

Abstract
The use of calcium indicators has greatly enhanced our understanding of neural dynamics and regulation. The nematode Caenorhabditis elegans, with its completely mapped nervous system and transparent anatomy, presents an ideal model for understanding real-time neural dynamics using calcium indicators. In combination with microfluidic technologies and experimental designs, calcium-imaging studies using these indicators are performed in both free-moving and trapped animals. However, most previous studies utilizing trapping devices, such as the olfactory chip described in Chronis et al., have devices designed for use in the more common hermaphrodite, as the less common male is both morphologically and structurally dissimilar. An adapted olfactory chip was designed and fabricated for increased efficiency in male neuronal imaging with using young adult animals. A turn was incorporated into the worm loading port to rotate the animals and to allow for the separation of the individual neurons within a bilateral pair in 2D imaging. Worms are exposed to a controlled flow of odorant within the microfluidic device, as described in previous hermaphrodite studies. Calcium transients are then analyzed using the open-source software ImageJ. The procedure described herein should allow for an increased amount of male-based C. elegans calcium imaging studies, deepening our understanding of the mechanisms of sex-specific neuronal signaling.
Keywords: Neuroscience, Issue 127, Microfluidics, calcium imaging, GCaMP, C. elegans, males, CEM neuron, ascaroside
Introduction
Microfluidic devices provide increased access to precisely controlled environments, wherein animals, such as the nematode C. elegans, can be experimentally manipulated1. These studies include behavioral assays, calcium imaging studies, or even screenings for specific phenotypes, resulting in more exact measurements of experimental outcomes1,2,3,4,5,6. Microfluidics provide small-scale liquid conditions through which detailed experiments can be run while utilizing minimal amounts of reagents. There is a constant production of new microfluidic device designs, and the use of each varies, from arenas that allow for the natural sinusoidal motion of C. elegans in behavioral assays and neural imaging studies, to trap devices used in neural imaging and olfactory studies, to devices that allow for high-throughput phenotypic analysis in genetic screens4,5,6,7. Following the fabrication of a master mold, microfluidic devices are inexpensive to construct—given the reusability of the master—and easy to use, allowing for rapid data generation via high-throughput studies. The fabrication of devices using polymers such as polydimethylsiloxane (PDMS) allows for the creation of new devices within hours.
Calcium imaging studies use genetically encoded calcium indicators (GECIs) expressed in target cells to measure the neural dynamics of those cells in real time8,9,10,11. The transparent nature of C. elegans allows for the recording of the fluorescent levels of these proteins in live animals. Traditionally, GECIs rely on the green fluorescent protein (GFP)-based sensor GFP-Calmodulin-M13 Peptide (GCaMP), although more recent studies have adapted these sensors to allow for better signal-to-noise ratios and red-shifted excitation profiles. Following the development of GCaMP3, proteins with these specifications have varied, including sensors such as GCaMP6s and GCaMP6f (slow and fast fluorescence off-rates, respectively), as well as RFP-Calmodulin-M13 Peptide (RCaMP), which has a red-shifted activation profile. The combination of these GECIs with C. elegans cell-specific gene promoter sequences can target cells of interest, particularly sensory neurons12,13,14,15,16.
While the ease of C. elegans use in microfluidic studies is apparent, almost all studies have focused on hermaphrodites. Despite males only accounting for 0.01-0.02% of the wild type population, invaluable findings can arise from their characterization. While the physical connectome of the hermaphrodite nervous system has been fully mapped for decades17, the male connectome remains incomplete, especially in the head region of the animal18. The use of calcium imaging in males will help to generate an understanding of the male nervous system and the differences that arise between the two sexes. The smaller size of C. elegans adult males prevents effective and reliable trapping in the loading ports of traditional olfactory devices designed for larger hermaphrodites. To address this, a modified version of the Chronis Olfactory Chip19 was developed with a narrower loading port, a lower channel height, and turns in the worm loading port (which rotate the animal), allowing for the visualization of bilateral left/right neuronal pairs. This design permits: (1) the effective trapping of young adult males, (2) a more reliable orientation of the animal for the visualization of both members of bilateral paired neurons, and (3) the precise imaging of neural activity in male neurons.
Increasingly, studies show that C. elegans males respond differently than hermaphrodites to a variety of ascarosides (ascr), or nematode pheromones20,21,22,23,24. Therefore, developing an understanding of the neural dynamics and representations within the male connectome has become even more pertinent. Male C. elegans contain 87 sex-specific neurons not present in the hermaphrodite25,26, altering the connectome in as-yet undetermined ways. Being able to image these unique neural dynamics will allow us to better understand sex-specific responses and neural representations.
