

Abstract
Regulation of gene expression in response to the changing environment is critical for cell survival. For instance, binding of macrolide antibiotics to the ribosome promote the translation arrest at the leader ORFs ermCL and ermBL necessary for inducing antibiotic resistance genes ermC and ermB. Cladinose-containing macrolides, like erythromycin (ERY), but not ketolides e.g., telithromycin (TEL), arrest translation of ermCL, while either ERY or TEL stall ermBL translation. How the ribosome distinguishes between chemically similar small molecules is unknown.
We show that single amino acid changes in the leader peptide switch the specificity of recognition of distinct molecules, triggering gene activation in response to only ERY, only TEL, to both antibiotics, or preventing stalling altogether. Thus, the ribosomal response to chemical signals can be modulated by minute changes in the nascent peptide, suggesting that protein sequences could have been optimized for rendering translation sensitive to environmental cues.
INTRODUCTION
Ribosome-targeting macrolide antibiotics act not only as inhibitors of translation but also as activators of resistance genes 1-4. Macrolides bind in the NPET approximately 10 Å away from the peptidyl transferase center (PTC)5-8. They inhibit protein synthesis by promoting peptidyl-tRNA drop-off during the early rounds of translation or by arresting ribosome progression in a context-specific manner 9-12. Antibiotic-dependent translation arrest has been evolutionarily co-opted to enable microbial antibiotic producers as well as resistant bacterial pathogens to rapidly activate expression of the resistance genes in response to the presence of the inhibitor 2,13,14. Macrolide-induced ribosome stalling at the leader regulatory ORFs of the inducible erm genes results in the isomerization of the mRNA secondary structure which allows activation of expression of the downstream resistance cistron 15,16 (Supplementary Results, Supplementary Fig. 1a). This mechanism takes advantage of the ability of macrolides to bind to the NPET, where the antibiotic molecule interacts both with the ribosome and the nascent chain 4,17.
The antibiotics of the macrolide family are built of a macrolactone scaffold augmented with drug-specific side chains (Fig. 1a). The macrolactone ring of the prototype macrolide ERY is rigged with desosamine and cladinose sugars, while in the drugs of the newest generation called ketolides (e.g. TEL), a keto group replaces cladinose at the C3 position of the ring and additional extended side chains are present. While the mechanisms of action of various macrolides are generally similar, individual erm genes can be induced only by a distinctive spectrum of antibiotics 3,4,18-20. Specific macrolides serve as efficient stalling cofactors and inducers of resistance, whereas other macrolide antibiotics fail to do so in spite of binding with comparable affinity to the same ribosomal site 8,21-23. Because the sequences of the leader peptides vary among the different erm genes 2,24, the specificity of the antibiotic response is likely encoded in the peptide structure. However, which features of the nascent peptide influence the ability of the ribosome to discriminate between inducing and non-inducing antibiotics remain unknown.
Figure 1. The C-terminal segment of the stalled ErmBL is critical for antibiotic cofactor specificity.
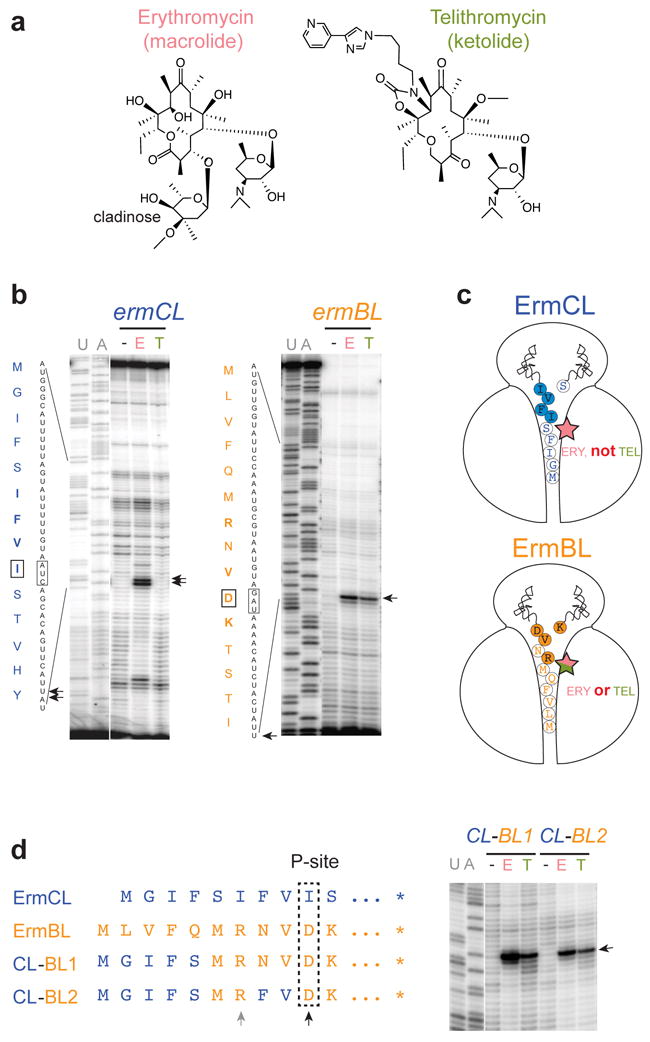
(a) Structures of the chemically distinct cofactors of translation arrest: macrolide erythromycin (ERY) and ketolide telithromycin (TEL). (b) ERY (E), but not TEL (T), induces arrest at the ermCL 9th codon (left panel), whereas both antibiotics promote ribosome stalling at the ermBL 10th codon (right panel). Translation arrest sites, detected by toeprinting, are indicated by arrows. The codons in the P site of the stalled ribosomes are boxed. Sequencing lanes are marked. Gels are representative of five independent experiments. (c) Cartoon representations of the ErmCL- and ErmBL- stalled ribosomal complexes with their nine or ten amino acid nascent peptides, respectively. The filled circles indicate the amino acids critical for stalling with ErmCL 3 or ErmBL 4 (see also Supplementary Fig. 3). Antibiotics recognized as stalling cofactors by the ribosome in each complex are represented by a star. (d) The ErmBL amino acids responsible for the ribosome ability to sense ERY and TEL. Left: N-terminal sequences of the peptides encoded in the templates used for cell free translation. The residues specified by the P site codons of the stalled ribosomes are boxed. Residues identical to those in ErmCL are colored blue; ErmBL-specific residues are orange. The ErmBL positions 7 and 10, which are responsible for antibiotic specificity, are indicated by grey and black arrows, respectively. Right: toeprinting analysis showing ERY- and TEL- mediated arrest (arrow) in the hybrid templates. Gels are representative of two independent experiments.
