

Abstract
Patient: Female, 88
Final Diagnosis: Adult onset still’s disease
Symptoms: Fever • rash
Medication: —
Clinical Procedure: —
Specialty: Rhaumatology
Objective:
Unusual clinical course
Background:
Adult-onset Still’s disease (AOSD) is a rare multi-systemic inflammatory disorder of unknown etiology characterized by spiking fever, characteristic rash, and arthritis. It often associates with high serum ferritin levels.
Case Report:
An 88-year-old woman had fever of over 39°C without response to extended-spectrum antibiotics for 6 days. She had non-specific erythema with infiltration on her trunk. She had leukocytosis with neutrophilia of 80%, mild hepatic dysfunction, normal level of rheumatoid factor and antinuclear antibody, thrombocytopenia, elevated d-dimer and soluble interleukin2 receptor, extremely high serum ferritin (78 662 ng/mL), and splenomegaly. Although she had no arthritis or specific erythema, we made the diagnosis of AOSD according to Yamaguchi’s criteria with disseminated intravascular coagulation (DIC) and hemophagocytic syndrome (HPS) after ruling out infections, malignancies, or other connective tissue diseases. Twelve percent of AOSD patients have HPS. The mean serum ferritin of AOSD with HPS was reported at 18 179 ng/mL, which supported the diagnosis of AOSD because only a few other diseases could show such extremely high serum ferritin. Although she was treated with prednisolone (30 mg/day), her condition deteriorated and her left pleural effusion increased. Therefore, methylprednisolone 500 mg/day for 3 days was started followed by prednisolone 30 mg/day and immunosuppressive agent (Cyclosporine 50 mg/day), which improved her general condition, elevated C-reactive protein levels, and extremely high serum ferritin levels.
Conclusions:
We report the case of an elderly patient with severe AOSD, who developed HPS and DIC, whose extremely high serum ferritin level was useful in diagnosis.
MeSH Keywords: Disseminated Intravascular Coagulation; Ferritins; Lymphohistiocytosis, Hemophagocytic; Still’s Disease, Adult-Onset
Background
Adult-onset Still’s disease (AOSD) is a rare multi-systemic inflammatory disorder of unknown etiology characterized by spiking fever, characteristic rash, and arthritis [1,2]. It often is associated with high serum ferritin levels. The annual incidence rate is 0.22 to 0.34 per 100 000 people in Japan, and it is more common in women than in men [1,3]. The typical rash of AOSD is salmon maculopapular erythema, which disappears with defervescence [4]. It is predominantly found on the proximal limbs and trunk [1]. However, various atypical rashes have been reported in some case studies of AOSD [5]. Moreover, there are some reports of AOSD without arthritis [6]. The diagnosis of atypical AOSD can be extremely difficult because of its non-specific serological and pathological findings in common with many other diseases, including infections, malignancies, and other connective tissue diseases, which should be excluded [1,5,7].
Twelve percent of AOSD patients have hemophagocytic syndrome (HPS) [8,9]. HPS is the condition caused by hypercytokinemia following genetic disorders with immunodeficiency, infections, malignancies, or other connective tissue diseases. The most frequent etiology of HPS of all connective tissue diseases is AOSD [8]. HPS can be diagnosed according to criteria including fever, splenomegaly, hepatic dysfunction, high serum ferritin level, high soluble interleukin-2 receptor, hypofibrinogenemia, and hyponatremia, even without the diagnostic findings of bone marrow [10].
We here report the case of an elderly woman with severe AOSD with HPS. Her extremely high serum ferritin level was a useful diagnostic clue in the absence of major symptoms, which made the diagnosis extremely difficult.