This protocol describes the use of a male-adapted olfactory chip for the neural imaging of male C. elegans chemosensation. The nociceptive neuron ASH responds reliably to 1 M glycerol in males, consistent with previous hermaphroditic studies27. Exposure to ascarosides may elicit responses that are variable from animal to animal, requiring a larger number of animals to be tested. The response of the male-specific CEM neurons has previously been shown, through both electrophysiology and calcium imaging studies, to respond variably to ascaroside #323.
Protocol
1. Device Fabrication
NOTE: See reference1.
NOTE: Silicon master molds were fabricated using standard photolithographic techniques for patterning SU-8 photoresist on a silicon master1,7. Photomasks for wafer patterning were printed at 25,000 dpi. The male-adapted device features a Chronis Olfactory Chip design19 with a change in the worm loading port, adapting a design obtained from M. Zimmer (personal correspondence, 2016). A turn is included to control the rotation of the animals. The width of the worm loading port channel is narrowed to 50 μm. All channels are 32 μm tall. Once a silicon master mold is available to the user, the user can follow the subsequent protocol, as described previously1.
Mix PDMS base and curing agent at a 10:1 ratio by weight.
Mix thoroughly with transfer pipettes.
Degas the mixture in a vacuum desiccator for 1 h, until all visible bubbles are removed.
Pour the mixture onto a silicon mold master in a 150 mm diameter dish until it is 5 mm thick (100 g). Use a Pasteur pipette to remove any bubbles or dust that have been introduced to the mixture.
Bake at 65 °C for at least 3 h, or overnight.
Cut the PDMS away from the mold using a scalpel and cut the separate devices apart using a razor blade.
Punch inlet and outlet holes with a 1 mm dermal punch.
Flush the holes with dH2O, ethanol, and again with dH2O to remove particles from the punches. Dry the device in an air stream pulse.
Clean both channel sides and the top side of the device with adhesive tape, removing any dust or debris remaining on the device to allow for successful bonding.
- Plasma-bond the device, channel-side down, to a no. 1 cover glass.
- Expose cover glass and device (channel-side up) to air plasma using conditions that allow for proper bonding, such as 100 W for 30 s or 24 W for 60 s. NOTE: Settings can be adjusted to improve the bonding efficiency. The plasma-bonding conditions are not as critical as proper cleaning when attempting to improve the bonding efficiency. An insufficiently cleaned device will not bond, even under ideal plasma conditions.
- Invert the cover glass onto the channel side of the device and press down with the thumb for 5 s.
2. Buffer Preparation
Dilute 1x S Basal (100 mM NaCl and 0.05 M KPO4, pH 6.0) from a sterile 10x stock.
Dilute 1 M tetramisole stock to a final concentration of 1 mM in 1x S Basal for all buffer solutions.
- Add fluorescein to both the "flow control" and "buffer" reservoirs.
- Create a 100-mg/mL stock of fluorescein in 1x S Basal.
- Dilute the stock to final concentrations of 1 µg/mL in the flow control and 0.1 µg/mL in the buffer.
- Create the stimuli.
- Dilute glycerol to a final concentration of 1 M in 1X S Basal.
- Dilute ascaroside #3 (ascr#3) to a final concentration of 1 µM into 1X S Basal.
3. Device Setup
NOTE: See1.
Figure 1. Microfluidic device setup. (A) Reservoirs and tubing. A 30 mL syringe without a plunger serves as the "reservoir." This is attached to a Luer valve with three flow options. One outlet is connected to a 3 mL syringe with a plunger, while the other is connected to a needle (orange) that is inserted into the tubing that connects to the microfluidic device. (B) The overall setup of the microfluidic imaging experiment. The device is placed on a stage of an inverted epifluorescence microscope, above the objective lenses. The "flow control" buffer travels through a 3-way valve that is controlled by a unit on the shelf above the setup. Lines containing buffers are then inserted into the appropriate device ports. (C) The ports of the microfluidic device. The "flow control" ports flank the other inlet ports: the "stimulus" and "buffer" ports. The "outlet" port is the right-most port. Due to the location of the worm loading arena, the "worm loading" port is the central-most port on the device. Please click here to view a larger version of this figure.
Prepare three fluid reservoirs by attaching a 30 or 60 mL syringe to 3-way Luer valve, with a 3 mL syringe and needle attached to the Luer valve as well (as in Figure 1A). Connect the needle to tubing that extends to the microfluidic device (as in Figure 1A-B).
