

Abstract
The interaction between NMDA receptors and μ-opioid receptors in primary afferent terminals was studied by using NMDA to induce substance P release, measured as neurokinin 1 receptor internalization. In rat spinal cord slices, the μ-opioid receptor agonists morphine, DAMGO and endomorphin-2 inhibited NMDA-induced substance P release, whereas the antagonist CTAP right-shifted the concentration response of DAMGO. In vivo, substance P release induced by intrathecal NMDA after priming with BDNF was inhibited by DAMGO. ω-Conotoxins MVIIC and GVIA inhibited about half of the NMDA-induced substance P release, showing that it was partially mediated by the opening of voltage-gated calcium (Cav) channels. In contrast, DAMGO or ω-conotoxins did not inhibit capsaicin-induced substance P release. In cultured DRG neurons, DAMGO but not ω-conotoxin inhibited NMDA-induced increases in intracellular calcium, indicating that μ-opioid receptors can inhibit NMDA receptor function by mechanisms other than inactivation of Cav channels. Moreover, DAMGO decreased the ω-conotoxin-insensitive component of the substance P release. Potent inhibition by ifenprodil showed that these NMDA receptors have the NR2B subunit. Activators of adenylyl cyclase and protein kinase A (PKA) induced substance P release and this was decreased by the NMDA receptor blocker MK-801 and by DAMGO. Conversely, inhibitors of adenylyl cyclase and PKA, but not of protein kinase C, decreased NMDA-induced substance P release. Hence, these NMDA receptors are positively modulated by the adenylyl cyclase-PKA pathway, which is inhibited by μ-opioid receptors. In conclusion, μ-opioid receptors inhibit NMDA receptor-induced substance P release through Cav channel inactivation and adenylyl cyclase inhibition.
Keywords: Calcium channel, neurokinin 1 receptor, mu-opioid receptor, NMDA receptor, primary afferent, protein kinase A
Chemical compounds: BVT948 (PubChem CID: 6604934), ω-conotoxin MVIIC (PubChem CID: 56841670), ω-conotoxin GVIA (PubChem CID: 73169082), CTAP (PubChem CID: 90479802), DAMGO (PubChem CID: 5462471), D-serine (PubChem CID: 71077), endomorphin-2 (PubChem CID: 5311081), KT5720 (PubChem CID: 454202), NMDA (PubChem CID: 22880), SQ22536 (PubChem CID: 5270)
NMDA receptors in the spinal dorsal horn play a key role in chronic pain and central sensitization (Kim et al., 2012; Randic et al., 1993; Woolf and Thompson, 1991). NMDA receptors are present in primary afferents (Li et al., 2004; Liu et al., 1994; Lovinger and Weight, 1988; Ma and Hargreaves, 2000; Marvizon et al., 2002; McRoberts et al., 2007) and evoke the release of substance P from their central terminals (Chen et al., 2010; Malcangio et al., 1998; Marvizon et al., 1997). Conversely, substance P release from primary afferents is inhibited by μ-opioid receptors (MORs) (Kondo et al., 2005; Yaksh et al., 1980; Zhang et al., 2010), δ-opioid receptors (DORs) (Zachariou and Goldstein, 1996a) and κ-opioid receptors (KORs) (Zachariou and Goldstein, 1996b, 1997). In the latent sensitization model of chronic pain, spinal cord hyperalgesia is maintained by NMDA receptors (Corder et al., 2013; Laulin et al., 2002; Richebe et al., 2005) and suppressed by MORs, DORs and KOR (Campillo et al., 2011; Corder et al., 2013; Walwyn et al., 2016). This suggests that the interaction between NMDA receptors and opioid receptors plays an important role in regulating nociceptive transmission between primary afferents and dorsal horn neurons.
Our goal was to investigate the mechanisms involved in the inhibition by MORs of NMDA receptor-evoked substance P release. We used neurokinin 1 receptor (NK1R) internalization in dorsal horn neurons as a measure in situ of substance P release and NK1R activation, an approach used and validated in numerous studies (Adelson et al., 2009; Liu et al., 1997; Mantyh et al., 1995; Marvizon et al., 1997; Marvizon et al., 2003a). In previous reports we have shown that NMDA-induced substance P release is mediated by NMDA receptors because it was inhibited by the NMDA receptor antagonist 2-amino-phosphopentanoic acid and by its channel blocker MK-801 (Chen et al., 2010; Marvizon et al., 1997). We also showed that substance P was released from primary afferents terminals, since it disappeared after depleting them of substance P with capsaicin. The NMDA receptors that induce substance P release are also present in the presynaptic terminals of primary afferents, because 1) the release was not affected by blocking action potentials with lidocaine (Chen et al., 2010), and 2) NMDA-induced substance P release was substantially decreased in mice with selective knockdown of NMDA receptors in nociceptive primary afferents expressing Nav1.8 channels (Chen et al., 2014).
We considered the following mechanisms that could mediate the inhibition of these NMDA receptors by MORs. 1) Inactivation of voltage-gated Ca2+ (Cav) channels (Heinke et al., 2011; Moises et al., 1994; Rusin and Moises, 1995), which would require that Cav channels activated by depolarization contribute to NMDA receptor-induced release. 2) Inhibition of the adenylyl cyclase (AC) - protein kinase A (PKA) pathway, since NMDA receptors with the NR2B subunit requires PKA phosphorylation at Ser1166 (Murphy et al., 2014). 3) Opening G protein-gated inwardly rectifying K+ (GIRK) channels (Gao et al., 2007; Marker et al., 2005), which would produce a hyperpolarization that would inhibit of the NMDA receptors. 4) The direct physical association of MORs and NMDA receptors, which increases morphine analgesia by inhibiting NMDA receptors (Garzón et al., 2012; Rodríguez-Muñoz et al., 2008; 2011; 2012).
We previously reported (Chen et al., 2014) that induction of substance P release by NMDA receptors in vivo requires the activation of tropomyosin-related kinase B (trkB) receptors by brain-derived neurotrophic factor (BDNF), which occurs after nerve injury. This is consistent with the finding that NMDA receptors increase primary afferent glutamatergic neurotransmission in nerve-injured rats (Yan et al., 2013) but not in normal rats (Bardoni et al., 2004; Yan et al., 2013; Zeng et al., 2006). BDNF triggers the phosphorylation of the NR2B subunit at Tyr1472 by Src family kinases (SFKs), which increases substance P release (Chen et al., 2010). Hence, we also studied how opioid agonists and BDNF interact to regulate NMDA receptor-induced substance P release.
Material and Methods
Animals
All animal procedures were approved by the Institutional Animal Care and Use Committee of the Veteran Affairs Greater Los Angeles Healthcare System and the Animal Research Committee of UCLA, and conform to NIH guidelines. Efforts were made to minimize the number of animals used and their distress. Rats used were male adult (2–4 months old) Sprague-Dawley (Harlan, Indianapolis, IND).
Chemicals
8-Br-cAMP, brain-derived neurotrophic factor (BDNF), BVT948, chelerythrine, ω-conotoxin (CTX) MVIIC, CTX GVIA, D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP), [D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin (DAMGO), endomorphin-2, forskolin, KT5720, phorbol 12-myristate 13-acetate, protein kinase inhibitor 14–22 (PKI 14–22), Ro-32-0432, SQ22536, tertiapin-Q and TPPB were from Tocris (Ellisville, MO); morphine sulfate was from Baxter; other compounds were from Sigma. Drugs were prepared as stock solutions of 1–10 mM in the appropriate solvent (DMSO or water) and then diluted in aCSF.
Spinal cord slices
Spinal cord slices were prepared as described (Adelson et al., 2009). The spinal cord was extracted from adult rats after euthanasia with pentobarbital (0.15–0.2 ml of Fatal Plus, Vortech Pharmaceuticals, Dearborn, Michigan, corresponding to 58–78 mg of pentobarbital per rat). Coronal slices (400 μm, 3–8 per rat) were cut from the lumbar spinal cord (L2–L4) with a vibratome (Integraslice 7550PSDS, Lafayette Instruments, Lafayette, IN) using low advance speed and fast vibration. For electrical stimulation of the dorsal root, slices had one dorsal root longer than 8 mm. Contact between the root and the slice was assessed with a stereomicroscope to ensure that at least 80% of the dorsal funiculus was continuous with dorsal root fibers. Slices were kept in artificial cerebrospinal fluid (aCSF), which contained (in mM) 124 NaCl, 1.9 KCl, 26 NaHCO3, 1.2 KH2PO4, 1.3 MgSO4, 2.4 CaCl2 and 10 glucose, and was bubbled with 95% O2/5% CO2. Vibratome cutting was done in sucrose-aCSF, which was the same medium with 5 mM KCl and 215 mM sucrose instead of NaCl. Slices were left to recover in oxygenated aCSF at 35°C for 1 h and were used within 3 h.
Slices were incubated with drugs using established procedures (Marvizon et al., 2003a; Marvizon et al., 2003b). The slices were placed on a nylon net glued to a plastic ring inserted halfway down a plastic tube containing 5 ml aCSF, which was kept at 35 °C using a metal block incubator (IncuBlock, Denville Scientific Inc., Metuchen, NJ). The aCSF was superficially gassed with 95% O2/5% CO2 delivered through a needle inserted through the cap of the tube. This arrangement ensured access of oxygenated aCSF and drugs from both sides of the slice. To change solutions, the ring and net with the slice was transferred to another tube. To avoid possible excitotoxic effects, NMDA and D-serine were applied to the slices only for 2 min.
Electrical stimulation of the dorsal root was performed as described (Adelson et al., 2009). It consisted of 1000 electrical square pulses of 20 V and 0.4 ms, delivered at 100 Hz to the L4 or L5 dorsal root entering the slice. These are optimal parameters to induce substance P release (Adelson et al., 2009). Electrical stimulation was generated by a Master-8 stimulator and an Iso-Flex stimulus isolating unit in constant voltage mode (AMP Instruments, Jerusalem, Israel). Slices were superfused in a custom-made chamber at 3–6 ml/min with aCSF at 35 °C. The dorsal root was drawn into a side compartment containing a bipolar platinum stimulation electrode (0.5 mm wire diameter, 1 mm separation) through a hole in a movable partition sealed with vacuum grease, and placed on top of the wires. Contact between the root and the electrode wires was monitored with a stereomicroscope. The electrode compartment was emptied of aCSF and filled with mineral oil. This arrangement ensured that the electrical pulses consistently stimulated the root and could not stimulate the dorsal horn.