Case Report
An 88-year-old Japanese woman, who was independent in her daily activities with no drinking history, had high fever. In March 2016, she had a fever over 38°C for 6 days without response to extended-spectrum antibiotics. On the sixth day of illness, she was transferred to our hospital because of her very low blood platelet count of 15×103/µL. On admission, her body temperature was 38.6°C, heart rate was 88 beats/min, blood pressure was 166/77 mmHg, respiratory rate was 16/min, and oxygen saturation was 97% on room air. Arthritis was not present in any of her joints. On the second hospital day, she had erythema annulare at the inner parts of both lower thighs, an edematous erythema on her back, and a granular-sized infiltrative erythema on the right knee (Figure 1). Her atypical erythema repeatedly appeared and disappeared regardless of the pattern of fever. Laboratory findings on admission are shown in Table 1. The patient had leukocytosis of 13.7×103/µL with neutrophilia of 87.3%, aspartate aminotransferase 484 IU/L, alanine aminotransferase 96 IU/L, C-reactive protein (CRP) 10.4 mg/dl, procalcitonin 0.85 ng/mL, and an extremely high serum ferritin levels of 78 662 ng/mL. In addition, she was diagnosed with complication of disseminated intravascular coagulation (DIC) scoring 6 on the Japanese Association for Acute Medicine (JAAM) DIC scoring system (thrombopenia of 21 000/µL and elevated d-dimer levels of 20.1 µg/mL) [11]. Rheumatoid factor, antinuclear antibody, several viral antibody titers, such as cytomegalovirus, Epstein-Barr virus, hepatitis B virus, hepatitis C virus and human T-cell leukemia virus type 1, tuberculosis-specific interferon-gamma release assay, and blood cultures were negative. An ultra-sound cardiogram revealed normal findings without vegetation on any heart valve. Bone marrow biopsy, skin biopsy of a single spot, and subsequent random skin biopsies were performed to rule out intravascular lymphoma. Bone marrow biopsy failed to show hemophagocytosis, and flow cytometry did not show any abnormalities, which allowed us to rule out myelodysplastic syndrome and aplastic anemia. Esophago-gastro-duodenoscopy was performed to exclude gastrointestinal malignancies, which showed normal results. Contrast-enhanced chest computed tomography revealed ground-glass opacity, small amounts of bilateral pleural effusions, and splenomegaly without lymphadenopathy. The imaging studies and laboratory data did not show the presence of liver cirrhosis, viral hepatitis, or autoimmune hepatitis. Even though it was extremely difficult to rule out the presence of infection, malignancy, and other connective tissue disease completely at this point, AOSD was strongly suspected according to the Yamaguchi criteria (fever over 39°C for more than a week, leukocytosis with neutrophilia of 80%, splenomegaly, hepatic dysfunction, and normal level of rheumatoid factor and antinuclear antibody) [7]. Her condition and results of examinations did not meet the diagnostic criteria of any other connective tissue diseases. We started treatment with the recommended dose of prednisolone of 30 mg/day (0.85 mg/kg/day) because her condition had deteriorated on the second hospital day [1]. Minocycline 200 mg/day was administered concurrently in case her skin lesions were caused by rickettsia. However, her body temperature increased above 38°C, and serum ferritin level and CRP remained high. Deterioration of her condition despite the treatment with minocycline made the diagnosis of rickettsia diseases unlikely. On the sixth hospital day, we performed thoracentesis because her left pleural effusion was increasing. The appearance of the pleural effusion was yellow and cloudy, and laboratory testing showed a white blood cell (WBC) count of 8 000/µL (neutrophils 94.6%, lymphocytes 2.4%), glucose 171 mg/dL, and pH 7.34. From these findings, the pleural effusion was classified as exudative according to Light criteria (ratio of pleural fluid and serum protein levels >0.5, ratio of pleural fluid and serum lactate dehydrogenase levels >0.6), but not compatible with empyema according to parapneumonic effusions criteria (moderate free-flowing effusion, negative culture and gram stain of pleural fluid, pH of pleural fluid >7.20). The negative finding of pleural fluid culture, which ruled out bacterial pleurisy, confirmed the diagnosis of AOSD. Additionally, a complication of HPS including hemophagocytic lymphohistiocytosis (HLH) was diagnosed based on HPS/HLH-2009 diagnostic criteria [fever, splenomegaly, hepatic dysfunction, serum ferritin level 78 662 ng/mL (≥500 ng/mL), soluble interleukin-2 receptor 4 140 U/mL (≥2 400 U/mL), hypofibrinogenemia 120 mg/dL (<150 mg/dL), and hyponatremia 129 mEq/L [10]. On the 20th hospital day, we started giving methylprednisolone 500 mg/day for 3 days, with which her fever came down, followed by prednisolone 30 mg/day, and cyclosporine 50 mg/day, after making the diagnosis of AOSD with HPS and DIC and ruling out infections, malignancies, and other connective tissue diseases. After her symptoms, including fever and intermittent rash, general condition, elevated CRP levels, and extremely high serum ferritin levels had improved, we tapered the dose of prednisolone to 22.5 mg/day and increased the dose of cyclosporine to 200 mg/day. On the 79th hospital day, she was transferred to another hospital (Figure 2) for further recuperation.