Remove the air bubbles from the reservoir and tubing.
Fill the 3 mL syringe with attached tubing with 1x S Basal and insert it into the outlet port.
Gently apply pressure to the syringe until the buffer appears at the top of the inlet holes.
Connect the flow control, buffer, and stimulus tubing to appropriate inlet holes (as in Figure 1B-C), ensuring that liquid drops are present on both the loading port hole and the buffer tubing to be attached.
Again, gently apply pressure to the syringe that is connected to the outlet port until droplets appear in the worm loading port inlet.
Insert a solid blocking pin into the worm loading port.
Remove the syringe from the outlet port and attach the outlet line connected to the house vacuum (-670 Torr).
- Inspect the device for any bubbles in the flow channels, visually and through video confirmation via a software compatible with the camera used, such as the open-source software Micro-Manger. See step 6 for tips on using Micro-Manager.
- If any bubbles are present, wait for them to dislodge or be absorbed into the PDMS wall prior to loading any animals; the presence of bubbles will disturb the proper flow of fluids through the device.
- Using a GFP filter, confirm proper flow dynamics within the device prior to worm loading by actuating the 3-way valve and observing the switching of buffers.
- After opening Micro-Manager, click on "Live" to observe a live image of the device. Turn on the fluorescent light source to observe the flow of buffers in the device (Figure 2D-2E).

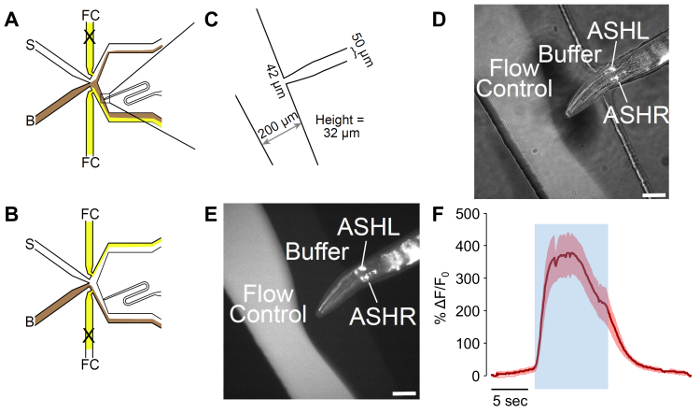
Figure 2: A male-adapted microfluidic olfactory chip. (A) The flow patterns of the device when the worm is exposed to buffer. Buffer (B) is shown in brown, and flow control (FC) is shown in yellow, with stimulus (S) in white. The worm loading port has been adapted to include a curve, which allows for better control of worm orientation. (B) The flow patterns of the device when the worm is exposed to stimulus. Buffer (B) is shown in brown, and flow control (FC) is shown in yellow, with stimulus (S) in white. (C) Measurements of the adapted device as fabricated. The worm loading port ends in a 42 µm opening, with a 50 µm channel designed for the male width. The measured height of the channels is 32 µm, despite a target of 25 µm in the design. (D-E) A trapped male expressing psra-6::GCaMP3. The sra-6 promoter is not ASH-specific, and some expression may be observed in the ASI neuron, although no calcium transients were observed in ASI. The image is (D) a combination of bright-field and fluorescent illumination, while (E) is fluorescent only. The scale bars denote 42 µm. (F) The ASH neuron responds to 1 M glycerol stimulation with robust neural activity. The blue area denotes the time of the 1 M glycerol stimulus. The shaded region denotes the standard error, with n = 20 pulses from seven worms. The red traces denote depolarizing responses. The Y-axes show ΔF/F0. The scale bar denotes 5 s. Please click here to view a larger version of this figure.
4. Animal Preparation
NOTE: See reference23.
- Imaging ASH responses to 1 M glycerol.
- Place approximately 20 C. elegans males that are positive for psra-6::GCaMP3 array expression onto a nematode growth medium (NGM) agar plate seeded with a lawn of OP50 E. coli. Use the expression of fluorescent GECI and/or a co-injection marker for the identification of array-positive animals. NOTE: Array positive animals will fluoresce according to the GECI used (i.e., animals expressing GCaMP will fluoresce green under blue-light stimulation, while RCaMP animals will fluoresce red under green-light stimulation). Co-injection markers can range from other fluorescent proteins, such as GFP and RFP, to phenotypic markers, such as rol-6, or can rescue a dominant phenotype, such as the pha-1 mutation28.
- If picking immediately prior to the assay, pick young adult males. If picking the day prior to the assay, pick L4 larval males.