At the end of the experiment, slices were fixed by immersion in ice-cold fixative (4% paraformaldehyde, 0.18% picric acid).
Intrathecal injections
To deliver drugs to the lumbar spinal cord, rats were surgically implanted with chronic intrathecal catheters inserted between the L5 and L6 lumbar vertebrae (Chen and Marvizon, 2009; Storkson et al., 1996). Rats (2–4 months old) were anesthetized with isoflurane (2–4% in oxygen, Halocarbon Laboratories, River Edge, NJ) and kept under anesthesia on a metal platform maintained at 35 °C by a feedback device. The skin and muscle were cut to expose vertebrae L5 and L6. A 20G needle was inserted between the L5 and L6 vertebrae to puncture the dura mater, which was inferred from a flick of the tail or paw and the backflow of spinal fluid. The needle was removed and the catheter (20 mm of PE-5 tube heat-fused to 150 mm of PE-10 tube) was inserted into the subdural space and pushed rostrally. The PE-10 catheter was then tunneled under the skin and externalized over the head. The skin was sutured, and the catheter was flushed with 10 μl saline and sealed. Rats were given an antibiotic (enrofloxacin) and an analgesic (carprofen) twice daily for 3 days after surgery. Rats were housed separately and used for the experiment 5–7 days after surgery. The presence of motor weakness or signs of paresis was established as criterion for immediate euthanasia of the rat, but this did not occur in any of the rats in this study.
Intrathecal injection volume was 10 μl of injectate plus 10 μl saline flush (Jensen and Yaksh, 1984; Kondo et al., 2005). This volume leads to the distribution of the injectate over most of the spinal cord, but not into the brain (Chen et al., 2007; Yaksh and Rudy, 1976). Solutions were preloaded, in reverse order of administration, into a tube (PE-10), and delivered with a 50 μl Hamilton syringe within 1 min. The position of the catheter was examined postmortem. We established the following exclusion criteria: 1) loss of the catheter, 2) termination of the catheter inside the spinal cord, and 3) occlusion of the catheter tip.
Characterization of the NK1R antiserum
The NK1R antiserum was either 94168 from CURE: Digestive Diseases Research Center (University of California Los Angeles) or AB5060 purchased from EDM Millipore (Billerica, MA). Both antiserums were generated in rabbits. Immunogens were peptides at the C-terminus of the rat NK1R, amino acids 393–407 for 94168 and amino acids 385–407 for AB5060. Antiserum 94168 labeled cells transfected with rat NK1R (but not non-transfected cells); its staining was eliminated by preadsorption with the immunizing peptide, and it yielded a single band in Western blots corresponding to a molecular weight of 100 kDa (Grady et al., 1996). Both antiserums produced the same labeling pattern of the rat dorsal horn, which was identical to that observed in numerous previous studies using these and other NK1R antibodies (Adelson et al., 2009; Allen et al., 1997; Kondo et al., 2005; Mantyh et al., 1995).
Immunohistochemistry
Rats were euthanized with pentobarbital (100 mg/kg) and fixed by aortic perfusion of 100 ml phosphate buffer (0.1 M sodium phosphate, pH 7.4) containing 0.01% heparin, followed by 400 ml of ice-cold fixative (4% paraformaldehyde, 0.18% picric acid in phosphate buffer). A segment of the lumbar spinal cord (L4–L6) was post-fixed, cryoprotected, frozen and sectioned at 25 μm in the coronal plane using a cryostat (Chen et al., 2007; Lao et al., 2008). Spinal cord slices were fixed and processed similarly (Adelson et al., 2009; Lao and Marvizon, 2005; Lao et al., 2003; Marvizon et al., 2003a). Sections were washed four times and then incubated overnight at room temperature with the NK1R antiserum diluted 1:3000 in phosphate-buffered saline containing 0.3% Triton X-100, 0.001% thimerosal and 5% normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA). After three washes, the secondary antibody (1:2000, Alexa Fluor 488 goat anti-rabbit, Molecular Probes-Invitrogen, Eugene, OR) was applied at for 2 hours at room temperature. Sections were washed four more times, mounted on glass slides, and coverslipped with Prolong Gold (Molecular Probes-Invitrogen).
Quantification of NK1R internalization
We used NK1R internalization as a measure in situ of substance P release. This method has higher sensitivity than immunoassay (Marvizon et al., 2003a), can be used non-invasively in vivo (Honore et al., 1999; Mantyh et al., 1995), allows the spatial location of substance P release (Abbadie et al., 1997; Hughes et al., 2007) and measures substance P release at the physiologically relevant concentrations that activate NK1Rs (Trafton et al., 1999).
The amount of NK1R internalization was quantified using a standard method (Mantyh et al., 1995) with minor modifications (Adelson et al., 2009). NK1R neurons in lamina I were visually counted while classifying them as with or without internalization, using a Zeiss Axio-Imager A1 (Carl Zeiss, Inc., Thornwood, NY) fluorescence microscope with a 63× (1.40 numerical aperture) objective. The criterion for having internalization was the presence in the neuronal soma of ten or more NK1R endosomes, defined as a small region of bright staining separated from the cell surface. The person counting the neurons was blinded to the treatment. Four sections per spinal segment or slice were used, counting all lamina I NK1R neurons in each section. Results were expressed as the percentage of the NK1R neurons in lamina I with NK1R internalization.
Confocal microscopy
Confocal images were acquired using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Inc., Thornwood, NY), with objectives of 20× (numerical aperture 0.8) and 63× oil (numerical aperture 1.4). Excitation light for the Alexa Fluor 488 fluorophore (emission peak 519 nm) was provided by the 488 nm line of an argon laser. The emission window was 500–560 nm. The pinhole was 1.0 Airy unit: 31.5 μm for the 20× objective and 50.7 μm for the 63× objective, as determined by the confocal microscope software. Images were acquired in grayscale as confocal stacks of sections of 1024×1024 pixels. Each section was averaged 2 times to reduce noise. The separation between confocal sections was 0.85 μm for the 20× objective and 0.38 μm for the 63× objective, as determined by the confocal microscope software using the Nyquist formula. Photomultiplier gain and offset were adjusted before acquiring each confocal stack to avoid pixel saturation.
Image processing
Images of the entire dorsal horn obtained with the 20× objective were used to show the location of each of the neurons imaged with the 63× objective. The program Imaris 6.1.5 (×64, Bitplane AG, Zurich, Switzerland) was used to crop the images in three dimensions. Images at 20× were cropped into the 10 brightest optical sections. Often, images of several NK1R neurons were acquired in a single confocal stack, in which case Imaris was used to crop each cell out of the stack into 4–6 optical sections through the middle of the cell. After cropping, a two-dimension projection picture was generated in Imaris and imported into Adobe Photoshop 5.5 (Adobe Systems Inc., Mountain View, CA), which was used to compose the multi-panel figures and to add text and arrows.
Dorsal root ganglion (DRG) neuron culture
Primary cultures of DRG neurons from spinal levels T12-S2 were prepared as previously described with minor modifications (Li et al., 2006). Neurons were plated on coverslips coated with Matrigel (BD Biosciences, San Jose, CA) in Neurobasal media containing B-27 supplement, 5 ng/ml nerve growth factor (all from Life Technologies, Grand Island, NY), 10% fetal bovine serum (Irvine Scientific, Santa Ana, CA) and 200 μM ketamine (Sigma). Ketamine in the culture medium is necessary to preserve NMDA receptor function in cultured DRG neurons (Chen et al., 2014).
Intracellular Ca2+ ([Ca2+]i) measurement
DRG neurons were used 2–3 days after plating. Neurons were loaded for 1 hour at 37°C on coverslips with 5 μM Fura-2 AM (Life Technologies) in Neurobasal media supplemented as described above, then washed and incubated for 30 min in Mg2+-free Hank’s buffer containing 10 mM Hepes, pH 7.2, 5 mM glucose, 1.2 mM CaCl2. During the last 15 min, the incubation buffer was supplemented 20 ng/ml BDNF with or without 1 μM DAMGO. In some experiments, the irreversible Cav2 channel blocker CTX GVIA (1 μM) was added to the neurons during the 1-hour incubation with Fura-2. Coverslips were mounted in an experimental chamber (volume 0.5 ml) and perfused (0.5 ml/min) with the same buffer. The chamber was placed on the stage of an inverted microscope (Axio Observer.A1) outfitted with a 40× objective and a digital camera (AxioCam MRm) and operated with associated software (AxioVision, Carl Zeiss, Thornwood, NY). Ratio images (340 nm/380 nm) were obtained at 1 sec intervals, and the average ratio values from each cell were stored for offline analysis. Neurons were stimulated by rapid addition of NMDA and glycine to a final concentration of 250 μM and 10 μM, respectively. KCl (50 mM final) was added at the end of the experiment a means to verify viable neurons. Responses were evaluated as the change in the 340 nm/380 nm ratio from the initial baseline value to the peak value. In a subset of cells, initial resting [Ca2+]i was estimated to be 91 ± 15 nM (n=20) using the method of Williams and Fay (1990).
Experimental Design and Statistical Analysis
Data were analyzed using Prism 7.03 (GraphPad Software, San Diego, CA). Statistical significance was set at 0.05. Data points and error bars indicate mean and standard error, except for [Ca2+]i measures where they show the median response and the interquartile range.
A power analysis (PASS software) of previous NK1R internalization data revealed that a sample size of 4 provides 81% power in a two-sample t test with 0.05 two-sided significance to detect effect sizes of 2.27 (in slices) or 2.57 (in vivo).