Figure 1.

Rash on the second hospital day. The figure shows edematous erythema on the patient’s back (A), erythema annulare at the inner aspect of both lower thighs (B), and a granular-sized infiltrative erythema (C), all of which are not typical rashes of adult-onset Still’s disease.
Table 1.
Laboratory data on admission (italic indicates abnormal values).
| Complete blood count | Biochemistry | Immune serum |
|---|---|---|
| White blood cell 13.7×103/µL | BUN 14.3 mg/dL | Rheumatoid factor 5 IU/mL |
| Neutrophil 87.3% | Creatinine 0.57 mg/dL | ANA <40 fold |
| Lymphocyte 9.7% | T-Bil 0.7 mg/dL | MPO-ANCA <1.0 U/mL |
| Red blood cell 393×104/µL | AST 434 U/L | PR3-ANCA <1.0 U/mL |
| Hemoglobin 12.2 g/dL | ALT 96 U/L | HBs-Ag 0.0 IU/mL |
| Hematocrit 34.5% | LDH 952 U/L | HCV-Ab 0.0 C.O.I |
| Platelet 2.1×104/µL | ALP 308 U/L | Tuberculosis IFN-γ (−) |
| Congealing system | CK 144 U/L | Viral antibodies* (−) |
| PT-INR 1.13 | Sodium 129 mEq/L | Bacteriological examination |
| APTT 71.1 second | Potassium 2.7 mEq/L | Blood culture (−) |
| Fibrinogen 120 mg/dL | Chlorine 98 mEq/L | Urinalysis (Qualitative) |
| FDP 46.6 µg/mL | Calcium 7.1 mEq/L | pH 6.5 |
| D-dimer 21.1 µg/mL | Glucose 100 mg/dL | Specific gravity 1.041 |
| Biochemistry | CRP 10.4 mg/dL | Protein (1+) |
| Total protein 5.4 g/dL | Ferritin 78 662 ng/mL | Occult blood (2+) |
| Albumin 2.5 g/dL | sIL-2R 4 140 IU/mL | White blood cell (−) |
PT-INR – prothrombin time-international normalized ratio; APTT – activated partial thromboplastin time; FDP – fibrin/fibrinogen degradation products; BUN – blood urea nitrogen; T-Bil – total bilirubin; AST – aspartate aminotransferase; ALT – alanine aminotransferase; LDH – lactate dehydrogenase; CK – creatine kinase; CRP – C-reactive protein; sIL-2R – soluble interleukin-2 receptor; ANA – antinuclear antibody; MPO-ANCA – myeloperoxidase-anti-neutrophil cytoplasmic antibody; PR3-ANCA – proteinase 3-anti-neutrophil cytoplasmic antibody; HBs-Ag – hepatitis B surface antigen; HCV-Ab – anti-hepatitis C virus antibody; tuberculosis IFN-γ – tuberculosis interferon-gamma release assays.
Viral antibodies including cytomegalovirus, Epstein-Barr virus, and human T cell leukemia virus type 1 were negative.
Figure 2.