- Imaging the CEM responses to 1 µM ascr#3.
- Pick approximately 20 L4 C. elegans males (fkEx98[ppkd-2::GCaMP::SL2::dsRED + pBX-1]; pha-1(e2123ts); him-5(e1490); lite-1(ce314)) that are positive for dsRed co-injection marker expression. NOTE: dsRed expression within the ray neurons of the male tale is easier to observe and confirm than GCaMP expression within the four CEM neurons.
- Isolate these males from hermaphrodites on an NGM agar plate seeded with a lawn of OP50 E. coli for 5-14 h before performing the imaging experiment. NOTE: Males not isolated for a minimum of 5 h do not behaviorally respond to ascr#3 and therefore may exhibit even fewer calcium transients to the ascaroside than observed here.
5. Animal Loading
NOTE: See refefence1.
- Pick one worm onto an unseeded NGM agar plate using standard worm maintenance techniques.
- Pick worms by flaming a pick (made of flattened platinum wire), picking bacteria onto the pick, and "dabbing" a worm to pick it up. Gently place the worm onto the new plate, allowing it to crawl off on its own.
Add approximately 5 mL of 1x S Basal to the unseeded plate, such that plate is flooded.
- Draw the worm into a loading syringe (i.e., 3 mL syringe with attached tubing) that has been pre-filled with 1x S Basal.
- Be sure to suck the worm only into the tubing, not all the way into the syringe. NOTE: If the worm travels into the syringe, it is near impossible to get it back into the tubing.
Turn off the vacuum to stop the flow by turning the outlet Luer valve.
Remove the solid pin blocking the worm loading port.
Turn the Luer valve connected to the outlet port (Figure 1B) so that it is venting. NOTE: Use a live video feed while loading the worm to confirm the location and orientation of the animal (steps 5.8-5.13).
Insert the worm loading tube into the worm loading port.
Gently apply pressure to the syringe until the worm appears in the loading channel.
If the worm starts to enter the channel tail-first, pull on the syringe plunger to prevent the worm from entering the channel.
Switch between applying and reversing pressure until the head enters the channel first.
Open the vacuum by turning the 3-way Luer valve connected to the outlet port to open it to vacuum instead of atmosphere.
Manually apply pressure by depressing the syringe plunger to orient and place the worm head such that it is exposed to the buffer flow channel, but not so far that the head can move around freely (Figure 2D-2E).
6. Stimulus and Acquisition
Using an open-source microscopy software, such as Micro-Manager, record by capturing images as a TIFF stack at 10 frames/s using blue-light excitation (470 nm) for 30 s.
- Set the exposure on the main menu to 100 ms.
- Open "Multi-D Acq." from the main menu of the software. Set the "number" to "300," and the "interval" to "0." Click "Acquire!" to acquire the video.
- Apply a 10 s pulse of the stimulus 5 s after initiating acquisition. Adjust the duration of stimulus application as desired.
- After acquiring 5 s of video, change the 3-way valve controlling the flow control buffer to apply the stimulus to the animal being tested. Click the left-most button on the valve link (Figure 1B).
- After 10 s of stimulus exposure (this time can be adjusted as desired by the user), alter the flow of buffers by again pressing the left-most button on the valve link.
Record under buffer only until the 30-s window is complete to allow the GECI fluorescence to return to baseline.
Repeat as desired. Wait 30 s between the end of acquisition and the initiation of the next trial.
7. Image Analysis
Open the TIFF stack with the open-source software, ImageJ, by dragging file into the ImageJ window.
Click using the cursor and drag to set the region of interest (ROI) around the neuron of interest. Set the region to include the soma of the neuron of interest (as in Figure 3A).
Plot the z-stack of the fluorescence intensity of the ROI across stacks by clicking Open -> Image -> Stacks -> Plot Z-axis Profile.
Click "List" in the window that opens. Click Edit -> Copy to copy the values. Paste the values into a spreadsheet program.
Analyze the background fluorescence for each pulse by dragging the ROI to a region of the worm that does not contain GCaMP expression.
Perform background subtraction for each pulse by subtracting the background fluorescence value from the neuron fluorescence intensity value.
- Calculate ΔF/F0 for each frame of each pulse.
- Calculate F0 as the average intensity value of the ROI for first 1 s of acquisition (e.g., frames 1-10).
- Calculate ΔF/F0 by dividing the background-subtracted value for the frame of interest by the calculated F0 value.
Repeat for every neuron imaged and every stimulus pulse.
For neurons with consistent response profiles, such as ASH, average all pulses for each neuron and calculate the SEM (as in Figure 2F).