Regulation of the ermC and ermB genes presents a striking example of idiosyncratic small molecule specificity of programmed ribosome stalling. Both ERY and TEL readily arrest the ribosome at the 10th codon of the ermBL leader ORF by inhibiting peptide bond formation between the MLVFQMRNVD nascent chain, which esterifies tRNA in the P site of the ribosome, and the incoming Lys-tRNA 4 (Fig. 1b). In contrast, ERY, but not TEL, halts translation at the 9th codon of ermCL after the ribosome has polymerized the MGIFSIFVI sequence 3,17,20 (Fig. 1b). Which segment of the nascent chain or particular amino acid residues define the specificity of the cofactor recognition remains unclear. This question prompted us to explore the idiosyncratic properties of the peptides that aid the ribosome in distinguishing between diverse stalling cofactors. We found that minute changes in the nascent peptide structure, especifically mutations of the C-terminal amino acid residue of the stalled peptide, are able to modulate the spectrum of small molecules recognized by the ribosome.
RESULTS
Nascent peptide C-terminus defines cofactor recognition
In order to discern which part of the protein nascent chain delimits the choice of antibiotic cofactor for ribosome stalling (Fig. 1c), we engineered a hybrid leader peptide (construct CL-BL1) with its N-terminal sequence (residues 1-5) corresponding to ErmCL, and the C-terminal segment (residues 6-11) representing ErmBL (Fig. 1d). In contrast to ErmCL, which completely lacks the ability to cooperate with TEL in halting translation (Fig. 1b), but similar to wt ErmBL, which can use ERY and TEL as a stalling co-factor (Fig. 1b), translation arrest at the Asp10 codon of the hybrid CL-BL1 was induced by both antibiotics (Fig. 1d and Supplementary Fig. 2), revealing that it is the C-terminal sequence of the stalling peptide that primarily defines the antibiotic specificity of the translation arrest.
Within the ErmBL segment of the CL-BL1 hybrid, the penultimate amino acid (Val9) is identical to that in ErmCL and thus could not be the cause of the different cofactor requirements of ErmCL and ErmBL (Fig. 1c). Neither could it be residues Met6 or Asn8, whose identities are not essential for ERY or TEL mediated stalling at ermBL 4 (Supplementary Fig. 3). Indeed, not surprisingly, the mutation of Asn8 of ErmBL to the ErmCL-specific Phe (construct CL-BL2) preserved the arrest in response to both antibiotics (Fig. 1d and Supplementary Fig. 2). Therefore, the determinants of the antibiotic specificity of the ErmBL stalling peptide could only be Arg7, which is located in the vicinity of the antibiotic molecule in the NPET of the arrested ribosome 4, and/or the C-terminal Asp10 that esterifies the P-site tRNA in the PTC4.
Arg7 of ErmBL is important for dual cofactor response
When Arg7 of ErmBL was mutated to all other 19 main amino acids (Fig. 2a) and translation arrest was analyzed by toeprinting (Supplementary Fig. 4), we observed a variety of effects. To simplify the analysis, we arbitrarily set the cut-off of 50% stalling efficiency relative to the ERY mediated stalling achieved with wt ErmBL. The majority of the mutations rendered stalling ERY-specific, abrogating an efficient response to TEL. Only Leu7 (and possibly Met7 and Thr7) mutant showed residual stalling with TEL, whereas Ala, Asp, Glu and Pro mutations significantly diminished ribosome stalling in response to any of the drugs. Altogether, these results (Fig. 2b-c and Supplementary Fig. 4) suggest that the mutations of Arg7 of ErmBL can alter the cofactor recognition properties of the ribosome-ErmBL complex by primarily narrowing the small molecule specificity or abolishing ribosome stalling. The importance of the Arg7 residue for translation arrest in response to both antibiotic cofactors is rationalized by its extensive interaction with the NPET wall, promoted by the peptide repositioning in the tunnel due to drug presence, as observed in recent cryo-EM reconstructions 4.
Figure 2. Identity of Arg7 of ErmBL modulates recognition of macrolide stalling cofactors.