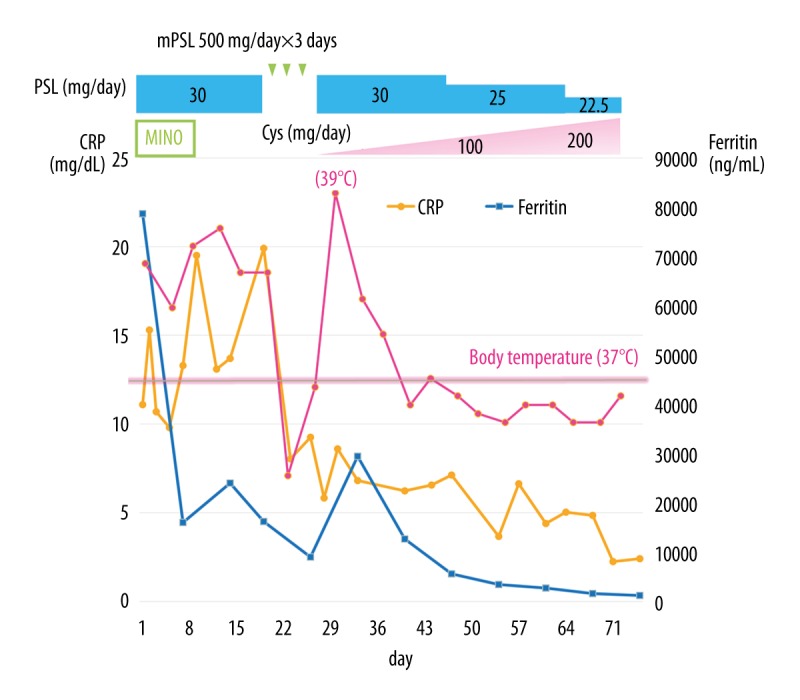
Clinical course. We started prednisolone 30 mg/day (0.85 mg/kg/day) because her condition deteriorated on the second hospital day. Minocycline 200 mg/day was administered concurrently in case her skin lesions were caused by rickettsia. On the 20th hospital day, we started methylprednisolone 500 mg/day for 3 days followed by prednisolone 30 mg/day, and cyclosporine 50 mg/day when we made the diagnosis of adult-onset Still’s disease with hemophagocytic syndrome and disseminated intravascular coagulation after ruling out infections, malignancies, or other connective tissue diseases, which improved her general condition, elevated C-reactive protein levels, and extremely high serum ferritin levels. CRP – c-reactive protein; PSL – prednisolone; mPSL – methylprednisolone; Cys – cyclosporine; MINO – minocycline.
The objective of this case report is show that extremely high serum ferritin level can be a useful diagnostic clue of AOSD complicated with HPS and DIC, even in very old patients.
Discussion
It was difficult to diagnose AOSD in this patient because of the lack of typical symptoms and signs and due to her exceptional old age. Infection, malignancy, and connective tissue disease should be excluded to accurately diagnose AOSD. The triad of AOSD is spiking fever, salmon-pink rash, and arthritis. According to a previous report in which 90 Japanese AOSD cases were analyzed, the incidence rates of spiking fever, rash, and arthritis were over 97%, 87% of which were typical salmon-pink rash appearing with fever and disappearing with defervescence [2]. Therefore, making a diagnosis of atypical AOSD as with our case, without arthritis or typical rash, could be extremely difficult because of its rarity. AOSD accounts for 4% of etiologies of fever of unknown origin, which is the most frequent condition (approximately 25%) among connective tissue disease-related disorders [4]. AOSD is a disease of rather younger patients, with mean age at onset of 36 years, with 2 peaks of distribution at 15–25 and 36–45 years [12,13]. However, patients over 70 years old have been reported, with the oldest patient being 83 years old [14–16]. At 88 years of age, our case is the oldest ever reported, which made the diagnosis still more difficult. However, an increase in AOSD in very elderly people is predicted with the unprecedented aging of the population in Japan.
