

Abstract
NMR of a uniformly 13C-labeled carbohydrate was used to elucidate the atomic details of a sugar-protein complex. The structure of the 13C-labeled Manα(1–2)Manα(1–2)ManαOMe trisaccharide ligand when bound to cyanovirin-N was characterized and revealed that in the complex the glycosidic linkage torsion angles between the two reducing-end mannoses are different from the free trisaccharide. Distances within the carbohydrate were employed for conformational analysis, and NOE-based distance mapping between sugar and protein revealed that Manα(1–2)Manα(1–2)ManαOMe is bound more intimately with its two reducing-end mannoses into the domain A binding site of CV-N than with the non-reducing end unit. Taking advantage of the 13C spectral dispersion of 13C-labeled carbohydrates in isotope-filtered experiments is a versatile means for a simultaneous mapping of the binding interactions on both, the carbohydrate and the protein.
Graphical abstract
INTRODUCTION
In contrast to proteins, labeling of carbohydrates with 13C is not commonly employed, despite the more severe resonance overlap in carbohydrate 1H NMR spectra, compared to proteins. One reason is the lack of easily available 13C-labeled material, which has to be prepared by chemical synthesis.1 In addition, NMR experiments that suppress the large 13C-13C couplings from the sugar rings have to be devised to extract useful conformational NMR parameters. Over the last few years, several NMR experiments that were originally developed for labeled proteins or nucleic acids have been adapted for uniformly 13C-labeled carbohydrates, and their use for structure determination2 and conformational analysis3 has been demonstrated. In particular, the insertion of constant-time periods (CT) in the indirect dimension or virtual decoupling in the direct dimension were implemented to avoid the splittings from 13C-13C couplings.2 Furthermore, using uniformly 13C-labeled carbohydrates permits access to the larger spectral dispersion of 13C, compared to 1H, and opens up the possibility to perform 3D or higher order NMR experiments from which information can be extracted more easily. Efforts are also under way to improve accessibility to 13C-labeled material either employing production of complex 13C-labeled oligosaccharides through overexpression using engineered yeast strains4 and/or chemoenzymatic strategies.5
Carbohydrate-protein interactions are crucial in many biological processes, where glycans can be considered as bioactive signals, with information-coding ability translated by carbohydrate-binding proteins (lectins).6 The molecular details of these interactions are critically important for understanding the molecular and structural basis for numerous diseases.7
Binding between carbohydrates and proteins can be studied by NMR spectroscopy, either by monitoring changes in the protein or the carbohydrate spectra. Titration of a carbohydrate into a 15N-labeled protein sample and observation of amide backbone chemical shift changes (chemical shift mapping) is commonly used to identify the sugar binding pocket on a protein.8 However, such experiments are blind to any effects that may be present on the carbohydrate ligand. Therefore, ligand-based NMR approaches need to be applied, including waterLOGSY, saturation-transfer difference (STD), transferred NOE (trNOE) and diffusion based methods.9 These types of experiments employ high excess of ligand over protein and yield information about the conformation of the ligand, but not about the protein binding pocket.
A few studies have been reported in which 13C-filtered NOESY experiments were used on unlabeled carbohydrates bound to 13C-labeled proteins to extract intramolecular ligand NOEs for determination of the bound ligand conforma-tion,10,11 and intermolecular NOEs for determination of ligand-protein interactions.12,13 However, the assignment of all proton resonances of the bound carbohydrate in slow exchange is not straightforward and involves connecting the free and bound ligand resonances via exchange peaks.13
Up to date, 13C-labeled carbohydrates have only been used in a small number of carbohydrate-protein interaction studies. The interaction between estrone-3-glucuronide, uniformly 13C-labeled in the glucuronic acid moiety, and an antibody Fv fragment was investigated, and intra- and intermolecular NOEs were extracted.14 Uniformly 13C-labeled α-methyl mannopyranoside binding to recombinant rat mannose binding protein was studied,15 the conformation of a 13C-labeled trisaccharide bound to a toxin was determined from residual dipolar couplings,16 and the bound conformation of 13C-labeled sialyl Lewisx bound to E-selectin was determined via trNOEs.17
If either the carbohydrate or the protein is labeled, 13C and/or 15N-filtered NOESY experiments are ideally suited to extract intermolecular interactions. However, the structural details of the unlabeled component are more difficult to de-termine. Here we show that using 13C-labeled carbohydrates and 15N (or 13C/15N) labeled proteins together NMR approaches can be devised, with which the details of the contact sites on the carbohydrate and the protein can readily be determined simultaneously.
As a model system, we used the interaction between 13C-labeled Manα(1–2)Manα(1–2)ManαOMe trisaccharide (Man3) and the cyanovirin-N (CV-N) P51G variant for establishing and evaluating suitable NMR spectroscopic approaches for detailed analysis of the sugar conformation. CV-N is a 11 kDa antiviral lectin with two sugar binding sites, with domain A exhibiting a slight preference for a trimannose unit and domain B for a dimannose unit.18,19 Earlier STD NMR studies with Man3 resulted in low STD signals,20 and STD NMR with Man3 for single binding site mutants did not yield STD signals because the off-rate was too small.21 NMR studies on a 19F-labeled Man3 revealed a single binding mode on domain A, but two binding modes in the binding site of domain B.22 Previous work on CV-N mannose binding primarily used the disaccharide Manα(1–2)Manα. This dimannoside preferentially binds to the site on domain B, which is the higher affinity site for Man2, but the lower affinity site for Man3.12,23–25 The binding site on domain A is much less explored and the only reported structures with a carbohydrate bound to this site of CV-N are two crystal structures of CV-N in complex with Man-9 or a hexamannoside by Botos et al.26
In this study we report on the use of uniformly 13C-labeled carbohydrates to investigate carbohydrate-protein interactions, taking advantage of the 13C spectral dispersion for the sugar in isotope-filtered experiments. This approach is applicable to systems that exhibit slow exchange on the NMR timescale, which are not amenable to trNOE or STD experiments. Chemical shifts for the bound sugar signals are easily extracted from 1H,13C-HSQC experiments, and 2D 13C-filtered NOESY-1H,13C-HSQC experiments permit the identification of protein contact sites on the sugar. On the protein site, amide resonances are identified in 1H,15N-HSQC experiments and carbohydrate contact sites on the protein are identified from 2D CNH-NOESY experiments. This approach complements the available methods for fast-exchange regime carbohydrate-protein interactions.
