therapeutic targets for cancer and chronic inflammatory diseases. The role of TLTs may be variable and be influenced by the stage of the disease, site of formation, and various environmental factors, all of which determine the impact of TLTs on disease progression. Further studies are required to determine the precise roles of TLTs in various pathologic conditions.
Conclusions
Dysfunction of resident fibroblasts leads to a series of clinically relevant pathological conditions that are common in CKD, indicating their importance in maintaining homeostasis under normal conditions. Beyond its conventional role as an executor of fibrosis, resident fibroblasts display more pro-inflammatory phenotypes and contribute actively to driving inflammation during kidney injury (Fig. 3), and intervention with anti-inflammatory agents has the potential to ameliorate kidney injury. Further studies are required to create novel therapeutic approaches, which may emerge as a consequence of a better understanding of the behavior of fibroblast under physiologic and pathologic conditions.
Acknowledgements
The authors would like to express sincere appreciation to Prof. Hideyuki Okano and Prof. Masataka Kuwano for giving us the opportunity to write this review article, to Dr. Hirosuke Nakata for drawing the illustrations, and to all lab members and collaborators.
Funding
YS receives funding from Mitsubishi Tanabe Pharma Corporation. MY receives research grants from Astellas, Chugai, Daiichi Sankyo, Fujiyakuhin, Kyowa Hakko Kirin, Mitsubishi Tanabe Pharma Corporation, MSD, Nippon Boehringer Ingelheim, and Torii.
Availability of data and materials
Not applicable. This review article included only published data.
Authors’ contributions
YS drafted the manuscript, and MY revised it. Both authors read and approved the final manuscript.
Competing interests
YS is employed by the TMK project. MY is on the advisory board of Astellas.
Consent for publication
Not applicable.
Ethics approval
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- AKI
Acute kidney injury
- CKD
Chronic kidney disease
- DAMPs
Damage-associated molecular patterns
- ECM
Extracellular matrix
- EPO
Erythropoietin
- ESRD
End stage renal disease
- FDC
Follicular dendritic cell
- iBALT
Inducible bronchus-associated lymphoid tissue
- P0
Myelin protein zero
- PG
Prostaglandin
- rMoPh
Resident renal mononuclear phagocytes
- TLR
Toll-like receptor
- TLT
Tertiary lymphoid tissue
- αSMA
Alpha smooth muscle actin
Contributor Information
Yuki Sato, Email: yukisato@kuhp.kyoto-u.ac.jp.
Motoko Yanagita, Phone: +81-75-751-3860, Email: motoy@kuhp.kyoto-u.ac.jp.
References
- 1.Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015;87(2):297–307. doi: 10.1038/ki.2014.287. [DOI] [PubMed] [Google Scholar]
- 2.Sato Y, Yanagita M. Renal anemia: from incurable to curable. Am J Physiol Renal Physiol. 2013;305(9):F1239–48. doi: 10.1152/ajprenal.00233.2013. [DOI] [PubMed] [Google Scholar]
- 3.Sato Y, Mii A, Hamazaki Y, Fujita H, Nakata H, Masuda K, Nishiyama S, Shibuya S, Haga H, Ogawa O, et al. Heterogeneous fibroblasts underlie age-dependent tertiary lymphoid tissues in the kidney. JCI Insight. 2016;1(11):e87680. doi: 10.1172/jci.insight.87680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boor P, Floege J. The renal (myo-)fibroblast: a heterogeneous group of cells. Nephrol Dial Transplant. 2012;27(8):3027–36. doi: 10.1093/ndt/gfs296. [DOI] [PubMed] [Google Scholar]
- 5.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008;130(2):247–62. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol. 2012;23(2):194–203. doi: 10.1681/ASN.2011070680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weisheit CK, Engel DR, Kurts C. Dendritic cells and macrophages: sentinels in the kidney. Clin J Am Soc Nephrol. 2015;10(10):1841–51. doi: 10.2215/CJN.07100714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang CK, Tanaka T, Inagi R, Fujita T, Nangaku M. Indoxyl sulfate, a representative uremic toxin, suppresses erythropoietin production in a HIF-dependent manner. Lab Invest. 2011;91(11):1564–71. doi: 10.1038/labinvest.2011.114. [DOI] [PubMed] [Google Scholar]
- 9.Leaf IA, Nakagawa S, Johnson BG, Cha JJ, Mittelsteadt K, Guckian KM, Gomez IG, Altemeier WA, Duffield JS. Pericyte MyD88 and IRAK4 control inflammatory and fibrotic responses to tissue injury. J Clin Invest. 2017;127(1):321–34. doi: 10.1172/JCI87532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 11.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124(6):2299–306. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asada N, Takase M, Nakamura J, Oguchi A, Asada M, Suzuki N, Yamamura K, Nagoshi N, Shibata S, Rao TN, et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest. 2011;121(10):3981–90. doi: 10.1172/JCI57301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S, Yamamura K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. 1999;212(1):191–203. doi: 10.1006/dbio.1999.9323. [DOI] [PubMed] [Google Scholar]