Experiments in spinal cord slices
Data were collected in weekly experiments of 12–24 slices prepared from 2–8 rats. Typically, 3–8 slices were obtained per rat. The number of slices for each data point is indicated in the figures. Replicates in each data point are from slices from more than one rat, so the statistical error represents both inter-slice and inter-animal variability. We set the target sample size at n = 6 per group, with the following exceptions. In concentration-response and time course experiments we used a minimum of 3 slices per data point because statistical significance depends on the sample size of the whole curve. Control data include a large number of slices because separate controls were included in each weekly experiment to assess consistency, and controls were later aggregated for the whole experiment. Likewise, a larger sample size resulted when the same data were collected in different experiments and then aggregated. In some critical experiments with atypical results, the sample size was increased to rule out the presence of outliers or experimental mistakes. Statistical analyses consisted of one-way ANOVA followed by Holm-Sidak’s post-hoc tests, except that non-linear regression analyses were used in the following experiments. Concentration-response data were fitted by a sigmoidal dose-response function: Y = bottom + (top-bottom)/(1 + 10ˆ(Log IC50-Log X)), where X is the concentration of drug and IC50 is the concentration of drug that produces half of the effect. The Hill coefficient was assumed to be 1.0. Baseline measures (zero concentration of drug) were included in the non-linear regression by assigning them a concentration value three log units lower than the lowest concentration of drug. Parameter constraints were: 0% < top < 100%, 0% < bottom. The statistical error of the IC50 is expressed as asymmetrical 95% confidence intervals (CI). Data from multiple concentration-response curves were fitted simultaneously and Akaike’s Information Criterion was used to test whether the two curves had the same or different parameter values. Concentration-responses of an agonist in the absence and presence of an antagonist were fitted using the Gaddum-Schild model (Colquhoun, 2007) to calculate the IC50 of the agonist and the KB of the antagonist. In this case, the top and bottom parameters were constrained to be the same for both curves. Time course data were fitted by non-linear regression to a one phase decay function: Y = (Y0 − plateau) * exp(−K * X) + plateau, where K is the rate constant, Y0 is the value at time 0 and “plateau” is the maximum value. Half-life was calculated as ln(2)/K.
Experiments in vivo
The target sample size was n = 8, but some rats were eliminated from data collection due to death or misplacement of the intrathecal catheter revealed by post-mortem examination. Rats were randomly assigned to treatment. The investigator quantifying the NK1R internalization data was blinded to treatment. Statistical analyses consisted of one-way ANOVA followed by Holm-Sidak’s post-hoc tests.
Experiments in cell cultures
Measures of [Ca2+]i presented a large variability due to cell heterogeneity, so they required using a large number of cells. Separate controls were used for batches of cells prepared under slightly different conditions. Results were analyzed with the Mann-Whitney test due to the non-Gaussian distribution of the [Ca2+]i data. Lines in the figure show the median response and error bars show the interquartile range.
Results and Discussion
MOR inhibition of NMDA-induced substance P release
Our first objective was to determine whether MORs inhibit substance P release in the spinal cord induced by NMDA receptor activation, which was accomplished by incubating rat spinal cord slices for 2 min with 10 μM NMDA and 10 μM D-serine (D-Ser). MOR agonists and antagonists were added to the slices during a 60 min incubation prior to the 2 min incubation with NMDA. All incubations were done at 35 °C in oxygenated and buffered aCSF. As we previously reported (Chen et al., 2014; Chen et al., 2010), NMDA induced NK1R internalization in about 50% of the NK1R neurons in lamina I (Fig. 1 A, ‘control’). The MOR agonists morphine, endomorphin-2 and DAMGO (all 1 μM) reduced NMDA-induced NK1R internalization to control levels (Fig. 1 A), showing that MORs are able to abolish NMDA-induced substance P release. The inhibition by DAMGO was eliminated by the MOR antagonists naloxone, naltrexone or CTAP (all 10 μM, Fig. 1 A), showing that it has the expected pharmacological profile of MORs. ANOVA of data in Fig. 1 A: p<0.0001, F9,58 = 51.25.
Figure 1. MOR inhibition of NMDA-induced substance P release in spinal cord slices.
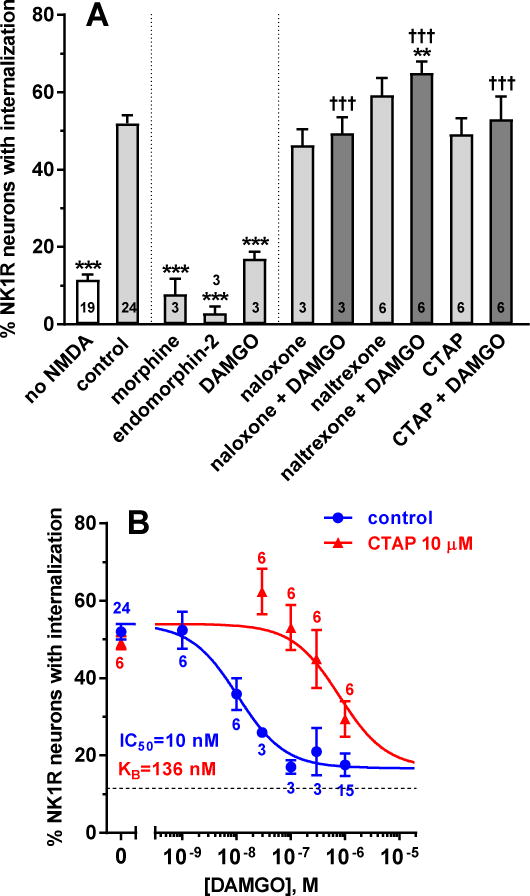
Spinal cord slices were incubated for 60 min with compounds and then for 2 min with NMDA and D-Ser (both 10 μM). The number of slices (n) is given inside the bars or next to the points. A. ‘no NMDA’: no compounds and no NMDA; ‘control’: no compounds followed by NMDA+D-Ser. Compounds were MOR agonists (1 μM morphine, 1 μM endomorphin-2, 100 nM DAMGO) and antagonists (10 μM naloxone, 10 μM naltrexone, 10 μM CTAP). ANOVA, p<0.0001. Holm-Sidak’s post-hoc tests: *** p<0.001, ** p<0.01 compared with control; ††† p<0.001 compared with DAMGO. B. DAMGO concentration-responses without (control) and with 10 μM CTAP. Data were analyzed by non-linear regression using the Gaddum-Schild model, with shared bottom and top parameters. Parameter values obtained were: IC50 (DAMGO) = 10 nM (95% CI 3.9 – 26.5 nM), KB (CTAP) = 136 nM (95% CI 43 – 393 nM), top = 54.0 ± 1.8%, bottom = 16.7 ± 2.7%, R2 = 0.64 (global fit).
Unexpectedly, DAMGO plus naltrexone produced a small but statistically significant increase of NMDA-induced NK1R internalization, and a trend in that direction was observed with naltrexone alone. Since naltrexone is an inverse agonist of MORs (Kenakin, 2001), this may reflect the presence of some MOR constitutive activity (Connor and Traynor, 2010; Walwyn et al., 2016).
To obtain further evidence of the involvement of MORs, we performed a Gaddum-Schild analysis by obtaining concentration-response curves for DAMGO in the absence and presence of the MOR-selective antagonist CTAP (10 μM). DAMGO produced a concentration-dependent inhibition of NMDA-induced substance P release with an IC50 of 10 nM (Fig. 1 B), similar to its potency to induce MOR internalization (30 nM) (Marvizon et al., 1999), to inhibit adenylyl cyclase (5–18 nM) (Keith et al., 1998; Yabaluri and Medzihradsky, 1997) and to increase [γ-35S]GTP binding (28 nM) (Yabaluri and Medzihradsky, 1997). CTAP produced a marked right-shift of the concentration-response curve, resulting in an equilibrium dissociation constant (KB) for CTAP of 136 nM in the Gaddum-Schild analysis. The corresponding pA2 value was 6.87 (95% CI = 6.40 – 7.36), similar with the pA2 reported for CTAP in guinea pig ileum: pA2 = 7.36 (95% CI = 6.83 – 7.78) (Kramer et al., 1989). The inhibition produced by DAMGO was complete, since the ‘bottom’ parameter of its concentration-response curve, 17 ± 3%, was similar to control NK1R internalization values in the absence of NMDA, 12 ± 1% (“no NMDA”, Fig. 1 A).
Time course of the inhibition by DAMGO of NMDA-induced substance P release
In the previous experiments spinal cord slices were incubated with MORs agonists for 60 min before the short (2 min) incubation with NMDA and D-Ser that evoked substance P release. In this experiment we studied the effect of the incubation time with DAMGO. First, we determined whether 1 μM DAMGO co-applied with NMDA and D-Ser to the slices for 2 min decreased NMDA-induced NK1R internalization. In this case DAMGO produced no inhibition (Fig. 2, “0 min”) compared with the effect of NMDA without DAMGO (“none”): p=0.317, Holm-Sidak’s post-hoc test of one-way ANOVA (global p<0.0001, F6,41 = 22). Second, we preincubated spinal cord slices with 1 μM DAMGO for various times prior to the 2 min incubation with NMDA and D-Ser (10 μM). As shown in Fig. 2, the inhibition by DAMGO was nearly complete at 15 min. Fitting of a one-phase decay function to these data yielded a time constant K = 0.18 ± 0.08 min−1, corresponding to a half-life of 3.8 min (95% CI 1.1 min – 11.5 min).
Figure 2. Time course of the inhibition of NMDA-induced NK1R internalization by DAMGO.