RESULTS AND DISCUSSION
Observation of bound Man3
A complex of 13C-labeled Man3 bound to 15N-labeled CV-N was prepared to investigate Man3 that is predominantly bound to the domain A site on CV-N. At the protein concentration used (0.8 mM) addition of equimolar amounts of sugar results in a sample in which no free Man3 is observed and only trace amounts of Man3 bound to the domain B site can be detected. Using the intensities of resonances in the 1H,15N-HSQC spectrum indicates that at most ~10% of the sugar is bound to domain B, whereas the majority is bound to the domain A binding site. As displayed in Figure 1, cross peaks for all free and bound 1H,13C correlations are easily identified. The bound Man3 resonances exhibit broader lines in the 1H,13C-CT-HSQC spectrum, with noticeable differences in linewidths and intensities for equivalent positions at the reducing vs non-reducing end (Table S1). This is most pronounced when comparing the C3H cross peaks of the non-reducing end sugar (73.4 ppm/3.53 ppm) with the reducing end one (72.8 ppm/3.84 ppm), whose linewidths are 20 and 30 Hz, respectively (Fig. 1c). The larger linewidths are due to the bigger size and slower tumbling of the protein-carbohydrate complex (11.5 kDa), compared to the free sugar (0.5 kDa), possibly also containing some contribution from exchange between free and bound ligand. The different bound linewidths for all the CH cross peaks are most likely caused by differences in the dynamics of the bound ligand. The flexibility of the (1′→2″) linkage for the non-reducing terminal sugar, Man′, compared to the reducing-end sugar, Man, is clearly an important factor.
Figure 1.
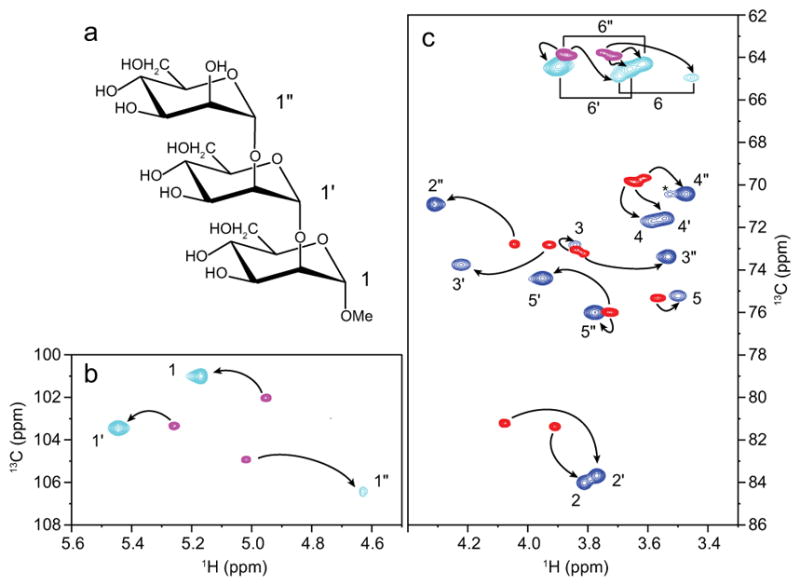
(a) Structural formula of Man3 (Manα(1–2)Manα(1–2)ManαOMe). (b) Anomeric region and (c) ring proton region of the 1H,13C-CT-HSQC spectrum of free Man3 (red/magenta) superimposed on the 1H,13C-CT-HSQC spectrum of CV-N-bound Man3 (blue/cyan). An unidentified cross-peak is marked with an asterisk (*).
Assignments of the bound Man3 1H and 13C resonances were obtained from 1H,13C-HSQC-TOCSY and HC(C)H-TOCSY experiments. Applying a C-C spin-lock in the HC(C)H-TOCSY experiment allowed correlations from H1 to H2, H3, and H4 in the mannose ring to be observed (Figure S1), which are difficult to detect in H-H spin-locked 1H,1H-TOCSY experiments, since 3JH1,H2 in mannose sugars are very small. All assignments are provided Figure 1.
Chemical shift differences (Δδ) between free and bound Man3 are listed in Table 1. ΔδH differences up to 0.4 ppm are seen (H6a; −0.30 ppm, H2″, −0.31 ppm; H1′, −0.39 ppm) and the largest ΔδC are ~ 2.5 ppm (C2, +2.6 ppm; C2″, +2.5 ppm). All chemical shifts for bound and free Man3 are provided in Table 1.
Table 1.