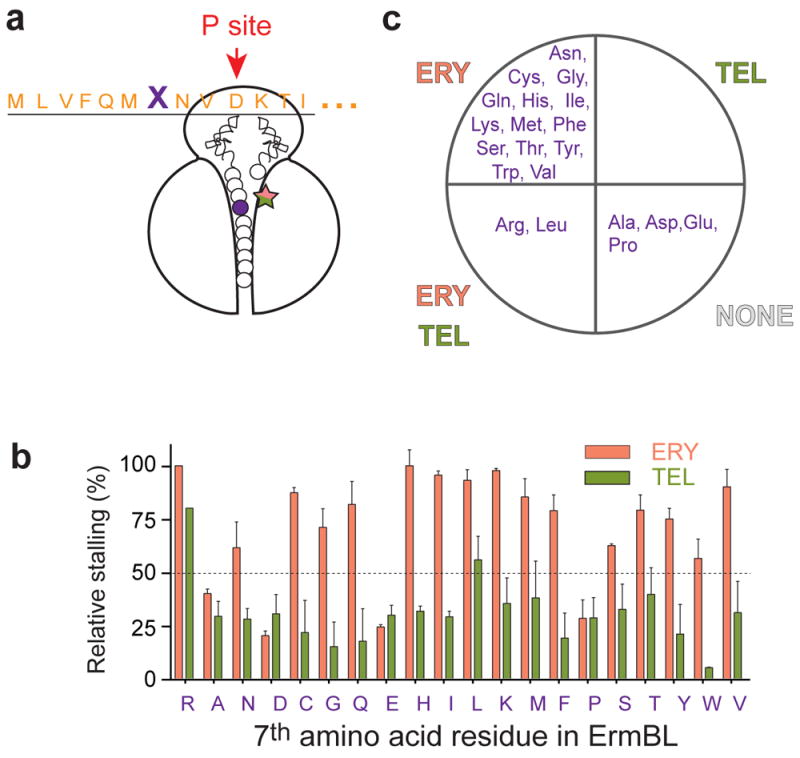
(a) The Arg7 residue of ErmBL (indicated as ‘X’), located in the proximity of the tunnel-bound antibiotic in the stalled ErmBL complex, was mutated to all other main 19 amino acids and mutant peptides were used in analysis of ERY-and TEL-specific translation arrest. (b) Bar graph of the stalling efficiency estimated by quantifying the relative intensities of the toeprint bands of translation arrest at codon 10 in the mutant templates relative to ERY-dependent stalling with wt ErmBL (see the gel in Supplementary Fig. 4). Error bars show deviation from the mean in two independent experiments. (c) Grouping of the ErmBL position 7 mutants according to their ability to efficiently (≥50%) support ERY- or TEL- directed stalling.
Last residue of stalled ErmBL as cofactor discriminator
Strikingly, the identity of the C-terminal residue of the stalling peptide (Asp10 in wt ErmBL), which is positioned in the PTC at some distance from the macrolide binding site, had the most dramatic impact on the discrimination between the stalling cofactors. When this residue was mutated (Fig. 3a), we observed that replacement of Asp10 with Gly or even structurally similar Glu, nearly abolished ketolide-mediated translation arrest at codon 10, but preserved ERY-dependent stalling. Unexpectedly, changes to Pro or Tyr had the opposite effect: the ribosome stalled at the 10th codon in response to TEL but not ERY. All other Asp10 mutations prevented sufficiently strong stalling at the 10th codon in response to any of the antibiotics. These data (Fig. 3b-c and Supplementary Fig. 5) show that the nature of the C-terminal residue of the stalled ErmBL nascent chain plays the major role in differentiation between chemically distinct stalling cofactors bound in the NPET.
Figure 3. The C-terminal amino acid of the stalled ErmBL directs differentiation between chemically distinct molecules and determines selectivity of antibiotic-mediated gene activation.
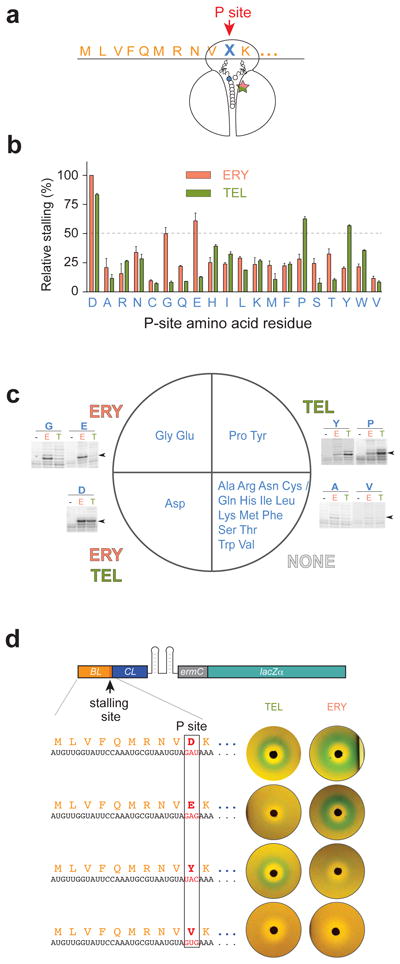
(a) The ermBL Asp10 codon (denoted with an X) in the P site of the stalled ribosome was mutated to code for all other 19 amino acids. (b) Bar plot representing the efficiency of ERY or TEL arrest estimated from the relative intensities of the stalled ribosomes toeprint bands. The band representing ERY-dependent stalling at the Asp10 codon of wt ermBL was taken as 100%. Error bars show deviation from the mean of two independent experiments. (c) Summary of the results shown in (b) with the ErmBL C-terminal amino acid mutants grouped by their ability to favor (relative stalling efficiency ≥50%) antibiotic-specific translation arrest. The sector marked as ‘NONE’ groups the mutants unable to promote efficient stalling with any of the antibiotics. Examples of toeprinting gels are shown for mutants exhibiting antibiotic-specific stalling and two ‘non-stalling’ mutants; the corresponding codon 10 mutations are indicated above the gels. Lanes are marked with ‘-‘ for no antibiotic, E for ERY and T for TEL. Arrowheads indicate the toeprint bands of the stalled ribosomes. Full gels are shown in Supplementary Fig. 5. (d) The expression of the pBLCLZα reporter where lacZ induction is controlled by ErmBL-dependent stalling, as visualized in the disc-diffusion tests. Antibiotic specificity of lacZ activation is dictated by the C-terminal residue of the ErmBL stalled peptide. Shown plates are representative of two independent experiments.
We wondered whether changes of the ErmBL Asp10 residue that affect drug-specific ribosome stalling in vitro make the activation of gene expression in vivo sensitive to distinct small molecules. To examine that, we tailored the ERY-inducible ermC-lacZα reporter plasmid 25 to make lacZ expression sensitive to ErmBL-controlled ribosome stalling (Fig. 3d, Supplementary Figs. 1c and 6). We then modified the resulting pBLCLZα construct to alter the wt Asp10 of ErmBL to Glu, Tyr or Val (Fig. 3d), in order to cover the entire spectra of the observed effects. Consistent with the dual antibiotic specificity of translation arrest directed by the wt ErmBL in vitro, both ERY and TEL activated the reporter containing the unaltered ErmBL (Asp10) stalling peptide sequence, as revealed by the appearance of a blue halo around the antibiotic disks on indicator plates (Fig. 3d). In agreement with the toeprinting analysis, the induction became either ERY-selective, when the Asp10 codon of ermBL was replaced with Glu, or TEL-selective with the Tyr10 mutant (Fig. 3d). Lastly, neither of the antibiotics was able to activate the reporter when Asp10 was mutated to Val (Fig. 3d). Thus, by solely changing the nature of the C-teminal residue of the ErmBL stalling peptide, it was possible to deliberately modify the ribosomal response to specific stalling cofactors and, as a result, render gene expression sensitive to the presence of distinct small molecules.