Extremely high serum ferritin level was one of the characteristics in our patient. Eighty percent of AOSD patients were reported to have increased ferritin levels, 70% of which were more than five times the normal upper limit [2]. Although different levels of serum ferritin denoted the various possibilities of diagnoses (Figure 3), a serum ferritin level above that was reported to be a strong indicator of AOSD [2,17]. However, patients with liver diseases such as Gaucher disease or hemochromatosis, infectious diseases such as HIV, or some malignancies sometimes have ferritin >1 000 ng/mL [18], with the minority being >3 000 ng/mL [19]. Because the median level of serum ferritin in AOSD patients was reportedly 4 752 ng/mL [18], mildly elevated serum ferritin levels from 1 000 to 3 000 ng/mL might not be so specific to AOSD. However, extremely high serum ferritin levels over 10 000 ng/mL were shown only in severe liver damage, multiple blood transfusions, or HPS [19,20]. Additionally, 12% of AOSD patients were reported to have HPS [8,9] with a mean serum ferritin level of 18 179 ng/mL [21]. There was another report asserting that patients with AOSD with leukopenia and/or thrombocytopenia and DIC could be diagnosed as being complicated with HPS [21]. Because our patient had extremely high serum ferritin level and DIC without blood transfusion or severe liver dysfunction, she was considered to be complicated with HPS.
Figure 3.
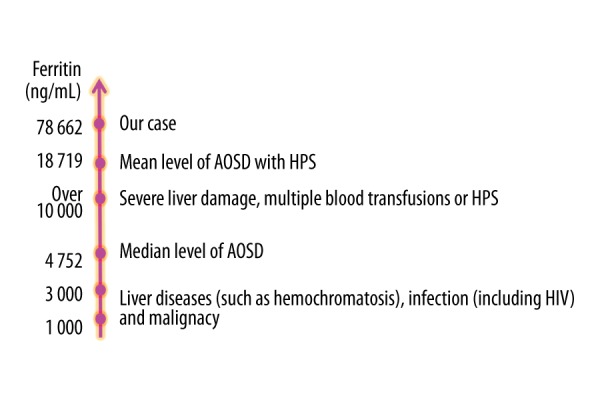
Ferritin levels. Suspected diagnosis varies according to the level of serum ferritin. Serum ferritin levels over 1 000 ng/mL suggest 3 possible diagnoses – liver diseases, infections, and malignancies – some of which exhibit ferritin levels over 3 000 ng/mL. The median level of serum ferritin in adult-onset Still’s disease (AOSD) patients is reportedly 4 752 ng/mL. Extremely high serum ferritin levels >10 000 ng/mL are seen only in severe liver damage, multiple blood transfusions, or hemophagocytic syndrome (HPS). Our patient had an extremely high serum ferritin level of 78 662 ng/mL, which is markedly higher than the mean serum ferritin level of 18 179 ng/mL in AOSD patients with HPS.
Various atypical rashes in AOSD have been reported, such as disseminated erythematous papular, verrucous plaques with vasculitic features, persistent papular characterized by noncaseating dermal granulomas, or peau d’orange-like skin infiltration due to cutaneous mucinosis, although Yamaguchi’s criteria includes only typical salmon-pink rash [5,22–24]. Our patient had only atypical rashes without the typical salmon-pink rash, which was a confusing aspect. Because only 23 cases of AOSD with atypical rash had been reported in the past five years [25], it is extremely difficult to diagnose AOSD correctly in such a patient. There is a report of a patient who died from delayed diagnosis because of the absence of typical salmon-pink rash [25]. It is therefore imperative not to exclude AOSD as a diagnosis only based on the absence of typical rash. Although a study analyzing 90 AOSD cases in Japan showed that all of their patients had arthritis [2], there was another study in which only 70% of cases had arthritis at diagnosis [6]. Moreover, arthritis may be mild and temporary at the early stage of the disease, and develop into severe poly-arthritis within several months after onset [1,26]. It is therefore also important not to exclude AOSD as a diagnosis merely based on absence of arthritis at the early stage of the disease.
Corticosteroids are usually used to treat AOSD, and the recommended initial dose of prednisolone ranges from 0.5 to 1.0 mg/kg/day [1,27]. The overall response rate of AOSD to corticosteroid treatments is over 60%, and 78% of patients with severe systemic complications such as DIC or HPS as in our case reportedly respond well [27]. Although our patient’s serum ferritin levels decreased after starting prednisolone 0.85 mg/kg/day (30 mg/day), her systemic condition worsened with the occurrence of pleuritis. About 55% of HPS cases were reported to be resistant to initial doses of corticosteroids [9], and some researchers insist that AOSD patients with life-threatening complications such as DIC or acute respiratory distress syndrome should be treated with methylprednisolone pulse therapy [28]. We used only 500 mg/day of methylprednisolone for 3 days, so-called half pulse therapy, because of her extreme old age, followed by oral prednisolone and cyclosporine, which was very successful.