- 14.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, Duffield JS. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176(1):85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, Liu X, Xu J, Wang P, Paik YH, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci U S A. 2014;111(32):E3297–305. doi: 10.1073/pnas.1400062111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, Gharib SA, Schnapp LM, Duffield JS. Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2013;188(7):820–30. doi: 10.1164/rccm.201212-2297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie T, Liang J, Liu N, Huan C, Zhang Y, Liu W, Kumar M, Xiao R, D'Armiento J, Metzger D, et al. Transcription factor TBX4 regulates myofibroblast accumulation and lung fibrosis. J Clin Invest. 2016;126(8):3063–79. doi: 10.1172/JCI85328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. 2012;18(8):1262–70. doi: 10.1038/nm.2848. [DOI] [PubMed] [Google Scholar]
- 19.Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP, et al. Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science. 2015;348(6232):aaa2151. doi: 10.1126/science.aaa2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell. 2015;16(1):51–66. doi: 10.1016/j.stem.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crisan M, Yap S, Casteilla L, Chen CW, Corselli M, Park TS, Andriolo G, Sun B, Zheng B, Zhang L, et al. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–13. doi: 10.1016/j.stem.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Stefanska A, Kenyon C, Christian HC, Buckley C, Shaw I, Mullins JJ, Peault B. Human kidney pericytes produce renin. Kidney Int. 2016;90(6):1251–61. doi: 10.1016/j.kint.2016.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bechtel W, McGoohan S, Zeisberg EM, Muller GA, Kalbacher H, Salant DJ, Muller CA, Kalluri R, Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16(5):544–50. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obara N, Suzuki N, Kim K, Nagasawa T, Imagawa S, Yamamoto M. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood. 2008;111(10):5223–32. doi: 10.1182/blood-2007-10-115857. [DOI] [PubMed] [Google Scholar]
- 25.Souma T, Yamazaki S, Moriguchi T, Suzuki N, Hirano I, Pan X, Minegishi N, Abe M, Kiyomoto H, Ito S, et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. J Am Soc Nephrol. 2013;24(10):1599–616. doi: 10.1681/ASN.2013010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brookhart MA, Schneeweiss S, Avorn J, Bradbury BD, Rothman KJ, Fischer M, Mehta J, Winkelmayer WC. The effect of altitude on dosing and response to erythropoietin in ESRD. J Am Soc Nephrol. 2008;19(7):1389–95. doi: 10.1681/ASN.2007111181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, Nameta M, Yamamoto T, Economides AN, Kohno K, et al. Severity and frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol. 2016;27(8):2393–406. doi: 10.1681/ASN.2015060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16(5):535–43. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Souma T, Yamazaki S, Moriguchi T, Suzuki N, Hirano I, Pan X, Minegishi N, Abe M, Kiyomoto H, Ito S, et al. Plasticity of renal erythropoietin-producing cells governs fibrosis. Journal of the American Society of Nephrology : JASN. 2013;24(10):1599–616. doi: 10.1681/ASN.2013010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campanholle G, Ligresti G, Gharib SA, Duffield JS. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol. 2013;304(7):C591–603. doi: 10.1152/ajpcell.00414.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka T, Nangaku M. Recent advances and clinical application of erythropoietin and erythropoiesis-stimulating agents. Exp Cell Res. 2012;318(9):1068–73. doi: 10.1016/j.yexcr.2012.02.035. [DOI] [PubMed] [Google Scholar]
- 32.Aoki T, Narumiya S. Prostaglandins and chronic inflammation. Trends Pharmacol Sci. 2012;33(6):304–11. doi: 10.1016/j.tips.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 33.Nakagawa N, Yuhki K, Kawabe J, Fujino T, Takahata O, Kabara M, Abe K, Kojima F, Kashiwagi H, Hasebe N, et al. The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int. 2012;82(2):158–71. doi: 10.1038/ki.2012.115. [DOI] [PubMed] [Google Scholar]
- 34.Stitt-Cavanagh EM, Faour WH, Takami K, Carter A, Vanderhyden B, Guan Y, Schneider A, Breyer MD, Kennedy CR. A maladaptive role for EP4 receptors in podocytes. J Am Soc Nephrol. 2010;21(10):1678–90. doi: 10.1681/ASN.2009121234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. 2012;33(6):297–305. doi: 10.1016/j.it.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6(3):205–17. doi: 10.1038/nri1786. [DOI] [PubMed] [Google Scholar]
- 37.Wu Q, Salomon B, Chen M, Wang Y, Hoffman LM, Bluestone JA, Fu YX. Reversal of spontaneous autoimmune insulitis in nonobese diabetic mice by soluble lymphotoxin receptor. J Exp Med. 2001;193(11):1327–32. doi: 10.1084/jem.193.11.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, Woodland DL, Lund FE, Randall TD. Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med. 2004;10(9):927–34. doi: 10.1038/nm1091. [DOI] [PubMed] [Google Scholar]