Plot the average ΔF/F0 with SEM over time for each neuron. NOTE: In this instance, it is common practice to include heatmaps of the individual neuronal responses of each trial as well. In neurons that do not exhibit consistent changes in calcium transients upon exposure to stimuli across repeated stimulations, or in different individuals23, it may be more applicable to show individual pulse traces (as in Figure 4). See the Discussion for details on determining how to display the data.


Representative Results
An example of the overall device setup can be seen in Figure 1A-B. Figure 1A depicts the proper reservoir construction and setup. Figure 1B shows the connections of the reservoirs to the microfluidic device. Figure 1C depicts a microfluidic device with individual ports labeled for clarity.
The design of the male-adapted microfluidic device contains a curve in the loading port, but flow dynamics are identical to the device designed by Chronis et al.19(Figure 2A-2C). The flow of buffers can be controlled by altering which flow control valve is open (Figure 2A-2B). The measurements of the device as fabricated vary from the designed file. The measurements provided in Figure 2C are "as fabricated" measurements.
After loading male C. elegans into the male-adapted olfactory device, their placement and orientation, as well as channel flow dynamics, can be verified via both bright-field and fluorescent imaging (Figure 2D-2E). The exposure of worms expressing GCaMP3 in the nociceptive neuron, ASH, to 1 M glycerol results in visible changes in fluorescence within the ASH neuron, indicative of neural activity (Figure 2F). Subtle changes in fluorescence may not be visible by eye, but software can be used to quantify these changes. The free ImageJ software can be used to analyze and quantify the fluorescent intensity of ASH neurons upon exposure to 1 M glycerol over time (Figure 2F). This is similar to what is observed in hermaphrodites27 and, due to the robustness of the ASH neuronal response to glycerol, this is observed in all animals tested.
A small amount of axial or rotational movement is expected in unparalyzed animals, often necessitating a neuron-tracking algorithm during video analysis (Figure 3A). The addition of a paralytic in the buffers (e.g., 1 mM tetramisole) nearly eliminates this effect, although some animals (~10%) still move during the trials. This can be circumvented by: (a) using older males, which are more efficiently trapped; (b) decreasing the width or thickness of the worm loading port even further; or (c) either increasing the concentration of the paralytic used or using another paralytic. This will also ensure that there is not too much of the head past the end of the trap and exposed to the odor channel. If a trial with worm movement occurs, the area of analysis can be moved and reread from the new neuronal location, starting at the frame after which the movement occurs (Figure 3B). Manual reconstruction of the neural traces by the user is required in this instance. Scripts that analyze the fluorescent changes within the neuron and that follow the neuron's center as it moves can also be written19.
Male C. elegans sense attractive biogenic pheromones called ascarosides via the four sex-specific CEM neurons23. When calcium transients are observed in males, the responses are variable in shape, sign, and magnitude between both neurons and animals (Figure 4A-B). However, male response to pheromones is not as reliably observed as calcium transients in many animals (Figure 4C). This is not discouraging, as most ascarosides do not elicit calcium transients upon sensation13,14,15.
Figure 3: Addressing worm movement during the image acquisition period. (A) ΔF/F0 (change in fluorescent intensity divided by the average background fluorescent intensity for first 1 s of acquisition) was calculated using ImageJ by defining an area where the neuron of interest was reliably static for 1 s. During the stimulus pulse (blue area), the animal moved, as seen in the images above the calcium trace, resulting in the neuron no longer being contained within the analyzed area (yellow box). The scale bars denote 42 µm. the dashed lines show the region of the trace that corresponds to the location of the worm in each image. (B) If the area of analysis is moved to follow the neuron during the trial and the individual traces are rebuilt to convey the actual neural dynamics, the trace appears as if the animal did not move. The dashed lines show the region of the trace that was corrected for worm movement. Traces for both ASH neurons, left and right (ASHL (red) and ASHR (blue), respectively) are shown. The Y-axes show ΔF/F0. The scale bar denotes 5 s. Please click here to view a larger version of this figure.