Antibiotics elicite misorientation of PTC substrates
Nascent peptide- and small molecule-dependent programmed translation arrest relies upon inefficient peptide bond formation at the stalling codon 3,4,26,27. In order to gain insights into the molecular mechanisms underlying peptide and antibiotic cooperation in interfering with peptidyl transfer, we employed molecular dynamics (MD) simulations to explore the behavior of representative ErmBL mutants in the drug-bound ribosome in the pre-reaction state. The atomic model, built upon the 4.5 Å resolution cryo-EM structure of the ERY/ErmBL-stalled ribosome carrying A site Lys-tRNA 4, was computationally modified to replace ERY with TEL, to remove the antibiotic, or to mutate Asp10 of ErmBL to Glu or Tyr to capture ERY- or TEL-specific stalling scenarios, respectively (see Methods for details). Proximity of the relative geometry of the closely-spaced (≤6 Å) donor and acceptor substrates derived from MD sampling to the orientation of the PTC substrates in the elongating ribosome in a pre-reaction state 28, was used as a measure of reactivity (Supplementary Fig. 7a). In the drug-free ribosome, the substrates frequently approach the ‘reactive’ orientation (Supplementary Fig. 7b-d). However, consistent with the scenario of dual antibiotic arrest, the productive conformations are sampled less frequently when ERY or TEL are bound to the ribosome carrying wt ErmBL-tRNA (Supplementary Fig. 7b-c). In agreement with the experimental data (Fig. 3b-c), only ERY skews the substrates towards unproductive constellations with the ErmBL(Glu10) mutant, whereas TEL, but not ERY, curtails the occurrence of the productive states in the ErmBL(Tyr10) complex (Supplementary Fig. 7b-c). Altogether, the results of MD simulations argue that misorientation of the reactive substrates is the key contributing factor to antibiotic-induced ribosome stalling. This inference resembles the previously proposed isomerization of the ribosome into a non-productive state when inadequate donor substrates are bound at the PTC 29,30.
Although MD simulations yielded a number of possible interpretations as to why specific peptides can trigger stalling only with a certain antibiotic molecule bound in the NPET, no consistent unifying theme has emerged. Nevertheless, two observations, which could offer fairly unorthodox potential clues for the cofactor specificity puzzle, are worth mentioning. Analyzing the C-terminal amino acid trajectories of the ErmBL(Glu10) mutant peptide, we detected occasional direct interaction between the γ-carboxyl of the C-terminal Glu side chain with the 4” hydroxyl of the C3 cladinose sugar of ERY (Supplementary Fig. 7e). Such an interaction, which restricts the donor substrate to acquire a non-productive orientation, would be impossible with the cladinose-lacking TEL bound in the NPET or if Asp10, with a shorter side chain, is at the ErmBL C-terminus and thus, could consistently explain the ‘Ery-only’ stalling response of the Glu10 mutant. MD analysis of the ErmBL(Tyr10) mutant emphasized a possible role of the conformational dynamics of the PTC substrates which could be influenced by the cofactor: a broader rotational freedom of the Tyr10 side chain (radius of gyration 4.87 ± 1.93 Å) (Supplementary Fig. 7f), afforded by the lack of cladinose in the TEL structure, allows the donor substrate to frequently wander away from the productive conformation. The movement of the Tyr10 side chain is more constrained in the ERY complex (radius of gyration 1.10 ± 0.70 Å (Supplementary Fig. 7f), thus accounting for the TEL-specific response.
Productive cofactor recognition is a complex phenomenon
While inhibition of peptide bond formation is a key requisite for antibiotic-specific translation arrest 3,4,17,26,27, productive stalling leading to activation of the downstream gene requires that two other conditions are met: i) translation of the leader peptide must proceed unobstructed until the ribosome reaches the site of the programmed arrest, and ii) the stalled ribosome should remain stably associated with mRNA to allow its isomerization into the ‘induced’ conformation (Supplementary Fig. 1a). In fact, some of the ErmBL mutant peptides, which were unable to support efficient formation of a stalled complex with one or both antibiotics as measured by the absolute intensity of the codon 10 toeprint band, were nevertheless only poorly transferred to the Lys-tRNA acceptor, as determined by the ratio of the ribosomes arrested at codon 10 to those able to bypass this codon (Supplementary Fig. 5). We believe that the inability of those mutants to produce a strong stalling band in toeprinting experiments stems from the poor stability of the stalled complexes, possibly due to rapid peptidyl-tRNA drop-off. Although the molecular bases of the drop-off mechanism are unknown, the rate of peptidyl-tRNA dissociation is likely affected by the structure of the NPET-bound antibiotic and the nature of the C-terminal residue of the nascent peptide. Thus, cofactor-dependent stalling peptides must have evolved to not only stop the ribosome when the cofactor is present but also to allow the ribosome to reach the site of the programmed arrest and provide sufficient stability for the stalled complex.
Cofactor C3 substituent is the main discriminatory trait
What chemical features of ERY and TEL are recognized by the ribosome and nascent peptides for differentiating between them as stalling cofactors? Although these two antibiotics differ from each other by several structural elements, cryo-EM reconstructions 4, as well as the MD simulation presented here (Supplementary Fig. 7), point specifically to the importance of C3 cladinose present in ERY but lacking in TEL (Fig. 1a). Thus in order to test the role of the C3 substituent in cofactor discrimination, we compared translation arrest directed by wt ErmBL or its three representative mutants (Glu10, Tyr10 and Val10) in response to ERY, TEL and the cladinose derivative of TEL, RU69874 (Supplementary Fig. 8a-b). The toeprinting data showed that none of the peptides could distinguish the cladinose version of TEL from ERY (Supplementary Fig. 8c-d). Therefore, ErmBL-assisted discrimination between the NPET-bound antibiotics is based upon the chemical nature of the macrolactone C3 chain.