- 39.Drayton DL, Liao S, Mounzer RH, Ruddle NH. Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol. 2006;7(4):344–53. doi: 10.1038/ni1330. [DOI] [PubMed] [Google Scholar]
- 40.Jones GW, Hill DG, Jones SA. Understanding immune cells in tertiary lymphoid organ development: it is all starting to come together. Front Immunol. 2016;7(401). doi:10.3389/fimmu.2016.00401. [DOI] [PMC free article] [PubMed]
- 41.Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG. BLC expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000;12(5):471–81. doi: 10.1016/S1074-7613(00)80199-5. [DOI] [PubMed] [Google Scholar]
- 42.Luther SA, Bidgol A, Hargreaves DC, Schmidt A, Xu Y, Paniyadi J, Matloubian M, Cyster JG. Differing activities of homeostatic chemokines CCL19, CCL21, and CXCL12 in lymphocyte and dendritic cell recruitment and lymphoid neogenesis. J Immunol. 2002;169(1):424–33. doi: 10.4049/jimmunol.169.1.424. [DOI] [PubMed] [Google Scholar]
- 43.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol. 2009;20(1):223–8. doi: 10.1681/ASN.2007080837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454(7203):428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 45.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110(1):159–73. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barone F, Gardner DH, Nayar S, Steinthal N, Buckley CD, Luther SA. Stromal fibroblasts in tertiary lymphoid structures: a novel target in chronic inflammation. Front Immunol. 2016;7(477). doi:10.3389/fimmu.2016.00477. [DOI] [PMC free article] [PubMed]
- 47.Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nat Rev Immunol. 2014;14(7):495–504. doi: 10.1038/nri3689. [DOI] [PubMed] [Google Scholar]
- 48.Krautler NJ, Kana V, Kranich J, Tian Y, Perera D, Lemm D, Schwarz P, Armulik A, Browning JL, Tallquist M, et al. Follicular dendritic cells emerge from ubiquitous perivascular precursors. Cell. 2012;150(1):194–206. doi: 10.1016/j.cell.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jarjour M, Jorquera A, Mondor I, Wienert S, Narang P, Coles MC, Klauschen F, Bajenoff M. Fate mapping reveals origin and dynamics of lymph node follicular dendritic cells. J Exp Med. 2014;211(6):1109–22. doi: 10.1084/jem.20132409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shimatani K, Nakashima Y, Hattori M, Hamazaki Y, Minato N. PD-1+ memory phenotype CD4+ T cells expressing C/EBPalpha underlie T cell immunodepression in senescence and leukemia. Proc Natl Acad Sci U S A. 2009;106(37):15807–12. doi: 10.1073/pnas.0908805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hwang JY, Randall TD, Silva-Sanchez A. Inducible bronchus-associated lymphoid tissue: taming inflammation in the lung. Front Immunol. 2016;7(258). doi:10.3389/fimmu.2016.00258. [DOI] [PMC free article] [PubMed]
- 52.Rangel-Moreno J, Carragher DM, de la Luz Garcia-Hernandez M, Hwang JY, Kusser K, Hartson L, Kolls JK, Khader SA, Randall TD. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol. 2011;12(7):639–46. doi: 10.1038/ni.2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rangel-Moreno J, Moyron-Quiroz JE, Hartson L, Kusser K, Randall TD. Pulmonary expression of CXC chemokine ligand 13, CC chemokine ligand 19, and CC chemokine ligand 21 is essential for local immunity to influenza. Proc Natl Acad Sci U S A. 2007;104(25):10577–82. doi: 10.1073/pnas.0700591104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishikawa S, Sato T, Abe M, Nagai S, Onai N, Yoneyama H, Zhang Y, Suzuki T, Hashimoto S, Shirai T, et al. Aberrant high expression of B lymphocyte chemokine (BLC/CXCL13) by C11b+ CD11c+ dendritic cells in murine lupus and preferential chemotaxis of B1 cells towards BLC. J Exp Med. 2001;193(12):1393–402. doi: 10.1084/jem.193.12.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, Pfeilschifter J, Young MF, Schaefer RM, Schaefer L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J Clin Invest. 2010;120(12):4251–72. doi: 10.1172/JCI42213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lehmann-Horn K, Wang SZ, Sagan SA, Zamvil SS, von Budingen HC. B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight. 2016;1(20):e87234. doi: 10.1172/jci.insight.87234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finkin S, Yuan D, Stein I, Taniguchi K, Weber A, Unger K, Browning JL, Goossens N, Nakagawa S, Gunasekaran G, et al. Ectopic lymphoid structures function as microniches for tumor progenitor cells in hepatocellular carcinoma. Nat Immunol. 2015;16(12):1235–44. doi: 10.1038/ni.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shinoda K, Hirahara K, Iinuma T, Ichikawa T, Suzuki AS, Sugaya K, Tumes DJ, Yamamoto H, Hara T, Tani-Ichi S, et al. Thy1 + IL-7+ lymphatic endothelial cells in iBALT provide a survival niche for memory T-helper cells in allergic airway inflammation. Proc Natl Acad Sci U S A. 2016;113(20):E2842–51. doi: 10.1073/pnas.1512600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable. This review article included only published data.