Conclusions
We here reported the oldest women with AOSD ever reported, whose extremely high serum ferritin helped diagnose AOSD without typical rash or arthritis. Serum ferritin level higher than 10 000 ng/mL can be a rather specific marker of AOSD if severe liver dysfunction and history of multiple blood transfusions are denied, but serum ferritin level itself is only a non-specific marker of inflammation.
Thus, physicians can use extremely high serum ferritin to make the diagnosis of AOSD, especially when the major symptoms such as arthritis and typical rash are absent, response to the recommended dose of steroid therapy is not sufficient, or it is difficult to make the definite diagnosis of AOSD.
Footnotes
Conflict of interest
None.
References:
- 1.Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014;13:708–22. doi: 10.1016/j.autrev.2014.01.058. [DOI] [PubMed] [Google Scholar]
- 2.Ohta A, Yamaguchi M, Tsunematsu T, et al. Adult Still’s disease: A multi-center survey of Japanese patients. J Rheumatol. 1990;17:1058–63. [PubMed] [Google Scholar]
- 3.Wakai K, Ohta A, Tamakoshi A, et al. Estimated prevalence and incidence of adult Still’s disease: findings by a nationwide epidemiological survey in Japan. J Epidemiol. 1997;7:221–25. doi: 10.2188/jea.7.221. [DOI] [PubMed] [Google Scholar]
- 4.Mert A, Ozaras R, Tabak F, et al. Fever of unknown origin: A review of 20 patients with adult-onset Still’s disease. Clin Rheumatol. 2003;22:89–93. doi: 10.1007/s10067-002-0680-3. [DOI] [PubMed] [Google Scholar]
- 5.Turkyilmaz AK, Devrimsel G, Topaloglu MS, et al. Atypical skin rash in a patient with adult-onset still’s disease: A case report. Oman Medical Journal. 2013:28. doi: 10.5001/omj.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colina M, Zucchini W, Ciancio G, et al. The evolution of adult-onset Still disease: An observational and comparative study in a cohort of 76 Italian patients. Semin Arthritis Rheum. 2011;41:279–85. doi: 10.1016/j.semarthrit.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424–30. [PubMed] [Google Scholar]
- 8.Arlet JB, Huong DLT, Marinho A, et al. Reactive haemophagocytic syndrome in adult-onset Still’s disease: A report of six patients and a review of the literature. Ann Rheum Dis. 2006;65:1596–601. doi: 10.1136/ard.2005.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukaya S, Yasuda S, Hashimoto T, et al. Clinical features of haemophagocytic syndrome in patients with systemic autoimmune diseases: Analysis of 30 cases. Rheumatology. 2008;47:1686–91. doi: 10.1093/rheumatology/ken342. [DOI] [PubMed] [Google Scholar]
- 10.Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Edu Program. 2009;2009:127–31. doi: 10.1182/asheducation-2009.1.127. [DOI] [PubMed] [Google Scholar]
- 11.Gando S, Saitoh D, Ogura H, et al. Natural history of disseminated intravascular coagulation diagnosed based on the newly established diagnostic criteria for critically ill patients: Results of a multicenter, prospective survey. Crit Care Med. 2008;36:145–50. doi: 10.1097/01.CCM.0000295317.97245.2D. [DOI] [PubMed] [Google Scholar]