Chemical shifts (ppm) of free Man3 and CV-N-bound Man3
| H1 | H2 | H3 | H4 | H5 | H6a, H6b | OMe | ||
|---|---|---|---|---|---|---|---|---|
| C1 | C2 | C3 | C4 | C5 | C6 | |||
| Man′ | Free | 5.02 | 4.04 | 3.81 | 3.61 | 3.73 | 3.72, 3.87 | |
| 104.9 | 72.8 | 73.2 | 69.6 | 76.0 | 63.9 | |||
| Bound | 4.63 | 4.31 | 3.53 | 3.47 | 3.77 | 3.62, 3.89 | ||
| 106.4 | 70.9 | 73.4 | 70.4 | 76.0 | 64.3 | |||
| Δδa | −0.39 | 0.27 | −0.28 | −0.14 | 0.05 | −0.10, 0.02 | ||
| 1.5 | −1.9 | 0.2 | 0.8 | 0.0 | 0.4 | |||
|
| ||||||||
| Man″ | Free | 5.26 | 4.08 | 3.93 | 3.65 | 3.73 | 3.72, 3.87 | |
| 103.3 | 81.2 | 72.8 | 69.8 | 76.0 | 63.9 | |||
| Bound | 5.45 | 3.77 | 4.22 | 3.55 | 3.95 | 3.66, 3.90 | ||
| 103.5 | 83.6 | 73.7 | 71.6 | 74.4 | 64.5 | |||
| Δδa | 0.19 | −0.31 | 0.29 | −0.11 | 0.23 | −0.06, 0.03 | ||
| 0.1 | 2.5 | 0.9 | 1.8 | −1.6 | 0.6 | |||
|
| ||||||||
| Man | Free | 4.95 | 3.91 | 3.84 | 3.64 | 3.57 | 3.75, 3.87 | 3.37 |
| 102.0 | 81.4 | 73.0 | 69.9 | 75.3 | 63.8 | 57.6 | ||
| Bound | 5.17 | 3.81 | 3.84 | 3.58 | 3.50 | 3.45, 3.70 | 3.42 | |
| 101.0 | 84.0 | 72.8 | 71.7 | 75.2 | 64.9 | 57.7 | ||
| Δδa | 0.22 | −0.09 | 0.01 | −0.06 | −0.07 | −0.30, −0.17 | 0.05 | |
| −1.0 | 2.6 | −0.2 | 1.8 | −0.1 | 1.2 | 0.1 | ||
Δδ = δbound − δfree
Conformation of bound Man3
The conformation of Man3 was elucidated from intramolecular NOEs, both for the free and bound trimannoside (Figure 2). 2D 1H,13C-HSQC-NOESY spectra (Figure 2d) with mixing times of 10–120 ms were recorded to extract NOEs for the bound Man3 and NOE build-up curves were used to extract distances (Figure 2f) for conformational analysis. Equivalent spectra were recorded for free Man3. However, only very small NOEs were detectable in NOESY spectra due to the small size and fast rotational correlation time of trimannoside. Therefore, 1D DPFGSE-T-ROESY spectra were recorded on a concentrated sample of unlabeled Man3 (Figure 2c), and distances were extracted from intensity difference in the 1D ROESY traces for increasing mixing times.
Figure 2.
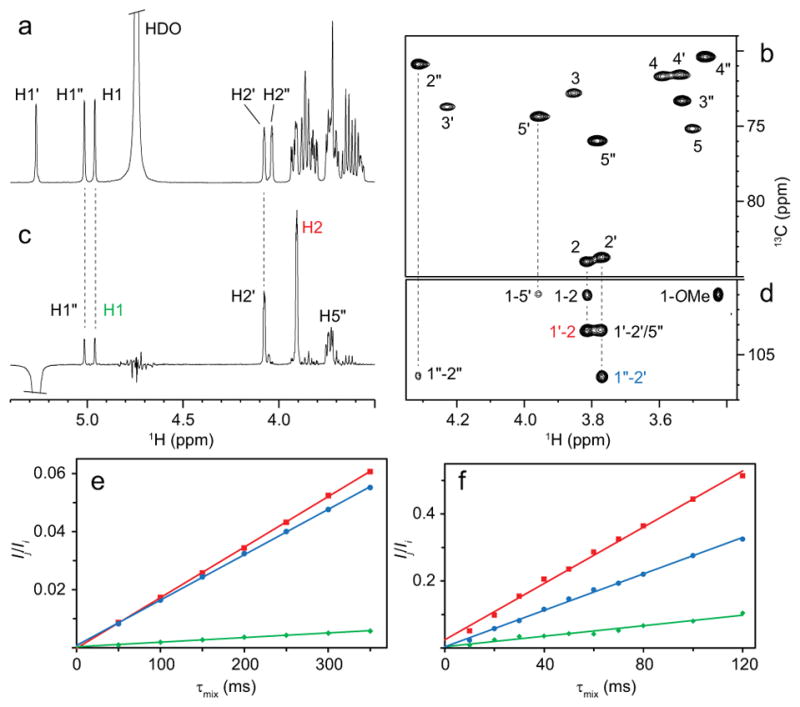
(a) 1H NMR spectrum of Man3 in D2O buffer. (b) Selected region of the 1H,13C-CT-HSQC spectrum of Man3 bound to CV-N. (c) 1D 1H,1H-DPFGSE-T-ROESY spectrum of Man3 with selective excitation on H1″ and a mixing time of 300 ms. (d) Selected region of a 1H,13C-HSQC-NOESY spectrum of CV-N-bound Man3 with a mixing time of 60 ms. (e-f) PANIC plots of Ij/Ii versus τmix, where Ij is the intensity of the NOE peak, -Ii is the intensity of the target peak (selective experiments) and Ii is the average intensity of the diagonal peaks (2D experiments). Plots are shown for H1″-H2 (red squares), H1′-H2″ (blue circles), and H1-H1″ (green diamonds) of (e) free and (f) bound Man3. Cross-relaxation rates were obtained from the slopes of the lines.
Representative intra-residue distances are very similar in free and bound Man3 (Table 2). Evaluating all intramolecular NOEs for bound Man3 (Table S2) shows that all sugar rings are close to normal 4C1 conformations. In particular, the H3-H5 distance is informative, given its 1,3-diaxial interaction in the 4C1 conformation. This distance would be considerably longer in most other possible conformations, including the 1C4 conformation. For the non-reducing terminal sugar this distance (H3′-H5′) is 2.62 Å, identical to the one in the reducing end sugar (H3-H5=2.62; see Table S2). The H3″-H5″ distance for the middle sugar could not be measured from the 2D 1H,13C-HSQC-NOESY spectrum because of 13C chemical shift overlap.
Table 2.
NOE-derived intramolecular distances (Å) of free Man3 and CVN-bound Man3a
| rfreeb | rboundc | |||
|---|---|---|---|---|
| Intra-residue | H1 | H2 | 2.59 | 2.54 |
| H1 | OCH3 | 2.94 | 2.71 | |
| H1″ | H2″ | 2.50 | 2.57 | |
| H2″ | H3″ | 2.37 | 2.52 | |
| H1′ | H2′ | 2.53d | 2.59 | |
| H2′ | H3′ | 2.44 | 2.55 | |
|
| ||||
| Inter-residue (1″→2) | H1 | H1″ | 3.26 | 2.87 |
| H1 | H5″ | 2.59 | 2.95 | |
| H1″ | H2 | 2.21 | 2.12 | |
|
| ||||
| Inter-residue (1′→2″) | H1″ | H1′ | 3.29 | >3.0e |
| H1″ | H5′ | 2.47 | 2.44 | |
| H1′ | H2″ | 2.22 | 2.30 | |
The complete list of all intramolecular NOEs is presented in Table S2.