Figure 4: Male C. elegans CEM response to 1 μM ascr#3 is variable. The male-specific CEM neurons display unique patterns of response to biogenic pheromones. (A) The responses observed in each CEM neuron for one pulse in one responsive animal are shown. There are four CEM neurons: dorsal right (CEM DR), dorsal left (CEM DL), ventral right (CEM VR), and ventral left (CEM VL). Three of the four neurons respond with depolarizations of varying shape and magnitude, with the fourth not responding to the ascaroside. (B) Approximately one-third of the trapped animals (2/7 tested in this study) result in only three neurons able to be imaged. The responses of a worm in this orientation resulted in CEM calcium transients that were different than those observed in the first worm. This was not due to the change in orientation of the worm, as CEM DR is visible in both orientations and exhibits variable response between animals. (C) Many worms do not respond with any detectable calcium transients (5/7 tested in this study). Individual traces shown are representative of the single animals shown in the images above the plots. The scale bars denote 42 µm. Traces: The blue area denotes the time of 1 μM ascr#3 exposure. The red traces denote the depolarizing response. The black traces denote no observed response. The Y-axes show ΔF/F0. The scale bar denotes 5 s. Please click here to view a larger version of this figure.
Discussion
The male-adapted olfactory chip incorporates a turn into a narrower loading port, which allows for more control of the orientation and for the efficient trapping of male C. elegans. This allows for the visualization of both the left and right members of neuronal bilateral pairs, without the need for z-stacking. This curve leads to an orientation away from vertical 100% of the time in worms where only one bilateral pair is targeted with a fluorescent marker, such as ASH (Figure 2D-E)29,30. However, in neuronal classes with four radially symmetric neurons, such as CEM, all four neurons are visible only one-third of the time. Another third of worms tested have only three of the four neurons visible, and for the remaining third, the only distinguishable difference is between the dorsal and ventral cell bodies, not the left-right asymmetry (data not shown). The narrower port is combined with a lower channel height to prevent worm fluctuation across the z-axis. This design allows for the imaging of males in future studies, which, when combined with the constantly increasing knowledge of the male connectome25,31, will allow for a better understanding of sex-specific neural function.
The analysis performed in this protocol uses the free software ImageJ to measure changes in fluorescence in the neuron of interest. With the current design, 1 mM tetramisole in the buffer effectively paralyzes the worms and prevents movement of the neurons being imaged. If movement is not preventable, or if the user wishes to avoid the use of a paralytic, more complex tracking scripts must be written that track the neurons as they move7. However, in this protocol, male worms only move when they were too small to be effectively constrained by the loading port and when presented with an extremely aversive stimulus, such as glycerol. Even in these instances, the movement is brief and does not require large amounts of tracking adjustments—setting an ROI around the new neuron location alleviates the incorrect fluorescent readouts (Figure 2).
A limitation of single-worm, trap-based imaging is that only one worm can be imaged at a time1. Another limitation of these traps is that worms can get stuck within the device, causing devices to be clogged and "used up" after imaging only a few worms. However, the quick turnaround time for the fabrication of new devices from a master mold alleviates this downside. Extended blue-light illumination has also been shown to induce photodamage in C. elegans32,33. The relatively short experimental time frame of this protocol (30 s) allows for imaging without measurable photobleaching. However, to avoid photobleaching and photodamage in longer experiments, the light source can be pulsed7. For example, during each 100-ms exposure, the light can be pulsed for 10 ms. This has been shown to eliminate increased body autofluorescence over time7.
In order to properly test males for their responses to ascarosides, larval-stage 4 (L4) males must first be isolated from hermaphrodites, for at least 5 h, in order to achieve a near-naïve response to the pheromones23. Isolation for less than this length of time may cause animals to fail to respond to the ascarosides. However, this isolation is not necessary when testing non-ascaroside cues, such as glycerol. For the sake of consistency, however, animals were always isolated at least 5 h prior to calcium imaging. In some neurons, such as the CEM, not every stimulation will elicit a neuronal response, and each CEM that does responds does so to generate a certain "code" of neural representation. This phenomenon in CEM has been observed via electrophysiology studies, as well as with calcium imaging studies like the ones described here23. Thus, measurable calcium transients in CEM neurons in every ascaroside-exposed animal are not guaranteed23. In fact, many of the ascarosides investigated to date do not elicit measurable calcium transients13,15,34,35. The successful elicitation of measurable transients was observed during only one of three pulses of pheromone in two of the five animals exposed to the ascaroside of interest (Figure 3). This matches the rate of success previously observed in other labs23. This variability is a limitation when studying pheromone response and is not due to the male-based focus of this protocol.