- 12.Gerfaud-Valentin M, Maucort-Boulch D, Hot A, et al. Adult-onset still disease: Manifestations, treatment, outcome, and prognostic factors in 57 patients. Medicine. 2014;93:91–99. doi: 10.1097/MD.0000000000000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magadur-Joly G, Billaud E, Barrier JH, et al. Epidemiology of adult Still’s disease: Estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54:587–90. doi: 10.1136/ard.54.7.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ichiki H, Shishido M, Nishiyama S. [Two cases of adult onset of Still’s disease in the elderly] Nihon Ronen Igakkai Zasshi. 1992;29(12):960–64. doi: 10.3143/geriatrics.29.960. [in Japanese] [DOI] [PubMed] [Google Scholar]
- 15.Steffe LA, Cooke CL. Still’s disease in a 70-year-old woman. JAMA. 1983;249:2062–63. [PubMed] [Google Scholar]
- 16.Uson J, Pena JM, Del Arco A, et al. Still’s disease in a 72-year-old man. J Rheumatol. 1993;20:1608. [PubMed] [Google Scholar]
- 17.Ota T, Higashi S, Suzuki H, Eto S. Increased serum ferritin levels in adult Still’s disease. Lancet. 1987;329:562–53. doi: 10.1016/s0140-6736(87)90204-2. [DOI] [PubMed] [Google Scholar]
- 18.Fautrel B. Ferritin levels in adult Still’s disease: Any sugar? Joint Bone Spine. 2002;69:355–57. doi: 10.1016/s1297-319x(02)00409-8. [DOI] [PubMed] [Google Scholar]
- 19.Bishara R, Braun-Moscovici Y, Dagan A, et al. Severe hyperferritinemia – a clue for severe hepatitis in a patient with adult-onset Still’s disease. Clin Rheumatol. 2016;35:795–800. doi: 10.1007/s10067-014-2829-2. [DOI] [PubMed] [Google Scholar]
- 20.Meijvis SC, Endeman H, Geers AB, Ter Borg EJ. Extremely high serum ferritin levels as diagnostic tool in adult-onset Still’s disease. Neth J Med. 2007;65:212–14. [PubMed] [Google Scholar]
- 21.Hot A, Toh ML, Coppéré B, et al. Reactive hemophagocytic syndrome in adult-onset Still disease: Clinical features and long-term outcome: A case-control study of 8 patients. Medicine. 2010;89:37–46. doi: 10.1097/MD.0b013e3181caf100. [DOI] [PubMed] [Google Scholar]
- 22.Kaur S, Bambery P, Dhar S. Persistent dermal plaque lesions in adult onset Still’s disease. Dermatology. 1994;188:241–42. doi: 10.1159/000247150. [DOI] [PubMed] [Google Scholar]
- 23.Luebbe I, Hofer M, Chavaz P, et al. Adult-onset Still’s disease with persistent plaques. Br J Dermatol. 1999;141:710–13. doi: 10.1046/j.1365-2133.1999.03115.x. [DOI] [PubMed] [Google Scholar]
- 24.Phillips WG, Weller R, Handfield Jones SE, Kobza-Black A. Adult Still’s disease. Br J Dermatol. 1994;130:511–13. doi: 10.1111/j.1365-2133.1994.tb03388.x. [DOI] [PubMed] [Google Scholar]
- 25.Ishiguro M, Kishimoto H, Nakashima H, et al. [A fatal case of adult-onset still’s disease showing two types of atypical eruptions] The Nishinihon Journal of Dermatology. 2016;78:19–23. [in Japanese] [Google Scholar]
- 26.Elkon KB, Hughes GRV, Bywaters EGL, et al. Adult-onset still’s disease. Arthritis Rheum. 1982;25:647–54. doi: 10.1002/art.1780250607. [DOI] [PubMed] [Google Scholar]
- 27.Franchini S, Dagna L, Salvo F, et al. Efficacy of traditional and biologic agents in different clinical phenotypes of adult-onset Still’s disease. Arthriti Rheum. 2010;62:2530–35. doi: 10.1002/art.27532. [DOI] [PubMed] [Google Scholar]
- 28.Iglesias J, Sathiraju S, Marik PE. Severe systemic inflammatory response syndrome with shock and ARDS resulting from Still’s disease: Clinical response with high-dose pulse methylprednisolone therapy. CHEST J. 1999;115:1738–40. doi: 10.1378/chest.115.6.1738. [DOI] [PubMed] [Google Scholar]