Standard errors are <0.01 Å.
Standard errors are <0.04, except H1-H5″, which is 0.07.
Reference distance obtained from molecular dynamics simulation data of a Manα(1–2)Manα(1–O)Ser.27
Contribution of spin diffusion prevented accurate determination.
NOEs across the glycosidic linkages exhibited large differences between free and bound Man3, suggesting that changes in the glycosidic torsion angles are present. For the (1″→2) linkage the measured H1-H1″ and H1-H5″ distances were clearly different, namely 3.26 Å and 2.59 Å in free Man3 and 2.87 Å and 2.95 Å in bound Man3, respectively. A smaller difference is seen for the H1″-H2 distance (2.21 Å versus 2.12 Å). For the connection between the middle and the non-reducing terminal sugar unit only small differences between free and bound Man3 are noted, with very similar distances for H1′-H2″ (2.22 Å versus 2.30 Å), H1″-H5′ (2.47 Å versus 2.44 Å) and H1″-H1′ (3.29 Å versus >3 Å). All inter-residue NOEs measured for free Man3 are similar for both linkages and are in agreement with molecular dynamics simulations on Manα(1→2)Man, which revealed glycosidic torsion angles ϕH = −40°, ψH = 33°.28 In the crystal structure of Man-9 with CV-N (PDB accession code 3GXZ) torsion angles of ϕH = −27°, ψH = 30° for (1″→2) and ϕH = −51°, ψH = 71° for (1′→2″) are found for the trimannose unit of the D1 arm.26 The (1″→2) linkage torsion angles are slightly different from the major conformation of the free Man3 and are more close to the solution conformation observed here. Interestingly, these glycosidic torsion angles are almost identical to the ones in a crystal structure of a CV-N complex with Manα(1→2)Man in the domain B binding site (ϕH = −27°, ψH = 31°).23 However, the glycosidic torsion angles for the (1′→2″) linkage, measured from the crystal structure, are different from the conformation in the solution NMR structure, as evidenced by a significantly shorter H1″-H5′ distance (2.44 Å) than in the crystal structure (3.32 Å). This bound sugar conformation is closer to the conformation of free Man3, since very similar inter-residue distances were measured for the (1′→2″) linkage.
Despite the difference between the two glycosidic linkages of Man3 bound to CV-N, with torsion angles close to ϕH = −27°, ψH = 30° for the (1″→2) linkage and ϕH = −40°, ψH = 33° for the (1′→2″) linkage, both conformations reside in the high probability region for Manα(1→2)Man, with ϕH around −40° and a positive ψH.28,29 Surprisingly, molecular dynamics simulations of the Manα(1→2)Man-CV-N complex found a negative ψH (ϕH = −45°, ψH = −20°) in the domain A binding site.29 This is a minor conformer of free Manα(1→2)Man.28,29 Experimentally, however, we found no evidence for this bound conformation in our current study.
Mapping the binding interface between Man3 and CV-N
A large number of intermolecular NOEs were obtained from 2D 1H,13C-HSQC-NOESY spectra, recorded on the complex between 13C-labeled Man3 and 15N-labeled CV-N, in conjunction with simultaneous 13C/15N 3D NOESY-HSQC experiments,30 recorded on a sample of 13C-labeled Man3 bound to 13C/15N-labeled CV-N. Build-up curves for intermolecular NOEs (Figure S2a) were used to extract distances by the isolated spin-pair approximation (Table 3). Note that significant spin diffusion was present in many cases for mixing times larger than 50 ms; in these cases only small mixing times devoid of spin diffusion were used in the fitting procedure. Despite these precautions, 23 out of 56 intermolecular NOEs are affected by spin diffusion. All intermolecular NOEs are provided in Table S3.
Table 3.
NOE-derived intermolecular distances (Å) of CV-N-bound Man3 and equivalent distances in the triman-noside unit of the D1 arm from the crystal structure of Man9 bound to CV-N (PDB accession code 3GXZ).a
| CV-N | Man3 | rexp | r3GXZ |
|---|---|---|---|
| G2 NH | H2′ | 2.82 | 2.44 |
| K3 Hβb | H4″ | 2.40 | 2.61 |
| K3 Hβb | H3′ | 3.06 | 3.01 |
| K3 Hδ | H3′ | 2.90 | 2.28 |
| Q6 Hβa | H4″ | 3.50 | 3.56 |
| Q6 Hβa | H6″ | 2.77b | 2.60/3.35 |
| Q6 Hβb | H6″ | 3.03b | 2.59/3.77 |
| T7 Hγ | H1 | 4.50 | 4.54 |
| T7 Hγ | H4″ | 4.01 | 3.98 |
| T7 Hγ | H5″ | 2.98 | 2.35 |
| T7 Hγ | H6″ | 2.67b | 1.82 |
| E23 Hβb | H6 | 2.29b | 2.40/3.09 |
| T25 Hα | H5 | 2.33 | 2.03 |
| T25 Hγ | H3 | 3.18 | 2.57 |
| T25 Hγ | H5 | 3.29 | 3.25 |
| A92 Hβ | H2″ | 3.44 | 3.46 |
| A92 Hβ | H1′ | 3.35 | 3.87 |
| A92 Hβ | H2′ | 2.99 | 2.57 |
| N93 NH | H2″ | 3.16 | 3.08 |
| N93 NH | H3″ | 2.58 | 2.66 |
| N93 Hβa | H2″ | 3.27 | 3.55 |
| N93 Hβa | H3″ | 2.32 | 1.86 |
| N93 δNHa | H4 | 2.97 | 3.26 |
| N93 δNHa | H6 | 2.52b | 2.56/2.69 |
| N93 δNHb | H4 | 3.12 | 3.40 |
| N93 δNHb | H6 | 2.58b | 2.55/3.46 |
| N93 δNHb | H3″ | 3.02 | 3.14 |
| I94 Hδ | H1″ | 4.42 | 4.44 |
| I94 Hδ | H2″ | 3.34 | 2.82 |
| I94 Hδ | H3″ | 4.87 | 4.82 |
| I94 Hδ | H1′ | 3.45 | 3.54 |
| D95 NH | H3 | 3.16 | 3.69 |
| D95 NH | H4 | 3.43 | 3.17 |
A complete list of intermolecular NOEs is provided in Table S3.