Broadening the spectrum of ErmCL cofactor response
Our experiments with the ErmBL peptide and its derivatives showed that minor variations in the peptide structure can modulate recognition of chemically distinct stalling cofactors. To examine the generality of this phenomenon, we aimed to broaden the specificity of cofactor recognition by the ERY-specific ErmCL peptide (Fig. 4a), whose sequence does not resemble that of ErmBL (Fig. 1c). In agreement with our previous results 3,26, changing the nature of the aminoacyl-acceptor of the ErmCL peptide from Ser-tRNA to Lys-tRNA did not affect ERY-specific stalling, nor allowed TEL to serve as a functional cofactor (Fig. 4b and Supplementary Fig. 9). However, when within this context, the C-terminal residue of the ErmCL stalling peptide was mutated from Ile to Asp, then translation arrest as well as the associated small molecule-controlled gene activation became responsive to both ERY and TEL (Fig. 4b-c and Supplementary Fig. 9). Thus, minimal sequence variations in the structures of unrelated arresting peptides (ErmBL and ErmCL) significantly change the nature and the spectrum of small molecules recognized as cofactors for regulation of translation arrest.
Figure 4. Mutations in the ErmCL peptide can change antibiotic specificity of ribosome stalling.
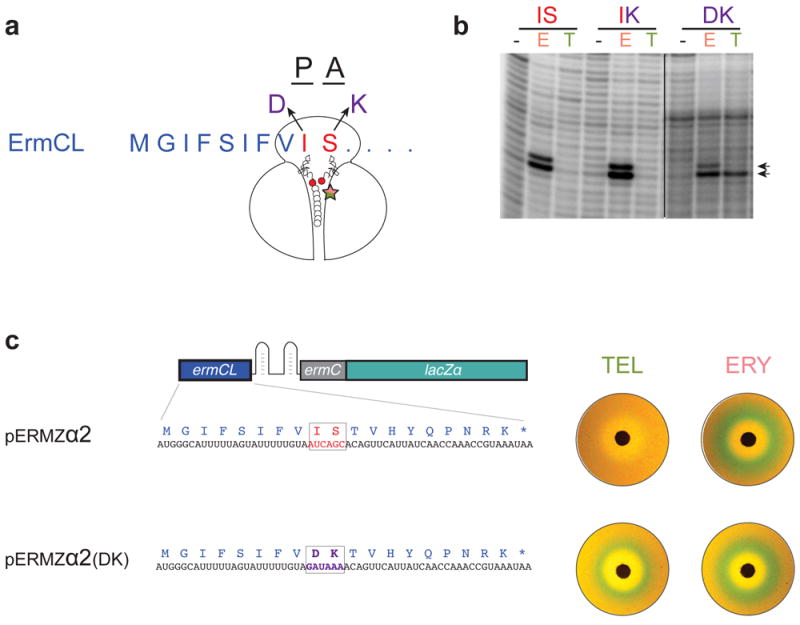
(a) Codons 9 (Ile) and 10 (Ser) of wt ermCL were mutated to specify for Asp and Lys, respectively, in order to mimic the identities of the P and A-site codons in ribosomes stalled at ermBL. (b) Toeprinting analysis of ERY (E) or TEL (T) mediated translation arrest with wt ErmCL (Ile-9 and Ser-10, IS), or its mutant variants IK (Ile9-Lys10) or DK (Asp9-Lys10). Arrows indicate the toeprint bands of the ribosomes stalled at the 9th codon of ermCL. Full gels, which are representative of two independent experiments, are shown in Supplementary Fig. 9. (c) Mutant ErmCL with expanded antibiotic sensitivity supports in vivo activation of the lacZ reporter in response to ERY and TEL as revealed by the disk-diffusion test. Plates are representative of two independent experiments.
DISCUSSION
The combined results of our biochemical and microbiological experiments, buttressed by computational MD simulations, have revealed the ribosome as a highly selective sensor of small molecules. Its ability to recognize and discriminate between them could be directly modulated by minor variations in the sequence of the nascent peptide. The mutation of a single amino acid of the nascent chain could be sufficient to not only broaden or narrow the spectrum of molecules, which can elicit the functional response of the ribosome, but also to switch the specificity of recognition from one chemical to another.
We carried our experiments with the regulatory stalling peptides, which control inducible resistance to macrolide antibiotics. Therefore, our findings have important medical implications. One of the clinical advantages of ketolides like TEL is their low capacity for activating some inducible resistance genes 31. Although pathogenic bacteria can gain resistance to non-inducing antibiotic by acquiring mutations that render the inducible resistance gene constitutive 32, such mutants are rare possibly because of the high fitness cost associated with constitutive expression of Erm methyltransferase 14. Our findings suggest that with the anticipated increase of clinical use of ketolides, mutants with an altered or broaden cofactor specificity of induction, which carry mutations in the regulatory stalling peptides, may become prevalent. In fact, the isolation of a strain with altered induction specificity due to a mutation in ermBL changing residue 7 of the peptide to Cys was previously reported 33, although the nature of the effect was not investigated at the time.
Our results may also have important evolutionary implications. The amazing ease with which sensing of small molecules can be switched, adjusted and optimized by only minute changes in the nascent peptide structure indicates that translation of many proteins could be rendered sensitive to the chemical composition of the cell milieu. While the known examples of small molecule-controlled translation arrest have been described so far only for the regulatory genes, these examples, which account for the most ‘extreme’ cases that require severe and prolonged translation arrest, likely represent only the tip of the iceberg. Conceivably, the amino acid sequences of many cellular proteins may have been evolutionarily selected not only for their proper function upon completion of their synthesis, but also for enabling their translation to be finely tunable by specific small molecules and thus, responsive to the changing environment.