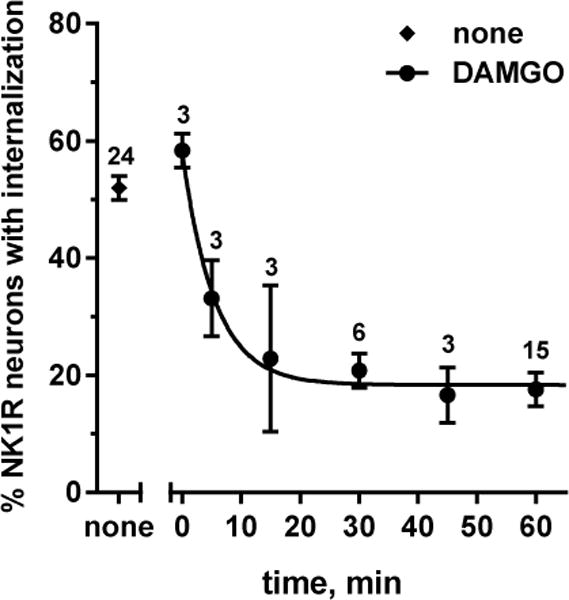
Spinal cord slices were incubated for the times indicated with 1 μM DAMGO and then for 2 min with 10 μM NMDA and D-Ser; “none”: no DAMGO incubation; 0 min: DAMGO included in the 2 min incubation with NMDA and D-Ser. Curve: fitting of a one-phase decay function: Y0 = 58 ± 6%, plateau = 18 ± 2%, K = 0.18 ± 0.08 min−1, t1/2 = 3.8 min (95% CI = 1.1 min – 11.5 min), R2 = 0.565. The number of slices (n) is given next to the points.
MORs inhibit NMDA-induced substance P release in vivo
Next, we studied whether MORs also inhibit substance P release induced by intrathecal NMDA in vivo. Whereas in slices NMDA readily evokes substance P release, in vivo NMDA does not consistently elicit substance P release (Chen et al., 2014; Nazarian et al., 2008). However, intrathecal NMDA evokes substance P release after nerve injury or intrathecal BDNF (Chen et al., 2014). This substance P release is mediated by NMDA receptors present in primary afferents because it disappeared in mice with selective NR1 subunit knockdown in nociceptive afferents (Chen et al., 2014). Since the effect of BDNF lasts more than 4 h, we devised a protocol consisting in three sequential intrathecal injections: 1) at 0 h, saline or BDNF (0.3 μg); 2) at 3 h, saline or DAMGO (3 nmol); 3) at 4 h, NMDA + D-Ser (10 nmol each). Rats were killed and fixed 10 min after the third injection. Intrathecal injections were delivered through catheters terminating at the L4–L5 spinal cord segments and NK1R internalization was measured in segment L4. Consistent with our previous results (Chen et al., 2014), after two injections of saline NMDA produced little NK1R internalization, except in three out of eight rats (Fig. 3, “NMDA”). Examples of lamina 1 neurons with NK1R surface staining in a rat from this group are shown in Fig. 4 A. Without BDNF, intrathecal DAMGO did not affect NMDA-induced NK1R internalization, except that outliers with high NK1R internalization were no longer observed (Fig. 3). Intrathecal BDNF resulted in a significant increase in NMDA-induced NK1R internalization (Fig. 3, “BDNF + NMDA”). Examples of five lamina 1 neurons with NK1R internalization in a rat injected with BDNF and NMDA are shown in Fig. 4 B. When injected 3 h after BDNF, DAMGO significantly inhibited NMDA-induced NK1R internalization (Fig. 3, one-way ANOVA: p=0.0011, F3,25 = 7.33, Holm-Sidak’s post-hoc tests are indicated in the figure). Examples of lamina 1 neurons with NK1R surface staining after DAMGO are shown in Fig. 4 C. Therefore, in vivo MORs inhibit substance P release induced by intrathecal NMDA only after priming with BDNF.
Figure 3. In vivo inhibition by DAMGO of NMDA-induced substance P release.
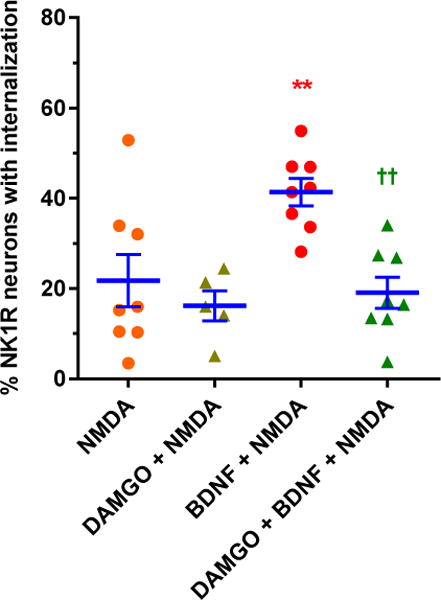
Rats received three intrathecal injections using the following time line: 0 h - saline or BDNF (0.3 μg); 3 h - saline or DAMGO (3 nmol); 4 h - NMDA + D-Ser (10 nmol each). Data are from spinal segment L4. NMDA induced NK1R internalization only after BDNF, and this was reduced by DAMGO. ANOVA: p=0.0011, F3,25 = 7.33. Holm-Sidak’s post-hoc test: ** p <0.01 compared with NMDA; †† p < 0.01 compared with BDNF + NMDA.
Figure 4. Confocal microscope images of lamina 1 neurons with and without NK1R internalization.
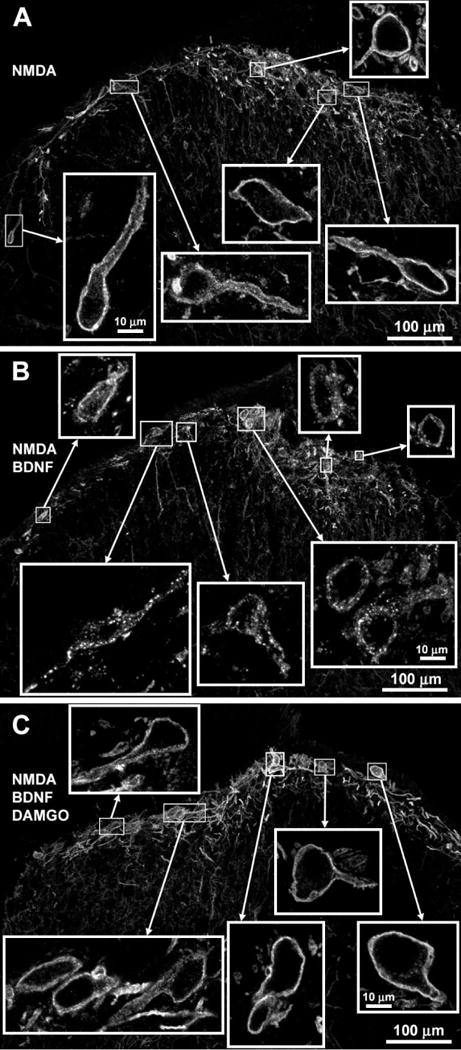
Images were taken from the L4 spinal segment of the rats used from the experiment in Fig. 3. Rats received three intrathecal injections using the following time line: 0 h - saline or BDNF (0.3 μg); 3 h - saline or DAMGO (3 nmol); 4 h - NMDA + D-Ser (10 nmol each). A. Rat injected with saline-saline-NMDA; there was no NK1R internalization. B. Rat injected with BDNF-saline-NMDA; NK1R internalization is clear in 5 out of 7 cells. C. Rat injected with BDNF-DAMGO-NMDA; there was no NK1R internalization. Images are 10 optical sections taken with a 20× objective (main panels, voxel size 692 × 692 × 854 nm, scale bar 100 μm) or a 63× objective (insets, voxel size 132 × 132 × 377 nm, scale bar 10 μm).
Contribution of Cav channels to NMDA-induced substance P release
Once we established that MORs inhibit NMDA-receptor induced substance P release, we investigated the mechanisms by which MORs may interact with these NMDA receptors. The substance P released in these experiments is from primary afferents because it disappears after depleting C-fibers of substance P by incubating the slices with capsaicin (Chen et al., 2010). Likewise, the NMDA receptors that induce the substance P release are present in primary afferents because NMDA-induced substance P release decreased considerably in mice with a selective deletion of NMDA receptors in nociceptive primary afferents (Chen et al., 2014). NMDA-induced substance P release was not affected when firing of action potentials was blocked by lidocaine (Chen et al., 2010), indicating that the NMDA receptors are present at the presynaptic terminals.
NMDA receptors may induce substance P release by two mechanisms: 1) direct Ca2+ entry through NMDA receptors, 2) Ca2+ entry through Cav channels activated by the depolarization they produce. Since MORs inhibit Cav2 channels in primary afferent (Moises et al., 1994; Raingo et al., 2007; Rusin and Moises, 1998; Strock and Diverse-Pierluissi, 2004), the second mechanism could explain their inhibition of NMDA receptor-induced substance P release. To evaluate the relative contribution of these two mechanisms, we determined whether NMDA-induced substance P release can be inhibited by the Cav channel blockers CTX MVIIC and CTX GVIA. CTX MVIIC blocks Cav2.2 (N type) channels with an IC50 18 nM and Cav2.1 (P/Q type) channels with an IC50 of 50 nM (McDonough et al., 1996; Rusin and Moises, 1995). CTX GVIA blocks Cav2.2 channels selectively and irreversibly (Hannon and Atchison, 2013; Keith et al., 1989; Rusin and Moises, 1995, 1998). The Cav channels that are present in primary afferent terminals and are inhibited by MORs are Cav2.2 and Cav2.1 (Rusin and Moises, 1995). To study the effect of CTXs on NMDA-induced substance P release, slices were incubated for 60 min with different concentrations of CTX MVIIC or CTX GVIA, and then for 2 min with 10 μM NMDA and D-Ser. The IC50s for CTX MVIIC and CTX GVIA were 117 nM and 374 nM, respectively (Table 1). Both CTXs produced a partial inhibition of the evoked NK1R internalization (Fig. 5, Table 1), to 34 ± 4% for CTX MVIIC and to 34 ± 6% for CTX GVIA, well above basal NK1R internalization in the absence of NMDA, which was 12±1% (dotted lines in Fig. 5).
Table 1.