H6(″)a and H6(″)b resonances could not be distinguished since NOEs were obtained in the 13C dimension of the 1H,13C-HSQC-NOESY spectrum.
Comparison of the intermolecular distances measured here with those in the crystal structure (PDB accession code 3GXZ)26 yielded a correlation coefficient of r2 = 0.68 (Figure S2b). The small deviations between the NMR-derived and X-ray distances for residues G2, K3, T7, T25, A92, N93, and I94 may relate to slightly altered side-chain orientations in the two complexes. Residues G2, K3, T7, T25, and N93 are all implicated in hydrogen bonding in the crystal structure, although these hydrogen bonds are inferred from the distances and not measured experimentally.
Contacts on the carbohydrate
13C-filtered NOESY-1H,13C-HSQC spectra were recorded to measure intermolecular carbohydrate-protein NOEs, filtering out all NOEs within the sugar. A 2D version, depicting the 1H,13C dimension is shown in Figure 3 and compared with the 1H,13C-CT-HSQC spectrum.
Figure 3.
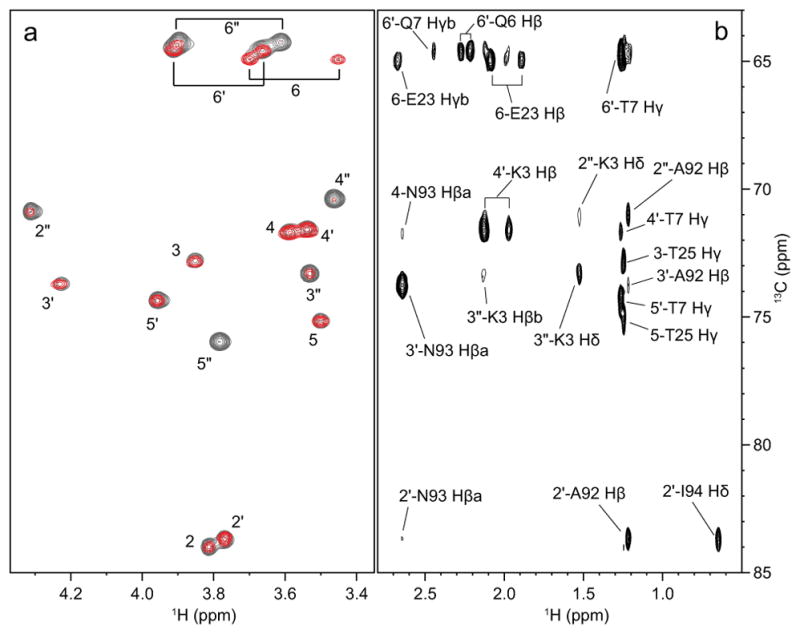
(a) Superposition of the 2D version of the 13C-filtered NOESY-HSQC (red, 80 ms mixing time) and the 1H,13C-CT-HSQC (grey) spectra of the CV-N/Man3 complex. (b) Side-chain region of the 1H,13C-HSQC-NOESY spectrum of the CV-N/Man3 complex (60 ms mixing time). Intermolecular NOEs between sugar ring protons (correlated through 13C in f1) and protein side-chain protons are labeled.
Intense cross-peaks correspond to several NOE contacts, while weak or absent cross-peaks are equivalent to few or no intermo-lecular NOEs. A 2D version of the 1H,1H dimension (not shown) revealed that most NOEs originated from protein side chains, whereas NOEs from backbone amide protons contributed to a lesser extent. The identity of the protein side chain of the different NOEs can be traced in the 13C dimension from a 1H,13C-HSQC-NOESY spectrum (Figure 3b). Note that the reducing-end mannose H2 and H5 cross-peaks reflect intramolecular NOEs originating from the OCH3 group, since the methyl group on the sugar was not 13C-labeled.
As can be appreciated from the spectrum in Figure 3a, the Man′ protons (H2′, H3′, H4′, H5′ and H6′a/b) of the non-reducing terminal sugar exhibit substantially less intermolecular NOEs than the Man and Man’ rings. Only three intermolecular distances < 3 Å involving Man′ were determined: G2 NH – H2′ (2.82 Å), A92 Hβ – H2′ (2.99 Å) and K3 Hδ – H3′ (2.90 Å) (Table 3 and S4). This lack of NOEs may be due to higher flexibility of the (1′→2″) linkage, consistent with the narrower lines that are observed for the associated cross-peaks in the 1H,13C-CT-HSQC spectrum (see above). The Man″ sugar is close to L1, G2, K3, and E101 at the N- and C-termini of the protein, a region of the protein that is more flexible than other parts.
At this point we would like to emphasize that the 1H,13C 2D version of a 13C-filtered NOESY-1H,13C-HSQC experiment, acquired in 4 hours, provided information about the contacts on the carbohydrate without the need to assign the protein spectrum. It only requires a 13C-labeled carbohydrate, and the protein can be at natural abundance.