METHODS
Antibiotics and other reagents
ERY was purchased from SIGMA (E5389), TEL was provided by Cempra Pharmaceuticals, RU69874 was provided by Roussel Uclaf, borrelidin was from Santa Cruz Biotechnologies (SC200379A), and mupirocin from Sigma (M7694). Antibiotics were dissolved in ethanol and when present in the in vitro translation assays were dried at the bottom of the reaction tube prior to addition of the rest of the components. AMV reverse transcriptase was from Roche (Cat No.10109118001). AccuPrime polymerase (Cat No.12344032) for PCR reactions, restriction enzymes, DNA ligase, and all other chemicals were from Thermo Fisher.
DNA oligonucleotides used in this work are listed in Supplementary Table 1.
Plasmids
The ermCL gene in the original reporter plasmid pERMZα 25 contains a slippery sequence near its 3’ end that causes ketolides to induce ermC expression through a mechanism unrelated to ribosome stalling 34. Therefore, for constructions of the reporters used in this work, the slippery sequence was mutated by site directed mutagenesis using the QuikChange Lightning Multi-site directed mutagenesis kit (Agilent Technologies, 210514) and the mutagenizing primer ermCL18(AAA:CGT) resulting in a plasmid pERMZα2.
The plasmids related to pBLCLZα (Supplementary Fig 1b) were prepared by first constructing the variant with the Tyr10 mutation. This was engineered by PCR-amplification of a segment of pERMZα2 using primers BL(Y10)-CL and lacZR, digestion of the resulting product with restriction enzymes NdeI and AflII and ligation of it into the plasmid pERMZα-Tet 35 digested with the same enzymes. The variants of the pBLCLZα plasmid with the 10th codon encoding Asp (the wild type codon of the original ErmBL), Glu or Val were engineered by site directed mutagenesis of the pBL(Y) CLZα plasmid using the corresponding mutagenizing primers BL(Y:D)-CL, BL(Y:E)-CL, and BL(Y:V)-CL, respectively.
Toeprinting assay
Toeprinting assays were performed as described previously (Vazquez-Laslop et al., 2008). Briefly, DNA templates (0.1 pmol) containing the desired gene under the control of the T7 RNA polymerase promoter were generated by PCR using primers described in Supplementary Table 1. The sequence of the wt template is shown in the Supplementary Fig. 4a. The transcription-translation reactions were carried out in a total volume of 5 μl of PURExpress cell-free transcription-translation system (New England Biolabs, Cat. No. E6800L) following the manufacturer’s protocol. When required, ERY or TEL were added to the final concentration of 50 μM. After 15 min incubation at 37°C, 10 pmol of the 5’ [32P]-labeled NV1 toeprinting primer (Supplementary Table1) and 2 U of reverse transcriptase were added and incubation continued for 20 min at 37°C. The reactions were stopped by the addition of 0.5 μl of 10 N NaOH and incubation for 15 min at 37°C. After phenol extraction and ethanol precipitation, cDNA products were resolved in 6% sequencing gels. Gels were dried and exposed overnight to a phosphoroimager screen.
The bands were quantified using the ImageJ software (http://rsbweb.nih.gov/ij/). The background intensity was subtracted and the resulting density values were normalized relative to intensity of the band in the wt sample containing ERY. In order to be able to compare the bands on different gels, the control sample (wt/ERY) was loaded on every gel.
In vivo testing of antibiotic-specific induction of the pBLCLZα reporters
E. coli TB1 cells 36 capable of α-complementation were transformed with wt (D10) or mutant (E10, Y10 and V10) variants of the reporter plasmid pBLCLZα (Supplementary Fig 1b). Inducibility of the reporters was tested by the disk-diffusion assay as previously described 25,34. Briefly, 108 exponentially-growing cells were plated in 5 ml soft agar on LB/agar plates containing 100 μg/ml ampicillin, 0.3 mM IPTG and 80 μg/ml X-gal. Antibiotic discs containing 300 μg ERY or 200 μg TEL were placed on the soft agar surface, plates were incubated overnight at 37°C and photographed together in a single image. The brightness and contrast of the entire image were adjusted in Graphic Converter (Lemke Software GmbH) to improve visualization of the color zones.
Molecular modeling
The atomic model of ErmBL-ribosome complex was prepared by Molecular Dynamics Flexible Fitting (MDFF) 37,38 using the fully solvated E. coli ribosome structure 39 as the initial structure which was fitted to the cryo-EM electron density map 4. The structures of the P-site tRNAAsp and A-site tRNALys were built according to their sequences 40 using tRNAs in the original model 39 as a template. The corresponding matching codons in mRNA were similarly modeled. The nascent ErmBL peptide in the ribosomal exit tunnel was modeled manually as a fully extended chain and later refined in the MDFF simulations (described below). The MDFF simulations were performed in a 278 × 336 × 331 Å explicit water box. The ribosome structure and that with ERY were fitted first followed by step-wise fitting of the ErmBL nascent chain as follows. Specifically, the side chains of the amino acids were removed to fit the backbone-only peptide into the ribosomal environment in a 3 ns MDFF run. Then, starting from the C-terminal residue, the side chains of each of the 10 amino acids of the nascent chain were fitted one at a time in 10 consecutive 2 ns-long MDFF simulations. Finally, the resulting ribosome structure, which included the ERY molecule and a full-atom ErmBL nascent chain, was refined with the cryo-EM map in a 5 ns long MDFF simulation.
The systems with the mutant ErmBL peptides were built based on the MDFF model of ERY-bound ribosome containing P-site ErmBL-tRNAAsp and A-site Lys-tRNALys. The P-site tRNAs in the mutant systems were modeled according to their sequences 40. For the TEL-bound ribosome systems, ERY was replaced with TEL by docking the drug molecule according to the reported crystallographic structure 8. Finally, the drug-free complex was obtained by removing the ERY compound in the model with wild type ErmBL. In total, 7 ribosome models were constructed: the ribosome with wt ErmBL (with ERY, TEL or drug-free), the ERY- or TEL-bound ribosome with ErmBL-Glu10 mutant, and the ERY- or TEL-bound ribosome with ErmBL-Tyr10 mutant.