Parameters for the concentration responses for ω-conotoxin MVIIC and GVIA.
| CTX | stimulus | IC50 (95% CI) | top | bottom | R2 |
|---|---|---|---|---|---|
| MVIIC | NMDA | 117 nM (19 – 556 nM) | 57 ± 2% | 34 ± 4% | 0.54 |
| MVIIC | dorsal root | 267 nM (72 – 833 nM) | 60 ± 2% | 28 ± 7% | 0.46 |
| GVIA | NMDA | 374 nM (70 – 2205 nM) | 58 ± 2% | 34 ± 6% | 0.52 |
Parameters obtained from non-linear regression fitting of data in Fig. 5 to a dose-response function. The Hill coefficient was assumed to be 1.0. 95% CI: 95% confidence interval (profile likelihood).
Figure 5. Inhibition by ω-conotoxins (CTX) of NK1R internalization evoked by NMDA or dorsal root stimulation. NMDA.
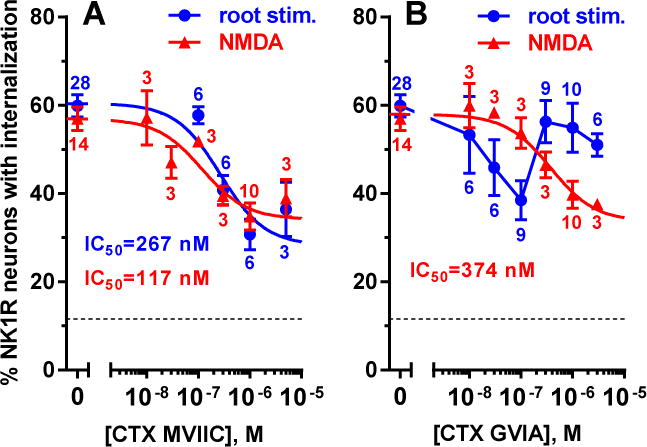
Spinal cord slices were incubated for 60 min with different concentrations of CTX MVIIC (A) or CTX GVIA (B), and then for 2 min with 10 μM NMDA and D-Ser. Root stimulation: Spinal cord slices with one dorsal root (L4–L5) were superfused for 2 min with CTX MVIIC (A) or incubated for 60 min with CTX GVIA and then superfused with it (B). Next, the dorsal root was stimulated with 1000 pulses (20 V, 0.4 ms) at 100 Hz, and the slice was superfused with aCSF for 10 min more. For controls (0 M), slices were incubated or superfused with aCSF. Parameters of the concentration-response curves are given in Table 1. The number of slices (n) is given next to the points. The dotted horizontal line represents basal values of NK1R internalization in the absence of NMDA or root stimulation.
As a positive control, we studied the inhibition by CTXs of substance P release induced by electrical stimulation of the dorsal root, which induces firing of action potentials in primary afferents (Adelson et al., 2009) leading to the opening of Cav channels in their central terminals. This electrical stimulation induced about the same amount of NK1R internalization as NMDA (Fig. 5; Table 1, top parameters). CTX MVIIC was applied to the slices for 2 min prior and during electrical stimulation. The concentration-response curve for CTX MVIIC using this stimulus was very similar to its concentration-response when NMDA was used as stimulus (Fig. 5 A). The inhibition was also partial, with a maximal inhibition (‘bottom’) of 28 ± 7% (Table 1), with a 65% probability of being the same as with NMDA (Akaike’s Information Criterion from global fitting of both curves). The IC50 was 267 nM (Table 1), with a 70% probability of being the same as with NMDA (Akaike’s Information Criterion). Therefore, CTX MVIIC had the same potency and efficacy to inhibit substance P release evoked by NMDA or by dorsal root stimulation, indicating that Cav2 channels are similarly involved in both processes.
Since the Cav2 blockers CTX MVIIC and CTX GVIA only partially inhibited NMDA-induced substance P release, both routes of Ca2+ entry seem to be involved. It is unlikely that the effect of the CTXs was due to direct inhibition of the NMDA receptors because CTX MVIIC inhibited NMDA-induced substance P release with the same potency as that elicited by electrical stimulation of the dorsal root. The later involves primary afferent firing, so it is likely to be mediated by Cav channel opening.
When using dorsal root stimulation, the concentration-response curve for CTX GVIA was anomalous (Fig. 5 B). Given its slow association kinetics (Hannon and Atchison, 2013), CTX GVIA was applied to the slices for 60 min and then superfused during root stimulation. The resulting concentration-response was V-shaped. Increasing the sample size of the data points showed that this was not due to the presence of outliers. Fitting to a V-shaped curve was not attempted, since this would be hard to interpret. It is possible that the V-shaped response is due to nonselective effects at the higher doses of CTX GVIA, such as effects on other channels activated by dorsal root stimulation. There is evidence that high frequency electrical stimulation leads to anomalous binding of CTX GVIA to Cav2 channels, resulting in partial inhibition of neurotransmitter release (Smith and Cunnane, 1998).
TRPV1-induced substance P release is not inhibited by MORs
Capsaicin is another powerful stimulus to evoke substance P release from primary afferents (Aimone and Yaksh, 1989; Lao et al., 2003; Marvizon et al., 2003a). This effect of capsaicin is mediated by the opening of TRPV1 channels, since it was blocked by the TRPV1 antagonist capsazepine (Lao et al., 2003). Since TRPV1 channels directly produce a strong influx of Ca2+, they may bypass the inhibition of Cav channels by MORs (Moises et al., 1994; Raingo et al., 2007; Rusin and Moises, 1995). To investigate this possibility, we investigated whether MORs inhibit capsaicin-induced substance P release. As in previous experiments, spinal cord slices were incubated for 60 min with the MOR agonist DAMGO (100 nM) and then for 2 min with capsaicin. Capsaicin at 1 μM elicited maximal NK1R internalization, which was not affected by DAMGO (Fig. 6 A). To determine whether DAMGO was able to inhibit sub-maximal substance P induced by capsaicin, we repeated the experiment lowering the concentration of capsaicin to 0.3 μM, which is near its EC50 to induce NK1R internalization (0.37 μM) (Marvizon et al., 2003a). This time 0.3 μM capsaicin produced 75% NK1R internalization and this was still unaffected by DAMGO (Fig. 6 B).
Figure 6. Capsaicin-induced NK1R internalization was not affected by DAMGO or ω-conotoxins (CTX).
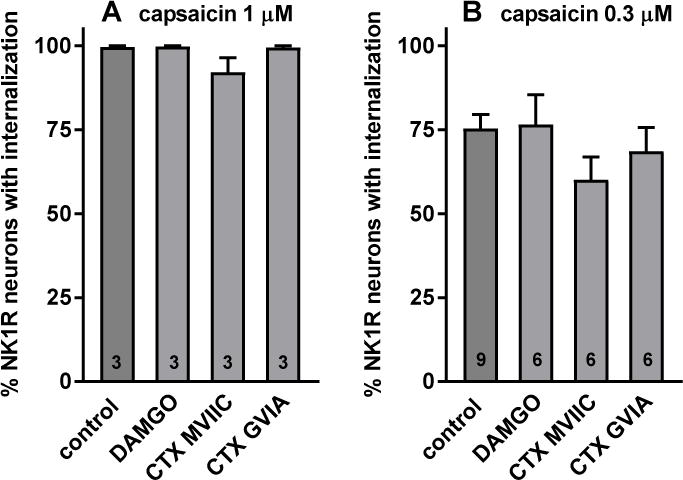
Spinal cord slices were incubated for 60 min with aCSF (“no capsaicin” and control), 100 nM DAMGO), 1 μM CTX-MVIIC or 1 μM CTX-GVIA. Then the slices (except for “no capsaicin”) were incubated for 2 min with 1 μM (A) or 0.3 μM (B) capsaicin. Numbers inside bars indicate the number of slices in each group. ANOVA (A): p=0.0957, F3,8 = 2.99; ANOVA (B): p=298, F3,23 = 1.3.
If capsaicin induces substance P release by allowing Ca2+ entry through TRPV1 channels, then it would not require Ca2+ entry through Cav channels. If this is true, then Cav channel blockers would not affect capsaicin-induced substance P release. Indeed, the Cav2 channel blockers CTX MVIIC and CTX GVIA (1 μM) did not inhibit NK1R internalization induced by 1 μM capsaicin (Fig. 6 A). When the concentration of capsaicin was lowered to 0.3 μM, CTX MVIIC showed a trend to inhibit NK1R internalization, but this effect was not statistically significant (p=0.27, Holm-Sidak’s post-hoc test). Therefore, MORs do not inhibit capsaicin-induced substance P release because Ca2+ entry through TRVP1 channels bypasses MOR inactivation of Cav2 channels.
These results appear to contradict early reports in which MOR inhibition of substance P release was demonstrated using capsaicin as the stimulus (Aimone and Yaksh, 1989). However, we have shown (Lao et al., 2003) that when capsaicin is applied to the dorsal root to open TRPV1 channels away from the presynaptic terminal, or when low concentrations of capsaicin are used, substance P release does require Cav2 channels and can be inhibited by the GABAB receptor agonist baclofen. Like DAMGO, baclofen did not inhibit substance P release when higher concentrations of capsaicin were used (Lao et al., 2003). Therefore, it is likely that the intrathecal superfusion with capsaicin used by Aimone and Yaksh opened a small number of TRPV1 channels, so that substance P release required opening of Cav2 channels in addition of TRPV1 channels.