Contacts on the protein
Binding site mapping of Man3 binding to CV-N has previously been reported from NMR titrations of Man3 into uniformly 15N-labeled CV-N.18,19 Using 13C-labeled Man3, a CNH-NOESY spectrum can be employed to extract NOEs between 13C-attached protons of the carbohydrate and 15N-attached protons of the protein. Traditionally, 1H,13C-HSQC-NOESY-1H,15N-HSQC experiments are used for resonance assignments of 13C,15N-labeled proteins,31 although here we used a 2D 1H,15N version for easy comparison with the 1H,15N-HSQC spectrum. As illustrated in Figure 4, protein amide protons involved in intermolecular NOEs can be easily identified when comparing the 1H,13C-HSQC-NOESY experiment (Figure 4a) with the 1H,15N 2D version of the CNH-NOESY (Figure 4b). Using a mixing time of only 20 ms (Figure 4b), no spin diffusion was detected and NOEs corresponding to distances <3.4 Å were observed. Longer mixing time (60 ms) resulted in significant spin diffusion from protein side chains to nearby amide protons (Figure S3). This second shell of the binding site, comprising amide protons, corresponds well to those amide protons that exhibited chemical shift changes upon Man3 addition in the 1H,15N-HSQC spectrum. Therefore, the 2D CNH-NOESY experiment can be employed to map amino acids in close proximity to the ligand, without the need for 13C,15N-labeled protein and full protein assignment. In contrast to chemical shift mapping, this NOE-based method clearly identifies whether direct contacts or ligand-induced conformational changes are involved. This can be critical, since it is well known, that remote conformational changes frequently contribute confounding aspects when binding sites are mapped by NMR; such issues are avoided in binding studies using a 13C-labeled ligand and the above approach. Additionally, for tight binding ligands, involving a large number of amino acids, chemical shift mapping often affects too many resonances, making it difficult to distinguish between the critical, primary contacts and less important secondary contacts. The 2D CNH-NOESY experiment with short mixing times only identifies resonances of residues in close proximity to the ligand and with a clear gradient of intensity due to the r−6 dependence of the NOE. Moreover, the binding interactions can be easily mapped from a 3D CNH-NOESY or a 2D 1H,13C-HSQC-NOESY experiment as illustrated in Figure 4b.
Figure 4.
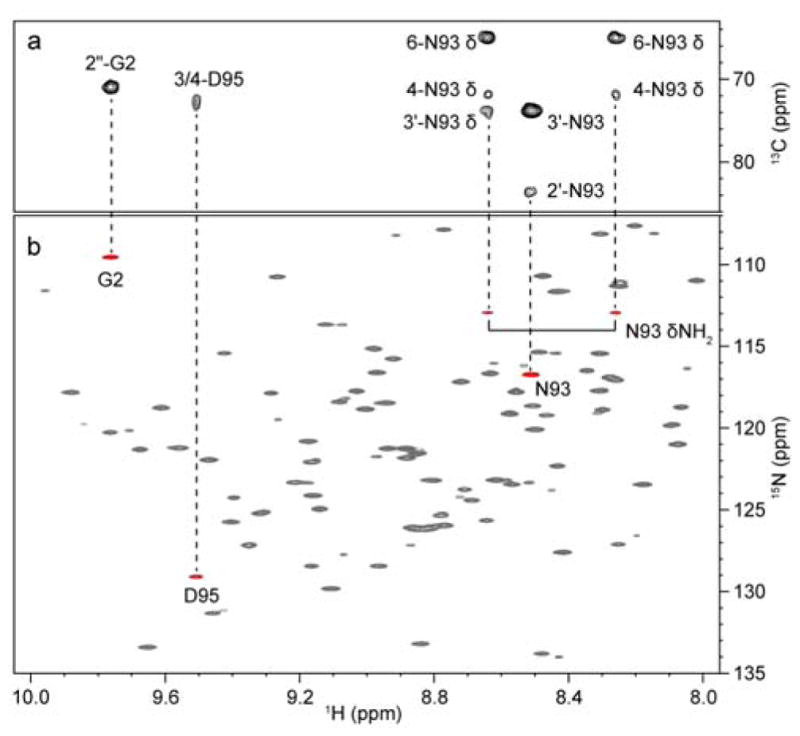
(a) Amide proton region of the 1H,13C-HSQC-NOESY spectrum (60 ms mixing time) of the CV-N/Man3 complex. Intermolecular NOEs between sugar ring protons and protein amide protons are labeled. (b) 1H,15N 2D version of the CNH-NOESY experiment (red, 20 ms mixing time) superimposed onto a 1H,15N-HSQC spectrum (grey). Amide proton resonances exhibiting NOEs to 13C-attached sugar ring protons are connected by dashed lines.
CONCLUSIONS
Here, we presented the effective use of 13C-labeled sugars to delineate the details of the bound ligand conformation as well as the binding site on a protein. The contacts on the carbohydrate are identified from a 2D 13C-filtered NOESY-1H,13C-HSQC experiment and contacts on the protein are detected via intermolecular NOEs in 2D CNH-NOESY spectra. These two 13C-filtered or edited NOESY experiments are powerful complements for investigating ligand-protein interactions by NMR, without the need for full protein assignment.
Using this approach on the Man3-CV-N complex we characterized the structure of the bound carbohydrate using intra-sugar NOEs and identified contacts with the protein via intermolecular NOEs. The (1″→2) glycosidic linkage was found to be similar to that of the trimannoside unit of Man-9 in the crystal with CV-N26 and the (1′→2″) glycosidic linkage is similar to that of free Man3. The density of intermolecular NOEs from the individual sugar units revealed that Man and Man″ are bound more intimately to the protein compared to the non-reducing terminal Man′ sugar.
EXPERIMENTAL PROCEDURES
Carbohydrate synthesis
Uniformly 13C-labeled Manα(1–2)Manα(1–2)ManαOMe (Man3) was synthesized from D-[13C6]-mannose. See supporting information for details.
Protein expression and purification
P51G CV-N, labeled with 15N or 13C/15N was expressed as described earlier.32 The protein was prepared from the soluble fraction after cell lysis by sonication. The cell debris was removed by centrifugation and the supernatant was dialyzed against 20 mM Tris-HCl buffer (pH 8.5), loaded onto a Q(HP) column (GE Healthcare) and protein was eluted using a linear gradient of 0–1 M NaCl. Protein-containing fractions were concentrated and further purified by gel filtration on a Superdex 75 column (GE Healthcare) in 50 mM Tris-HCl, 100 mM NaCl (pH 8.5). Fractions containing pure P51G CV-N were collected and concentrated using Centriprep and Amicon devices (Millipore). Samples for NMR were buffer-exchanged into 10 mM phosphate buffer, 3 mM NaN3, 95/5% H2O/D2O (pH 6.6).
NMR spectroscopy
NMR spectra were recorded at 20 °C or 25 °C on Bruker 600, 700, 800, and 900 MHz AVANCE spectrometers equipped with 5-mm-triple-resonance, z axis gradient cryoprobes. Parameter settings for the NMR experiments are summarized in Table S4.