When investigating calcium transients elicited in response to ascarosides, one should not dismiss a lack of consistent response without further investigation. This can be tested through experiments such as electrophysiology studies to confirm the variable response within a neuronal class. For neurons, such as ASH, that respond reliably, a lack of consistent response could be indicative of larger experimental problems, such as errors in stimulus control. The peak intensity of the responses can also be investigated if variability is expected in the response. The traces can be plotted with the standard deviation or standard error (as in Figure 2F). If the standard deviation is small, the traces can be plotted and analyzed as such. If there is noticeable amount of variation leading to moderate standard deviations, the data can be plotted the same, with accompanying heatmaps sorted by response "type" to show the response-by-response variation. If there is significant variation (Figure 3), wherein the peaks cannot be distributed in a Gaussian manner, responses can be categorized into response "types" (e.g., depolarizing, hyperpolarizing, or non-polarizing)23. Responses that fall into a certain "type" can be plotted and analyzed together. Similarly, heatmaps should accompany this analysis as well.
Moving forward, this device can be adapted to allow for the imaging of larval-staged nematodes by narrowing the loading port even further. Further narrowing of the end of the loading port will allow for the constraint of the animal to allow for imaging of just the cilia of the sensory neurons, as opposed to the cell body. While other devices are designed for the more commonly studied hermaphrodite, this adapted olfactory chip allows for the imaging of neural activity in male neural circuits. As the connectome of the male is still being elucidated, being able to measure neural dynamics in sex-specific networks is critical to fully understanding neuronal signaling. Differences between hermaphroditic and male responses can now be tested and measured using this device.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Manuel Zimmer for providing us with the initial design file that was adapted for use with males; Frank Schroeder for the synthesis and supply of ascr#3; Ross Lagoy for the insight and assistance with imaging and analysis; and Laura Aurilio for the master fabrication and who, alongside Christopher Chute, contributed to the review of this manuscript. Funding for this work was provided under the National Institutes of Health grant 1R01DC016058-01 (J.S.), the National Science Foundation grant CBET 1605679 (D.R.A.), and the Burroughs Wellcome Career Award at the Scientific Interface (D.R.A.).
References
- Lagoy RC, Albrecht DR. Microfluidic Devices for Behavioral Analysis, Microscopy, and Neuronal Imaging in Caenorhabditis elegans. Methods Mol Biol. 2015;1327:159–179. doi: 10.1007/978-1-4939-2842-2_12. [DOI] [PubMed] [Google Scholar]
- Ben-Yakar A, Chronis N, Lu H. Microfluidics for the analysis of behavior, nerve regeneration, and neural cell biology in C. elegans. Curr Opin Neurobiol. 2009;19(5):561–567. doi: 10.1016/j.conb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis N. Worm chips: Microtools for C. elegans biology. Lab on a Chip. 2010;10(4):432–437. doi: 10.1039/b919983g. [DOI] [PubMed] [Google Scholar]
- Lee H, Crane MM, Zhang Y, Lu H. Quantitative screening of genes regulating tryptophan hydroxylase transcription in Caenorhabditis elegans using microfluidics and an adaptive algorithm. Integr Biol (Camb) 2013;5(2):372–380. doi: 10.1039/c2ib20078c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockery SR, et al. A microfluidic device for whole-animal drug screening using electrophysiological measures in the nematode C. elegans. Lab Chip. 2012;12(12):2211–2220. doi: 10.1039/c2lc00001f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S, et al. Large-scale microfluidics providing high-resolution and high-throughput screening of Caenorhabditis elegans poly-glutamine aggregation model. Nat Commun. 2016;7:13023. doi: 10.1038/ncomms13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsch J, Ventimiglia D, Bargmann CI, Albrecht DR. High-throughput imaging of neuronal activity in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2013;110(45):E4266–E4273. doi: 10.1073/pnas.1318325110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerboom J, et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neuro. 2013;6:2. doi: 10.3389/fnmol.2013.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badura A, Sun XR, Giovannucci A, Lynch LA, Wang SSH. Fast calcium sensor proteins for monitoring neural activity. Neurophotonics. 2014;1(2):025008. doi: 10.1117/1.NPh.1.2.025008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatro ET. Brain-wide imaging of neurons in action. Front Neural Circuits. 2014;8:31. doi: 10.3389/fncir.2014.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6(12):875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JS, et al. Balancing selection shapes density-dependent foraging behaviour. Nature. 