NMDA-induced increases in [Ca2+]i in cultured DRG neurons
To study Ca2+ entry through NMDA receptors we measured increases in [Ca2+]i induced by NMDA in cultured DRG neurons (Li et al., 2004; McRoberts et al., 2001; McRoberts et al., 2007). Rat DRG neurons were isolated and cultured for 2 days in media containing 200 μM ketamine in order to preserve NMDA receptor function (Chen et al., 2014). The DRG neurons were loaded for 1 h with Fura-2 AM (5 μM) and preincubated for 15 min with BDNF (20 ng/ml) to boost NMDA receptor function (Chen et al., 2014). Then [Ca2+]i was measured with a fluorescence microscope while the cells were superfused with medium. Addition of NMDA (250 μM) plus glycine (10 μM) produced [Ca2+]I increases in some of the neurons, which varied from slow-developing (Fig. 7 A) to small and rapid (Fig. 7 B, C). [Ca2+]i returned to baseline upon washout (Fig 10). These experiments were done in small to medium sized DRG neurons. Large DRG neurons generally did not respond to NMDA with detectable increases in [Ca2+]i. Addition of 1 μM DAMGO during the preincubation with BDNF resulted in smaller increases in [Ca2+]i produced by NMDA + glycine (Fig. 7 D–F). Fig. 8 A shows a quantitative analysis of the effect of DAMGO on NMDA-induced increases in [Ca2+]i. DAMGO did not change the number of DRG neurons that responded to NMDA + glycine (35% for control, 37% for DAMGO, p = 0.405, Fisher’s exact contingency test). In the neurons that did respond to NMDA + glycine, DAMGO decreased the magnitude of the peak response by 56% (Fig. 8 A; Mann-Whitney test p=0.012). This finding is consistent with an inhibition of NMDA receptors by MORs.
Figure 7. Representative traces of increases in [Ca2+]i produced by NMDA in DRG neurons.
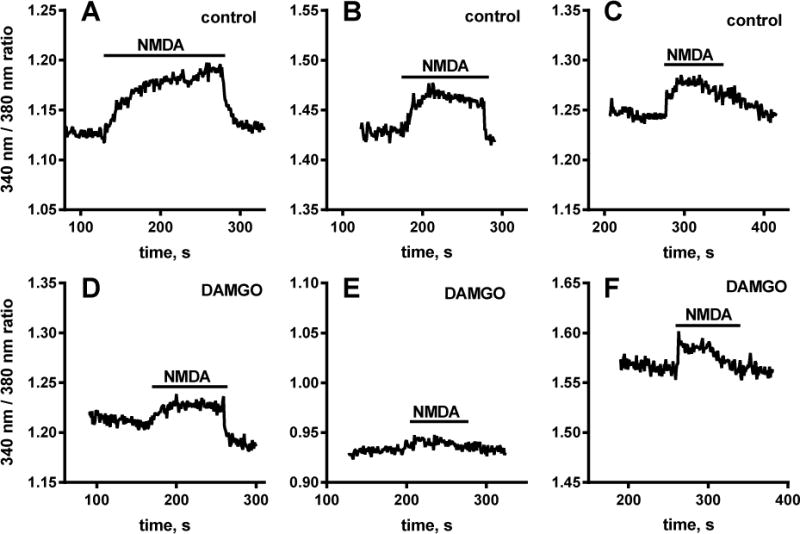
Cultured DRG neurons were loaded for 1 h with 5 μM Fura-2 AM and then incubated for 15 min with 20 ng/ml BDNF alone (A–C) or with 1 μM DAMGO (D–F). [Ca2+]i was measured with a fluorescence microscope while the cells were superfused with medium. Addition of 250 μM NMDA + 10 μM glycine (“NMDA”) produced increases in [Ca2+]i. Preincubation with DAMGO resulted in smaller increases in [Ca2+]i induced by NMDA + glycine.
Figure 10. Involvement of AC, PKA and PKC on substance P release.
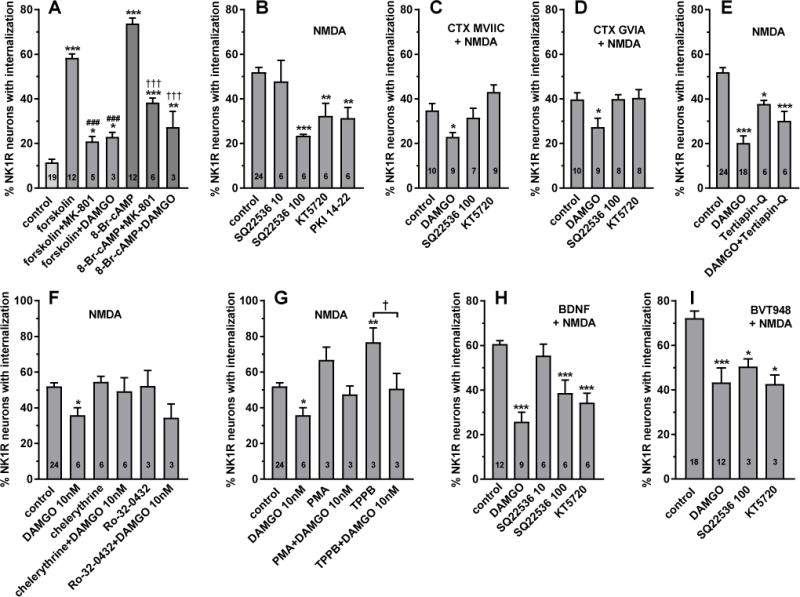
A. Spinal cord slices were incubated for 60 min with 10 μM forskolin, 0.5 mM 8-Br-cAMP, or nothing (control). MK-801 (10 μM) or DAMGO (1 μM) were added at the same time as forskolin or 8-Br-cAMP. B–I: Substance P release was induced by incubating spinal cord slices for 2 min with 10 μM NMDA and 10 μM D-Ser. Before that, the slices were incubated for 60 min with the other compounds indicated. Compounds listed inside the panels above “+ NMDA” were applied to all of the slices in that panel, and were: 1 μM CTX MVIIC (C), 1 μM CTX GVIA (D), 20 ng/ml BDNF (H), and 10 μM BVT948 (I). In these panels, ‘control’ refers to slices incubated with these compounds followed by NMDA+D-Ser. Other compounds were: 10 μM chelerythrine (PKC inhibitor), 1 μM DAMGO, 10 μM KT5720 (PKA inhibitor), 10 μM PKI 14–22 myristolated (PKA inhibitor), 1 μM phorbol 12-myristate 13-acetate (PMA, PKC activator), 1 μM Ro-32-0432 (PKC inhibitor), 10 μM or 100 μM SQ 22536 (AC inhibitor), 100 nM tertiapin Q (GIRK inhibitor), and 1 μM TPPB (PKC activator). ANOVA: A, p<0.0001, F4,49=112; B, p<0.0001, F5,48=9.1; C, p=0.0007, F3,31=7.4; D, p=0.02, F3,31=3.8; E, p<0.0001, F3,49=30; F, p=0.0179, F5,42=1.826; G, p<0.0001, F5,36=1.869; H, p<0.0001, F4,34=17; I, p=0.0003, F3,32=8.48. Holm-Sidak’s post-hoc tests: * p<0.05, ** p<0.01, *** p<0.001, compared with control; ### p<0.001 compared to forskolin, ††† p< 0.001 compared to 8-Br-cAMP, † p< 0.05 as indicated. Numbers inside the bars indicate the number of slices in each group (n).
Figure 9. Ifenprodil inhibition of NMDA-induced NK1R internalization.
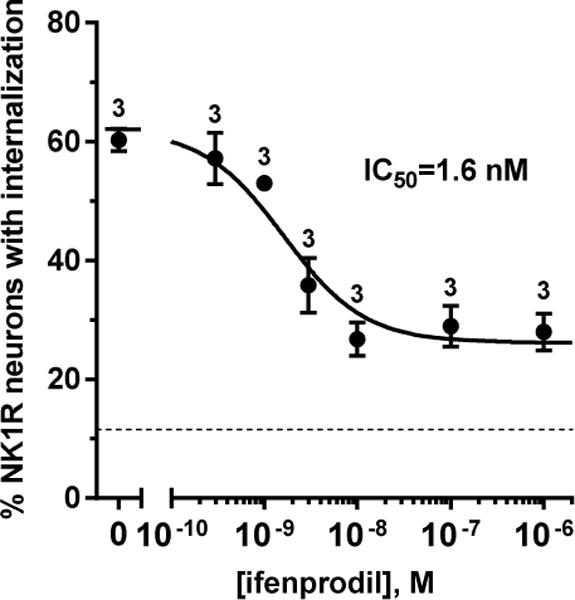
Concentration-responses for the NR2B-selective antagonist ifenprodil were obtained by incubating spinal cord slices with ifenprodil alone for 45 min and with NMDA, D-Ser, captopril and thiorphan (all 10 μM) for 2 min. The number of slices (n) is given next to the points. IC50 = 1.6 nM (95% CI = 0.7 – 3.6 μM), top = 62 ± 3%, bottom = 26 ± 2%, R2 = 0.86. The dotted horizontal line represents basal values of NK1R internalization in the absence of NMDA.
Figure 8. DAMGO, but not CTX MVIIC, decreases NMDA-induced [Ca2+]i responses in DRG neurons.
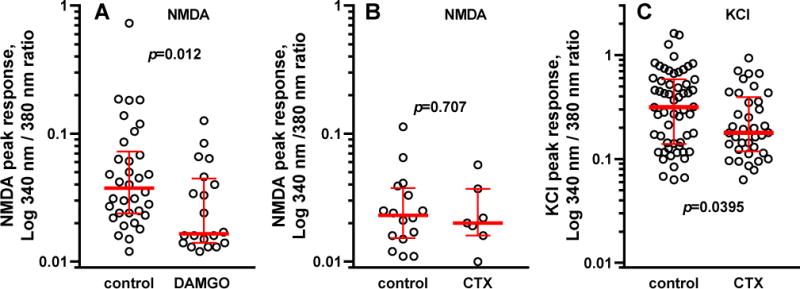
A. Cultured DRG neurons were loaded for 1 h with 5 μM Fura-2 AM and then incubated for 15 min with 20 ng/ml BDNF with or without 1 μM DAMGO. Responses were evoked by rapid infusion of 250 μM NMDA + 10 μM glycine. B. DRG neurons were loaded for 1 h with Fura-2 AM without (control) or with 1 μM CTX GVIA (CTX), and then incubated with BDNF as above before stimulation with 250 μM NMDA + 10 μM glycine. C. Neurons loaded with Fura-2 AM for 1 h with or without CTX GVIA were stimulated with 50 mM KCl to induce the opening of Cav channels. Bars show the median response and the interquartile range. A Mann-Whitney test was used to obtain the p values given in each panel.