For carbohydrate resonance assignments of bound Man3, a sample of 13C-labeled Man3 and 15N-labeled CV-N (1:1 molar ratio, 0.8 mM) was prepared. 2D 1H,13C-HSQC and 2D versions of HC(C)H-TOCSY, 1H,13C-HSQC-NOESY and a constant-time version of 1H,13C-HSQC-TOCSY spectra were recorded to obtain complete 1H and 13C assignments. Spectra were referenced to internal DSS (δH = 0.00 ppm, δC = 0.00 ppm).
For protein resonance assignments, a sample of 13C-labeled Man3 and 13C/15N-labeled CV-N (1:1 molar ratio, 0.8 mM) was prepared. Three-dimensional HNCACB and CBCA(CO)NH spectra were recorded for protein backbone assignment. 1H and 13C assignment of aliphatic side-chains was carried out using 2D 1H,13C-HSQC, 3D HC(C)H-TOCSY and 3D NOESY-HSQC spectra with simultaneous evolution of 13C and 15N chemical shifts in t2 and a mixing time of 80 ms.
Measurements of proton-proton cross-relaxation rates of free Man3 (without 13C-labeling) were carried out at 25 °C on a 35 mM sample in10 mM phosphate buffer in D2O (pD 7.0, equivalent to pH 6.6) on a 600 MHz spectrometer. 1D DPFGSE 1H,1H-T-ROESY spectra were recorded with 80 ms Gaussian shaped pulses, selective on H1, H1″, H1′, H2″, and H2′. Seven different mixing times ranging from 50 to 350 ms were used for excitation of each individual resonance. The recovery delay was set to 8 s to ensure >5 × T1.
Measurements of proton-proton cross-relaxation rates of CV-N-bound Man3 (with 13C-labeling) were carried out at 20 °C on a 0.9 mM sample (1:1 molar ratio of CV-N and sugar) on a 900 MHz spectrometer. 2D 1H,13C-HSQC-NOESY experiments were recorded for ten different mixing times, ranging from 10 to 120 ms. In addition, a 3D NOESY-HSQC spectrum with Watergate suppression was recorded on the same sample. This spectrum was used to measure the relaxation rate between H6a and H6b for calibration of distances with respect to the H6a-H6b reference distance (1.78 Å). Experimentally determined distances were compared to equivalent distances in the D1 arm trimannoside of Man-9 in the X-ray structure with CV-N (PDB accession code 3GXZ).26 Protons were added to the X-ray coordinates by PyMOL (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
Mapping of the binding contacts on the 13C-labeled Man3 was carried out at 20 °C on a 0.9 mM sample (1:1 molar ratio of CV-N and sugar) on a 900 MHz spectrometer. 2D versions of NOESY-1H,13C-HSQC spectra with 13C-filtering in t1 (not evolved) were recorded and compared with a 1H,13C-HSQC spectrum. The binding contacts on 15N-labeled CV-N were investigated at 25 °C on the same 0.9 mM sample on a 600 MHz spectrometer. 2D versions of 1H,13C-HSQC-NOESY-1H,15N-HSQC (CNH-NOESY) experiments were used to identify contacts on the protein by comparison with a 1H,15N-HSQC spectrum.
NMR spectra were processed with Topspin 3.1 (Bruker) and ccpNMR was used for resonance and NOE cross-peak assignments.33 NOE build-up curves were constructed using the PANIC approach.34,35 In the 1D DPFGSE 1H,1H-T-ROESY spectra, each NOE enhancement was normalized with respect to the target signal in the same spectrum. In 2D 1H,13C-HSQC-NOESY spectra, each NOE cross-peak intensity was normalized with respect to the average intensity of the two diagonal signals (intramolecular Man3 NOEs) or the Man3 diagonal signal intensity (intermolecular NOEs) in the same spectrum. Error values were obtained from the linear fitting of the NOE build-up curves. Lower errors were obtained by the PANIC approach, compared to exponential fitting, since normalization is performed within the same spectrum. The isolated spin-pair approximation (ISPA) was used to obtain distances relative to a known reference distance.
Supplementary Material
Acknowledgments
This work was supported by The Carl Trygger Foundation (G.N.), a Science Foundation Ireland Grant 13/IA/1959 (S.O.), and a National Institutes of Health Grant RO1GM080642 (A.M.G.). We thank Mike Delk for NMR technical support and Dr. Elena Matei for helpful discussions.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Linewidths and intensities of free Man3 and CV-N bound Man3 resonances (Table S1), complete lists of intramolecular (Table S2) and intermolecular (Table S3) distances of Man3 and the CV-N/Man3 complex, parameter settings for all NMR experiments (Table S4), a 2D HC(C)H-TOCSY spectrum of CV-N-bound Man3 (Figure S1), plots of intermolecular NOE build-up curves and correlation plot of distances from NOEs vs. X-ray structure (Figure S2), a 2D CNH-NOESY overlay on a 1H,15N-HSQC spectrum (Figure S3), experimental for the synthesis of 13C-labeled mannosides and NMR spectra for determination of the synthesized products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Homans SW. Biochem Soc Trans. 1998;26:551–560. doi: 10.1042/bst0260551. [DOI] [PubMed] [Google Scholar]
- 2.Fontana C, Kovacs H, Widmalm G. J Biomol NMR. 2014;59:95–110. doi: 10.1007/s10858-014-9830-6. [DOI] [PubMed] [Google Scholar]