Molecular dynamics simulations
To achieve enough sampling at an acceptable computational cost, we constructed reduced simulation systems of the above-mentioned 7 systems by including only 23S rRNA nucleotides located within 50 Å of the drug binding site or the PTC (as defined by the position of the U2585 residue). Atoms more than 50 Å away from these locations were not included in the MD simulations. Water boxes of dimension 125 × 125 × 150 Å, including solvating ions, were modeled using the previously described protocol 39. To reduce the simulation dependence on initial conditions, 10 independent MD simulations were performed for each of the 7 systems. For each simulation, a 65-ns-long trajectory was calculated. The trajectory of the first 50 ns was considered sufficient for equilibration 27 and the last 15 ns were used for analysis. To sum up, for each model 15 ns × 10 = 150 ns sampling data was collected with 1 frame / 5 ps recording rate for the subsequent analysis using VMD 41.
All simulations, including MDFF, were performed using NAMD 2.9 42 with the AMBER99SB force field 43,44, which includes parameters for modified nucleosides. The equations of motion were integrated with a 1 fs time step and bonded, non-bonded short- and non-bonded long-range interactions were calculated every 1, 2, and 4 fs, respectively. The particle mesh Ewald (PME) method 45,46 was used to handle the non-bonded long-range electrostatic interactions. All simulations were performed in the NpT ensemble (T = 310K; p = 1 atm).
The force field parameters of the α-amino group of the A-site lysine and the ERY and TEL compounds were optimized as described previously 27. The resulting parameter files for simulations with NAMD are available at http://www.ks.uiuc.edu/~boliu/ErmBL
The center structures of each simulation were determined as follows. First, all RNA residues within 20 Å of the drug binding site, the nascent peptide chain or the A-site amino acid were extracted from the last 15 ns of the 10 individual simulations for each system (which amounted to 15 ns × 10 replicas = 150 ns per system). Then, the Gromos algorithm 47 implemented in the g_cluster program from Gromacs 48 was used to determine the center structures, assuming there is a single cluster of structures by setting the cluster RMSD cutoff at 10 Å.
The figures showing atomic models were generated using PYMOL (www.pymol.org).
Supplementary Material
Acknowledgments
We thank Rhea Bovee for help with some experiments and Prabha Fernandes (Cempra Pharmaceuticals) for providing telithromycin. This work was supported by a grant from the National Science Foundation (MCB 1244455) to ASM and NV-L and 9P41GM104601 (to K.S.) and the National Science Foundation (PHY0822613) (to K.S.). MD modeling was facilitated by a grant from The Great Lakes Consortium for Petascale Computation on the Blue Waters Computer, financed by the National Science Foundation (OCI 07-25070) and the Oak Ridge Leadership Computing Facility at Oak Ridge National Laboratory, which is supported by the Office of Science of the Department of Energy under Contract DE-AC05-00OR22725.
Footnotes
AUTHORS CONTRIBUTIONS
P.G., A.S.M., and N.V.-L. designed research; P.G, B.L., D.K, and V.G. performed research; P.G., B.L., K.S., A.S.M., and N.V.-L., analyzed data; P.G., N.V.-L., and A.S.M. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests
References
- 1.Weisblum B. Erythromycin resistance by ribosome modification. Antimicrob Agents Chemother. 1995;39:577–585. doi: 10.1128/AAC.39.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramu H, Mankin AS, Vazquez-Laslop N. Programmed drug-dependent ribosome stalling. Mol Microbiol. 2009;71:811–824. doi: 10.1111/j.1365-2958.2008.06576.x. [DOI] [PubMed] [Google Scholar]
- 3.Vazquez-Laslop N, Thum C, Mankin AS. Molecular mechanism of drug-dependent ribosome stalling. Mol Cell. 2008;30:190–202. doi: 10.1016/j.molcel.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 4.Arenz S, et al. Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat Commun. 2014;5:3501. doi: 10.1038/ncomms4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlunzen F, et al. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 6.Tu D, Blaha G, Moore PB, Steitz TA. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell. 2005;121:257–270. doi: 10.1016/j.cell.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Bulkley D, Innis CA, Blaha G, Steitz TA. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Natl Acad Sci USA. 2010;107:17158–17163. doi: 10.1073/pnas.1008685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dunkle JA, Xiong L, Mankin AS, Cate JH. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci USA. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenson T, Lovmar M, Ehrenberg M. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J Mol Biol. 2003;330:1005–1014. doi: 10.1016/s0022-2836(03)00662-4. [DOI] [PubMed] [Google Scholar]
- 10.Kannan K, Vazquez-Laslop N, Mankin AS. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012;151:508–520. doi: 10.1016/j.cell.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 11.Davis AR, Gohara DW, Yap MN. Sequence selectivity of macrolide-induced translational attenuation. Proc Natl Acad Sci USA. 2014;111:15379–15384. doi: 10.1073/pnas.1410356111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannan K, et al. The general mode of translation inhibition by macrolide antibiotics. Proc Natl Acad Sci USA. 2014;111:15958–15963. doi: 10.1073/pnas.1417334111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weisblum B. Insights into erythromycin action from studies of its activity as inducer of resistance. Antimicrob Agents Chemother. 1995;39:797–805. doi: 10.1128/aac.39.4.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta P, Sothiselvam S, Vazquez-Laslop N, Mankin AS. Deregulation of translation due to post-transcriptional modification of rRNA explains why erm genes are inducible. Nat Commun. 2013;4:1984. doi: 10.1038/ncomms2984. [DOI] [PubMed] [Google Scholar]