To determine whether the increase in [Ca2+]i produced by NMDA + glycine could be due in part to Ca2+ entry through Cav2 channels, the Cav2.2 channel blocker CTX GVIA (1 μM) was added during the 1 h incubation of the DRG neurons with Fura-2. CTX GVIA did not affect the responses to NMDA + glycine (Fig 11 B, p=0.7), indicating that the Ca2+ entry produced by NMDA was through the NMDA receptors themselves and not through Cav2 channels. One possibility is that Cav2 channels are present in the presynaptic terminals but not in the cell bodies of DRG neurons. As a positive control, at the end of the experiment we induced Ca2+ entry in the DRG neurons by depolarizing them with 50 mM KCl. This produced rapid and large increases in [Ca2+]i in all DRG neurons. Incubating these neurons for 1 h with 1 μM CTX GVIA decreased the KCl-evoked raises in [Ca2+]i (Fig. 8 C, p=0.0395), showing that the Ca2+ entry induced by KCl depolarization was mediated by Cav2.2 channel opening. Therefore, the lack of effect of CTX GVIA on NMDA-induced Ca2+ entry was not due to the absence of Cav2 channels in the soma of DRG neurons. Taken together, these results indicate that MORs are able to inhibit NMDA receptors in primary afferents by a mechanism other than inactivation of Cav2 channels.
Most NMDA receptors that induce substance P release contain the NR2B subunit
Besides inactivating Cav2 channels, MORs also inhibit adenylyl cyclase (AC). Protein kinase A (PKA), which is activated by cAMP produced by AC, phosphorylates NMDA receptors at Ser1166 of their NR2B subunit, and this is required for NMDA receptor function (Lau et al., 2009; Murphy et al., 2014). Therefore, inhibition of AC could provide an additional pathway by which MORs inhibit NMDA receptors in primary afferent terminals. Indeed, the NR2B subunit is widely expressed in DRG neurons (Marvizon et al., 2002). To confirm that the NMDA receptors that induce substance P release have the NR2B subunit we assessed whether they are inhibited with high potency by the NR2B-selective antagonist ifenprodil. Spinal cord slices were incubated with different concentrations of ifenprodil for 45 min and then with NMDA and D-Ser for 2 min. The resulting concentration-response curve had an IC50 of 1.6 nM (95% CI = 0.7 – 3.6 nM, Fig. 9), a potency higher than that reported for ifenprodil to inhibit currents through NR1a/NR2B NMDA receptors in Xenopus oocytes, 340 nM (Williams, 1993), or the KD for ifenprodil binding to rat brain membranes, 25 nM (Grimwood et al., 2000). This shows that the NMDA receptors that induce substance P release have the NR2B subunit. Maximal inhibition by ifenprodil (‘bottom’) was 26 ± 2% NK1R internalization, whereas NK1R internalization in the absence of NMDA was 12±1% (dotted line in Fig. 9). This partial inhibition suggests that a small fraction of the substance P release may be induced by NMDA receptors without the NR2B subunit.
The AC/PKA pathway facilitates NMDA receptor-induced substance P release
One of the main signaling mechanisms of MORs is inhibition of AC (Childers, 1991; Piros et al., 1996). NMDA receptors containing the NR2B subunit are sensitive to AC inhibition because they require NR2B phosphorylation at Ser1166 by PKA (Murphy et al., 2014). The NR2B subunit is expressed in all types of DRG neurons (Marvizon et al., 2002) and is involved in the pro-nociceptive effects of NMDA receptors (Tan et al., 2005). The potent inhibition produced by ifenprodil demonstrates that most NMDA receptors that induce substance P release do have the NR2B subunit, so their function probably requires PKA phosphorylation.
To explore this possibility, we first studied the effects of activating AC or PKA on substance P release. To activate AC, spinal cord slices were incubated for 60 min with 10 μM forskolin. To directly activate PKA, slices were incubated for 60 min with the cell-permeable cAMP analog 8-Br-cAMP (0.5 mM) (Brown et al., 2000; Yang et al., 2005). Surprisingly, both compounds elicited an amount of NK1R internalization greater than NMDA+D-Ser, with 8-Br-cAMP producing the larger effect (Fig. 10 A). Addition of the NMDA receptor blocker MK-801 (10 μM) during the incubation with forskolin or 8-Br-cAMP substantially decreased their effect on substance P release, although not to control levels. This shows that the induction of substance P release by forskolin and 8-Br-cAMP is mediated by NMDA receptors. Likewise, addition of the MOR agonist DAMGO (1 μM) during the incubation with forskolin or 8-Br-cAMP markedly decreased the evoked substance P release (Fig. 10 A), but not to control levels. Since activation of PKA by 8-Br-cAMP is downstream of MOR inhibition of AC, this probably reflects MOR inactivation of Cav2 channels. These results indicate that in basal conditions NMDA receptors in primary afferent terminals are only partly phosphorylated at Ser1166 by PKA, and that their complete phosphorylation in the presence of forskolin or 8-Br-cAMP enables their activation by background amounts of glutamate and glycine. Hence, activation of the AC-PKA pathway in primary afferents would result in a large increase in NMDA receptor function, leading to hyperalgesia.
Next, we studied whether the NMDA receptors that induce substance P release depend on the activation of AC and PKA, by using the AC inhibitor SQ22536 and the PKA inhibitors KT5720 and PKI 14–22. NK1R internalization induced by applying NMDA + D-Ser (10 μM) to spinal cord slices was decreased by the AC inhibitor SQ22536 at 100 μM, but not at 10 μM (Fig. 10 B). SQ22536 is normally used at 50 μM or 100 μM (Chang et al., 2010; Evans et al., 2001; Shi and Bunney, 1992). NMDA-induced NK1R internalization was also decreased by the PKA inhibitors KT 5720 (10 μM) and PKI 14–22 myristolated (10 μM). These results show that complete elimination of PKA phosphorylation of these NMDA receptors at Ser1166 suppresses their activation even by saturating concentrations of NMDA and D-Ser. Therefore, the well-established inhibition of AC by MORs would decrease NMDA receptor-induced substance P release.
MORs inhibit the CTX-insensitive component of substance P release
CTX MVIIC and CTX GVIA only blocked part of the NMDA-induced substance P release (to 34%, compared to 12% basal NK1R internalization, Fig. 5, Table 1) while MOR agonists completely abolished it (Fig. 1). This suggests that MORs are able to inhibit NMDA-induced substance P by mechanisms other than inactivation of Cav2 channels. To confirm this, we determined whether 1 μM DAMGO produced an additional inhibition of substance P release in the presence of maximal concentrations (1 μM) of CTX MVIIC (Fig. 10 C) or CTX GVIA (Fig. 10 D). As usual, spinal cord slices were incubated for 60 min with CTX and DAMGO and then with for 2 min NMDA and D-Ser. Indeed, DAMGO produced a significant decrease in the presence of either CTX. Since this additional inhibition cannot be attributed to Cav2 inactivation, MOR inhibition of NMDA receptor-induced substance P release has to be mediated by an additional mechanism.
Then we explored whether the AC inhibitor SQ22536 (100 μM) or the PKA inhibitor KT5720 (10 μM) could similarly inhibit the CTX-insensitive component of substance P release. We found that these two inhibitors did not produce any further inhibition in the presence of CTXs (Fig. 10 C, D). However, unlike DAMGO, these two inhibitors did not completely abolish NMDA-induced NK1R internalization (Fig. 10 B). This, together with the small inhibition produced by DAMGO in the presence of CTX, make the interpretation of these results difficult. One possibility is that the MOR inhibition in addition to Cav channel inactivation is not due to AC inhibition. Another possibility is that PKA phosphorylates the same population of NMDA receptors that trigger the opening of Cav2 channels. This implies that there is a third mechanism of MOR inhibition of NMDA receptor-induced substance P release.
MOR inhibition is not mediated by GIRK channels
One of such mechanisms could be the opening of GIRK channels (Marker et al., 2005; Schneider et al., 1998), which would produce hyperpolarization and thus increase the Mg2+ blockade of NMDA receptors. GIRK channels are present in primary afferent neurons of rats and humans (but not mice), where they mediate the analgesia produced by peripherally-restricted opiates (Nockemann et al., 2013). This hypothesis predicts that DAMGO inhibition of NMDA-induced substance P release would be reversed by the GIRK inhibitor tertiapin-Q (Jin et al., 1999). Therefore, we incubated spinal cord slices for 60 min with 1 μM DAMGO, 100 nM tertiapin Q or DAMGO + tertiapin Q. Unexpectedly, tertiapin Q alone produced a small but significant inhibition of NMDA-induced NK1R internalization (Fig. 10 E). In any case, tertiapin Q did not affect the inhibition by DAMGO: results in the presence of DAMGO alone and DAMGO + tertiapin Q were not statistically different (p = 0.108, Holm-Sidak’s post-hoc test). Therefore, GIRK opening does not seem to mediate the MOR inhibition of NMDA receptor-induced substance P release. Consistent with our results, MOR inhibition of Aδ- and C-fiber-evoked EPSCs in rat spinal cord slices was not affected by the K+ channels blockers Ba2+ and Cs+ (Heinke et al., 2011).
MOR inhibition is not mediated by the physical association of MORs with NMDA receptors disrupted by protein kinase C (PKC)
Studies in the periaqueductal gray by the group of Javier Garzón (Garzón et al., 2012; Rodríguez-Muñoz et al., 2008; 2011; 2012) have shown that MORs are able to physically associate with NMDA receptors at postsynaptic sites. This promotes the analgesic effect of morphine by inhibiting the NMDA receptors. This physical association is disrupted by PKC phosphorylation of either the MOR or the NR1 subunit of the NMDA receptor, in both cases leading to a decrease in MOR analgesia. To determine whether this mechanism mediates MOR inhibition of NMDA receptors in primary afferent terminals, we studied the effect of PKC inhibitors and activators on the inhibition by DAMGO of NMDA-induced substance P release.