- 3.Battistel MD, Shangold M, Trinh L, Shiloach J, Freedberg DI. J Am Chem Soc. 2012;134:10717–10720. doi: 10.1021/ja300624j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamiya Y, Yamamoto S, Chiba Y, Jigami Y, Kato K. J Biomol NMR. 2011;50:397–401. doi: 10.1007/s10858-011-9525-1. [DOI] [PubMed] [Google Scholar]
- 5.Zhu T, Yamaguchi T, Satoh T, Kato K. Chem Lett. 2015;44:1744–1746. [Google Scholar]
- 6.Gabius HJ, Andre S, Jimenez-Barbero J, Romero A, Solis D. Trends Biochem Sci. 2011;36:298–313. doi: 10.1016/j.tibs.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Varki A, Freeze HH. In: Essentials of Glycobiology. 2. Varki ACR, Esko JD, et al., editors. Cold Spring Harbor Laboratory Press; New York: 2009. [PubMed] [Google Scholar]
- 8.Williamson MP. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 9.Meyer B, Peters T. Angew Chem, Int Ed. 2003;42:864–890. doi: 10.1002/anie.200390233. [DOI] [PubMed] [Google Scholar]
- 10.Canales A, Angulo J, Ojeda R, Bruix M, Fayos R, Lozano R, Gimenez-Gallego G, Martin-Lomas M, Nieto PM, Jimenez-Barbero J. J Am Chem Soc. 2005;127:5778–5779. doi: 10.1021/ja043363y. [DOI] [PubMed] [Google Scholar]
- 11.Nieto L, Canales A, Gimenez-Gallego G, Nieto PM, Jimenez-Barbero J. Chem—Eur J. 2011;17:11204–11209. doi: 10.1002/chem.201101000. [DOI] [PubMed] [Google Scholar]
- 12.Bewley CA. Structure (Oxford, U K) 2001;9:931–940. doi: 10.1016/s0969-2126(01)00653-0. [DOI] [PubMed] [Google Scholar]
- 13.Schubert M, Bleuler-Martinez S, Butschi A, Walti MA, Egloff P, Stutz K, Yan S, Wilson IBH, Hengartner MO, Aebi M, Allain FHT, Kunzler M. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Low DG, Probert MA, Embleton G, Seshadri K, Field RA, Homans SW, Windust J, Davis PJ. Glycobiology. 1997;7:373–381. doi: 10.1093/glycob/7.3.373. [DOI] [PubMed] [Google Scholar]
- 15.Sayers EW, Weaver JL, Prestegard JH. J Biomol NMR. 1998;12:209–222. doi: 10.1023/a:1008220522409. [DOI] [PubMed] [Google Scholar]
- 16.Shimizu H, Donohue-Rolfe A, Homans SW. J Am Chem Soc. 1999;121:5815–5816. [Google Scholar]
- 17.Harris R, Kiddle GR, Field RA, Milton MJ, Ernst B, Magnani JL, Homans SW. J Am Chem Soc. 1999;121:2546–2551. [Google Scholar]
- 18.Bewley CA, Kiyonaka S, Hamachi I. J Mol Biol. 2002;322:881–889. doi: 10.1016/s0022-2836(02)00842-2. [DOI] [PubMed] [Google Scholar]
- 19.Shenoy SR, Barrientos LG, Ratner DM, O’Keefe BR, Seeberger PH, Gronenborn AM, Boyd MR. Chem Biol (Oxford, U K) 2002;9:1109–1118. doi: 10.1016/s1074-5521(02)00237-5. [DOI] [PubMed] [Google Scholar]
- 20.Sandström C, Berteau O, Gemma E, Oscarson S, Kenne L, Gronenborn AM. Biochemistry. 2004;43:13926–13931. doi: 10.1021/bi048676k. [DOI] [PubMed] [Google Scholar]
- 21.Sandström C, Hakkarainen B, Matei E, Glinchert A, Lahmann M, Oscarson S, Kenne L, Gronenborn AM. Biochemistry. 2008;47:3625–3635. doi: 10.1021/bi702200m. [DOI] [PubMed] [Google Scholar]
- 22.Matei E, Andre S, Glinschert A, Infantino AS, Oscarson S, Gabius HJ, Gronenborn AM. Chem—Eur J. 2013;19:5364–5374. doi: 10.1002/chem.201204070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fromme R, Katiliene Z, Giomarelli B, Bogani F, Mc Mahon J, Mori T, Fromme P, Ghirlanda G. Biochemistry. 2007;46:9199–9207. doi: 10.1021/bi700666m. [DOI] [PubMed] [Google Scholar]
- 24.Ramadugu SK, Li Z, Kashyap HK, Margulis CJ. Biochemistry. 2014;53:1477–1484. doi: 10.1021/bi4014159. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Bolia A, Maxwell JD, Bobkov AA, Ghirlanda G, Ozkan SB, Margulis CJ. Biochemistry. 2015;54:6951–6960. doi: 10.1021/acs.biochem.5b00635. [DOI] [PubMed] [Google Scholar]
- 26.Botos I, O’Keefe BR, Shenoy SR, Cartner LK, Ratner DM, Seeberger PH, Boyd MR, Wlodawer A. J Biol Chem. 2002;277:34336–34342. doi: 10.1074/jbc.M205909200. [DOI] [PubMed] [Google Scholar]
- 27.Lycknert K, Helander A, Oscarson S, Kenne L, Widmalm G. Carbohydr Res. 2004;339:1331–1338. doi: 10.1016/j.carres.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 28.Säwén E, Massad T, Landersjö C, Damberg P, Widmalm G. Org Biomol Chem. 2010;8:3684–3695. doi: 10.1039/c003958f. [DOI] [PubMed] [Google Scholar]
- 29.Margulis CJ. J Phys Chem B. 2005;109:3639–3647. doi: 10.1021/jp0406971. [DOI] [PubMed] [Google Scholar]
- 30.Sattler M, Maurer M, Schleucher J, Griesinger C. J Biomol NMR. 1995;5:97–102. doi: 10.1007/BF00227475. [DOI] [PubMed] [Google Scholar]
- 31.Diercks T, Coles M, Kessler H. J Biomol NMR. 1999;15:177–180. doi: 10.1023/A:1008367912535. [DOI] [PubMed] [Google Scholar]
- 32.Barrientos LG, Lasala F, Delgado R, Sanchez A, Gronenborn AM. Structure (Oxford, U K) 2004;12:1799–1807. doi: 10.1016/j.str.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 33.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. Proteins: Struct, Funct, Bioinf. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 34.Macura S, Farmer BT, Brown LR. J Magn Reson. 1986;70:493–499. [Google Scholar]
- 35.Hu HT, Krishnamurthy K. J Magn Reson. 2006;182:173–177. doi: 10.1016/j.jmr.2006.06.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.