2016;539(7628):254–258. doi: 10.1038/nature19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JS, Dobosiewicz M, Butcher RA, McGrath PT, Bargmann CI. Regulatory changes in two chemoreceptor genes contribute to a Caenorhabditis elegans QTL for foraging behavior. Elife. 2016;5 doi: 10.7554/eLife.21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, et al. Two Chemoreceptors Mediate Developmental Effects of Dauer Pheromone in C. elegans. Science. 2009;326(5955):994–998. doi: 10.1126/science.1176331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath PT, et al. Parallel evolution of domesticated Caenorhabditis species targets pheromone receptor genes. Nature. 2011;477(7364):321–325. doi: 10.1038/nature10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C, Schultheis C, Husson SJ, Liewald JF, Gottschalk A. Specific Expression of Channelrhodopsin-2 in Single Neurons of Caenorhabditis elegans. PLoS ONE. 2012;7(8):e43164. doi: 10.1371/journal.pone.0043164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The Structure of the Nervous System of the Nematode Caenorhabditis elegans. Phil Trans of the Royal Soc of Lon. 1986;314(1165):1. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- White JQ, et al. The sensory circuitry for sexual attraction in C. elegans males. Curr Biol. 2007;17(21):1847–1857. doi: 10.1016/j.cub.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Chronis N, Zimmer M, Bargmann CI. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat Meth. 2007;4(9):727–731. doi: 10.1038/nmeth1075. [DOI] [PubMed] [Google Scholar]
- Chute CD, Srinivasan J. Chemical mating cues in C. elegans. Semin Cell Dev Biol. 2014;33:18–24. doi: 10.1016/j.semcdb.2014.06.002. [DOI] [PubMed] [Google Scholar]
- Izrayelit Y, et al. Targeted metabolomics reveals a male pheromone and sex-specific ascaroside biosynthesis in Caenorhabditis elegans. ACS Chem Biol. 2012;7(8):1321–1325. doi: 10.1021/cb300169c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludewig AH, Schroeder FC. Ascaroside signaling in C. elegans. WormBook. 2013. pp. 1–22. [DOI] [PMC free article] [PubMed]
- Narayan A, et al. Contrasting responses within a single neuron class enable sex-specific attraction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2016;113(10):E1392–E1401. doi: 10.1073/pnas.1600786113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan J, et al. A blend of small molecules regulates both mating and development in Caenorhabditis elegans. Nature. 2008;454(7208):1115–1118. doi: 10.1038/nature07168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammut M, et al. Glia-derived neurons are required for sex-specific learning in C. elegans. Nature. 2015;526(7573):385–390. doi: 10.1038/nature15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Albertson DG, Thomson JN. The Caenorhabditis elegans male: postembryonic development of nongonadal structures. Dev Biol. 1980;78(2):542–576. doi: 10.1016/0012-1606(80)90352-8. [DOI] [PubMed] [Google Scholar]
- Hilliard MA, et al. In vivo imaging of C. elegans ASH neurons: cellular response and adaptation to chemical repellents. The EMBO Journal. 2005;24(1):63–72. doi: 10.1038/sj.emboj.7600493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans TC. Transformation and microinjection. WormBook. 2006.
- Cáceres IdC, Valmas N, Hilliard MA, Lu H. Laterally Orienting C. elegans Using Geometry at Microscale for High-Throughput Visual Screens in Neurodegeneration and Neuronal Development Studies. PLoS ONE. 2012;7(4):e35037. doi: 10.1371/journal.pone.0035037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodel T, Prevedel R, Aumayr K, Zimmer M, Vaziri A. Brain-wide 3D imaging of neuronal activity in Caenorhabditis elegans with sculpted light. Nat Methods. 2013;10(10):1013–1020. doi: 10.1038/nmeth.2637. [DOI] [PubMed] [Google Scholar]
- García LR, Portman DS. Neural circuits for sexually dimorphic and sexually divergent behaviors in Caenorhabditis elegans. Curr Opin Neurobiol. 2016;38:46–52. doi: 10.1016/j.conb.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clokey GV, Jacobson LA. The autofluorescent "lipofuscin granules" in the intestinal cells of Caenorhabditis elegans are secondary lysosomes. Mech Ageing Dev. 1986;35(1):79–94. doi: 10.1016/0047-6374(86)90068-0. [DOI] [PubMed] [Google Scholar]
- Coburn C, et al. Anthranilate Fluorescence Marks a Calcium-Propagated Necrotic Wave That Promotes Organismal Death in C. elegans. PLoS Biology. 2013;11(7):e1001613. doi: 10.1371/journal.pbio.1001613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, et al. A hub-and-spoke circuit drives pheromone attraction and social behaviour in C. elegans. Nature. 2009;458(7242):1171–1175. doi: 10.1038/nature07886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, et al. Interaction of structure-specific and promiscuous G-protein-coupled receptors mediates small-molecule signaling in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2012;109(25):9917–9922. doi: 10.1073/pnas.1202216109. [DOI] [PMC free article] [PubMed] [Google Scholar]