- 15.Horinouchi S, Weisblum B. Posttranscriptional modification of mRNA conformation: mechanism that regulates erythromycin-induced resistance. Proc Natl Acad Sci USA. 1980;77:7079–7083. doi: 10.1073/pnas.77.12.7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gryczan TJ, Grandi G, Hahn J, Grandi R, Dubnau D. Conformational alteration of mRNA structure and the posttranscriptional regulation of erythromycin-induced drug resistance. Nucleic Acids Res. 1980;8:6081–6097. doi: 10.1093/nar/8.24.6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arenz S, Meydan S, Starosta A, Berninghausen O, Beckmann R, Vázquez-Laslop N, Wilson DN. Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol Cell. 2014;56:446–452. doi: 10.1016/j.molcel.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayford M, Weisblum B. The ErmC leader peptide: amino acid alterations leading to differential efficiency of induction by macrolide-lincosamide-streptogramin B antibiotics. J Bacteriol. 1990;172:3772–3779. doi: 10.1128/jb.172.7.3772-3779.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamimiya S, Weisblum B. Induction of ermSV by 16-membered-ring macrolide antibiotics. Antimicrob Agents and Chemother. 1997;41:530–534. doi: 10.1128/aac.41.3.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vazquez-Laslop N, et al. Role of antibiotic ligand in nascent peptide-dependent ribosome stalling. Proc Natl Acad Sci USA. 2011;108:10496–10501. doi: 10.1073/pnas.1103474108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiong L, Shah S, Mauvais P, Mankin AS. A ketolide resistance mutation in domain II of 23S rRNA reveals proximity of hairpin 35 to the peptidyl transferase centre. Mol Microbiol. 1999;31:633–639. doi: 10.1046/j.1365-2958.1999.01203.x. [DOI] [PubMed] [Google Scholar]
- 22.Hansen LH, Mauvais P, Douthwaite S. The macrolide-ketolide antibiotic binding site is formed by structures in domains II and V of 23S ribosomal RNA. Mol Microbiol. 1999;31:623–631. doi: 10.1046/j.1365-2958.1999.01202.x. [DOI] [PubMed] [Google Scholar]
- 23.Llano-Sotelo B, et al. Binding and action of CEM-101, a new fluoroketolide antibiotic that inhibits protein synthesis. Antimicrob Agents Chemother. 2010;54:4961–4970. doi: 10.1128/AAC.00860-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian SL, Ramu H, Mankin AS. In: Antibiotic Drug Discovery and Development. Dougherty TJ, Pucci MJ, editors. Springer; 2011. pp. 455–484. [Google Scholar]
- 25.Bailey M, Chettiath T, Mankin AS. Induction of ermC expression by ‘non-inducing’ antibiotics. Antimicrob Agents Chemother. 2008;52:866–874. doi: 10.1128/AAC.01266-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramu H, et al. Nascent peptide in the ribosome exit tunnel affects functional properties of the A-site of the peptidyl transferase center. Mol Cell. 2011;41:321–330. doi: 10.1016/j.molcel.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 27.Sothiselvam S, et al. Macrolide antibiotics allosterically predispose the ribosome for translation arrest. Proc Natl Acad Sci USA. 2014;111:9804–9809. doi: 10.1073/pnas.1403586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nature Struct Mol Biol. 2009;16:528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaher HS, Shaw JJ, Strobel SA, Green R. The 2’-OH group of the peptidyl-tRNA stabilizes an active conformation of the ribosomal PTC. EMBO J. 2011;30:2445–2453. doi: 10.1038/emboj.2011.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Englander MT, et al. The ribosome can discriminate the chirality of amino acids within its peptidyl-transferase center. Proc Natl Acad Sci USA. 2015;112:6038–6043. doi: 10.1073/pnas.1424712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnefoy A, Girard AM, Agouridas C, Chantot JF. Ketolides lack inducibility properties of MLS(B) resistance phenotype. J Antimicrob Chemother. 1997;40:85–90. doi: 10.1093/jac/40.1.85. [DOI] [PubMed] [Google Scholar]
- 32.Sutcliffe J, Leclercq R. In: Macrolide Antibiotics. Schönfeld W, Kirst HA, editors. Birkhäuser Verlag; 2002. pp. 281–318. [Google Scholar]
- 33.Oh TG, Kwon AR, Choi EC. Induction of ermAMR from a clinical strain of Enterococcus faecalis by 16-membered-ring macrolide antibiotics. J Bacteriol. 1998;180:5788–5791. doi: 10.1128/jb.180.21.5788-5791.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS-only references
- 34.Gupta P, Kannan K, Mankin AS, Vazquez-Laslop N. Regulation of gene expression by macrolide-induced ribosomal frameshifting. Mol Cell. 2013;52:629–642. doi: 10.1016/j.molcel.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vazquez-Laslop N, Ramu H, Klepacki D, Kannan K, Mankin AS. The key role of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J. 2010;29:3108–3117. doi: 10.1038/emboj.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnston TC, Thompson RB, Baldwin TO. Nucleotide sequence of the luxB gene of Vibrio harveyi and the complete amino acid sequence of the beta subunit of bacterial luciferase. J Biol Chem. 1986;261:4805–4811. [PubMed] [Google Scholar]
- 37.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods. 2009;49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trabuco LG, et al. The role of L1 stalk-tRNA interaction in the ribosome elongation cycle. J Mol Biol. 2010;402:741–760. doi: 10.1016/j.jmb.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Juhling F, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–162. doi: 10.1093/nar/gkn772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 42.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cornell WD, et al. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 44.Aduri R, et al. AMBER Force Field Parameters for the Naturally Occurring Modified Nucleosides in RNA. J Chem Theory Comput. 2007;3:1464–1475. doi: 10.1021/ct600329w. [DOI] [PubMed] [Google Scholar]
- 45.Darden Y, York D, Pedersen L. Particle mesh Ewald: An N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 46.Essmann U, et al. A smooth particle mesh ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 47.Daura X, et al. Peptide Folding: When Simulation Meets Experiment. Angew Chem -Int Edit. 1999;38:236–240. [Google Scholar]
- 48.Pronk S, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.