First, the Garzón mechanism predicts that inhibiting PKC will promote MOR/NMDA receptor association, increasing the inhibitory effect of DAMGO on NMDA-induced substance P release. To detect effects on the potency of DAMGO we used it at 10 nM, a concentration near its IC50 (Fig. 1 B). The PKC inhibitors chelerythrine (10 μM) and Ro-32-0432 (1 μM) did not affect NMDA-induced substance P release and did not increase its inhibition by DAMGO (Fig. 10 H). In fact, chelerythrine showed a trend to decrease the inhibition by 10 nM DAMGO, contrary to the prediction.
Second, the Garzón mechanism predicts that activating PKC will promote the dissociation of the MOR/NMDA receptor complex, decreasing MOR inhibition of NMDA receptor-induced substance P release. We found that the PKC activator TPPB (1 μM) strongly increased NMDA-induced substance P release (Fig. 10 I), an effect consistent with the reported potentiating effect of PKC on NMDA receptors in primary afferent terminals (Xie et al., 2017). TPPB did not affect inhibition by 10 nM DAMGO (Fig. 10 I). Phorbol 12-myristate 13-acetate (1 μM) produced similar results, although not statistically significant (Fig. 10 I). Again, these results do not support the prediction made by the Garzón mechanism.
Therefore, our results do not support the idea that PKC-regulated physical association of MORs and NMDA receptors mediates MOR inhibition of NMDA receptors in primary afferent terminals. However, we found evidence that PKC activation increases NMDA receptor function in primary afferents, which needs to be addressed in future studies.
An additional possibility is an effect of MORs on substance P release machinery, suggested by the finding that DAMGO moderately decreased vesicle exocytosis from Aδ-fiber terminals, but not C-fiber terminals (Heinke et al., 2011). However, the fact that DAMGO did not inhibit capsaicin-induced substance P release argues against MORs having any major effect on the release machinery.
Interaction between the NMDA receptor potentiating effects of PKA and BDNF
We previously reported that NMDA-induced substance P release is increased by BDNF (Chen et al., 2014) acting on a truncated form of trkB receptors (Baxter et al., 1997; Fenner, 2012) present in DRG neurons (Chen et al., 2014; Ernfors et al., 1993; Lee et al., 1999). This effect of BDNF seems to be mediated by Src family kinase (SFK) phosphorylation of the NMDA receptors at Tyr1472 of the NR2B subunit (Chen et al., 2010), because incubating DRG neurons with BDNF increased Tyr1472 phosphorylation (Chen et al., 2014).
To investigate how this potentiating effect of BDNF relates to the inhibition by MORs and phosphorylation by PKA, spinal cord slices were preincubated for 60 min with 20 ng/ml BDNF prior to the 2 min incubation with NMDA + D-Ser. This increased NMDA-induced NK1R internalization (compare the controls of Fig. 10 G and Fig. 10 H), although the effect was not as pronounced as that produced by BDNF in vivo (Fig. 3). This potentiating effect of BDNF did not preclude the inhibition by 1 μM DAMGO, 100 μM SQ22536 or 10 μM KT5720, when they were added during the 60 min incubation with BDNF (Fig. 10 H).
The inhibitor of protein tyrosine phosphatases BVT948 (Liljebris et al., 2004) also increases NMDA-induced substance P release, likely by reducing the dephosphorylation of Tyr1472 and thus opposing the effect of SFKs. Again, we determined whether this effect of BVT948 precludes the inhibition by MORs or by AC and PKA inhibitors. Preincubating the slices for 60 min with 10 μM BVT948 substantially increased NMDA-induced NK1R internalization (compare the controls of Fig. 10 G and Fig. 10 I). Inhibition by 1 μM DAMGO, 100 μM SQ22536 and 10 μM KT5720 (added during the 60 min preincubation with BVT948) still occurred (Fig. 10 I), although to a lesser extent than in the absence of BVT948.
Taken together, results in Figs. 10 H and I indicate that NR2B phosphorylation at Ser1166 by PKA and at Tyr1472 by SFKs potentiate NMDA receptor function independently of each other.
Physiological role
The interaction between NMDA receptors and AC-PKA signaling in primary afferents plays a major role in the maintenance of chronic pain. Thus, in the latent sensitization model of chronic pain (Marvizon et al., 2015) NMDA receptors, AC and PKA in the spinal cord are required for the maintenance of hyperalgesia (Corder et al., 2013). In this model, substance P and NK1Rs also contribute to hyperalgesia (Rivat et al., 2009). Conversely, MORs in the spinal cord continuously suppress the hyperalgesia in latent sensitization (Walwyn et al., 2016). The fact that α2A adrenergic receptors, which are exclusively located in primary afferent terminals (Stone et al., 1998), also suppress the hyperalgesia suggests the mechanisms of latent sensitization are located in primary afferents. All this suggests that suppression of hyperalgesia by MORs during chronic pain involves their inhibition of NMDA receptors in primary afferent terminals.
Conclusions
This study shows that MORs strongly inhibit NMDA receptor-induced substance P release from the central terminals of primary afferents. This inhibition is mediated by two mechanisms. The first is inactivation of Cav2 channels, which partly mediate the release of substance P triggered by the NMDA receptors. The second is inhibition of AC, since AC and PKA signaling is required by these NMDA receptors. Most of them have the NR2B subunit, which undergoes activating phosphorylation by PKA. In contrast, TRPV1-induced substance P release is not inhibited by MORs because it occurs independently of Cav2 channel opening. MOR inhibition and PKA activation of these NMDA receptors occurs independently of the potentiating effect of BDNF and SFKs on them.
Highlights.
NMDA receptor-induced substance P release is inhibited by μ-opioid receptors.
This release involves the opening of voltage-gated calcium channels.
These NR2B-containing NMDA receptors require protein kinase A phosphorylation.
The μ-opioid receptors act by inhibiting calcium channels and adenylyl cyclase.
Capsaicin-induced substance P release is not inhibited by μ opioid receptors.
Acknowledgments
Intracellular calcium imaging was performed at the Imaging and Stem Cell Biology Core (ISCB), CURE: Digestive Diseases Research Center (P30DK41301). This study was done under the umbrella of the following UCLA institutes: Brain Research Institute, Center for the Study of Opioid Receptors and Drugs of Abuse, CURE: Digestive Diseases Research Center and the Oppenheimer Family Center for Neurobiology of Stress and Resilience.
Funding: The research described here was supported by the Department of Veterans Affairs, Veterans Health Administration, Rehabilitation Research & Development Service (award numbers 1I01RX000378, 1I01RX001646A), and by the National Institute of Health (grant number R01-DA033059).
Abbreviations
- 8-Br-cAMP
8-Bromoadenosine-3′,5′-cyclic monophosphate
- AC
adenylyl cyclase
- ANOVA
analysis of variance
- BDNF
brain-derived neurotrophic factor
- BVT948
4-hydroxy-3,3-dimethyl-2H-benz[g]indole-2,5(3H)-dione
- Cav channel
voltage-gated calcium channel
- CI
confidence interval
- CTX
ω-conotoxin
- CTAP
D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2
- DAMGO
[D-Ala2, NMe-Phe4, Gly-ol5]-enkephalin
- D-Ser
D-serine
- KT5720
(9R,10S,12S)-2,3,9,10,11,12-Hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester
- MK-801
dizocilpine, (5S,10R)-(+)-5-Methyl-10,11-dihydro-5H-dibenzo[a, d]cyclohepten-5,10-imine maleate
- NK1R
neurokinin 1 receptor
- NMDA
N-methyl-D-aspartate
- PKA
protein kinase A
- PKC
protein kinase C
- PKI 14–22
protein kinase inhibitor 14–22
- Ro-32-0432
3-[(8S)-8-[(Dimethylamino)methyl]-6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl]-4-(1-methyl-1H-indol-3-yl)-1H-pyrrole-2,5-dione hydrochloride
- SEM
standard error of the mean
- SFK
Src family kinase
- SQ22536
9-(Tetrahydro-2-furanyl)-9H-purin-6-amine
- TPPB
(2E4E)-N-[(2S,5S)-1,2,3,4,5,6-Hexahydro-5-(hydroxymethyl)-1-methyl-2-(1-methylethyl)-3-oxo-1,4-benzodiazocin-8-yl]-5-[4-(trifluoromethyl)phenyl]-2,4-pentadienamide
- TRPV1
transient receptor potential vanilloid 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbadie C, Trafton J, Liu H, Mantyh PW, Basbaum AI. Inflammation increases the distribution of dorsal horn neurons that internalize the neurokinin-1 receptor in response to noxious and non-noxious stimulation. J Neurosci. 1997;17:8049–8060. doi: 10.1523/JNEUROSCI.17-20-08049.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelson DW, Lao L, Zhang G, Kim W, Marvizón JC. Substance P release and neurokinin 1 receptor activation in the rat spinal cord increases with the firing frequency of C-fibers. Neuroscience. 2009;161:538–553. doi: 10.1016/j.neuroscience.2009.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimone LD, Yaksh TL. Opioid modulation of capsaicin-evoked release of substance P from rat spinal cord in vivo. Peptides. 1989;10:1127–1131. doi: 10.1016/0196-9781(89)90003-x. [DOI] [PubMed] [Google Scholar]
- Allen BJ, Rogers SD, Ghilardi JR, Menning PM, Kuskowski MA, Basbaum AI, Simone DA, Mantyh PW. Noxious cutaneous thermal stimuli induce a graded release of endogenous substance P in the spinal cord: imaging peptide action in vivo. J Neurosci. 1997;17:5921–5927. doi: 10.1523/JNEUROSCI.17-15-05921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci. 2004;24:2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter GT, Radeke MJ, Kuo RC, Makrides V, Hinkle B, Hoang R, Medina-Selby A, Coit D, Valenzuela P, Feinstein SC. Signal transduction mediated by the truncated trkB receptor isoforms, trkB.T1 and trkB.T2. J Neurosci. 1997;17:2683–2690. doi: 10.1523/JNEUROSCI.17-08-02683.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM. Long-term potentiation induced by theta frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci. 2000;20:7880–7887. doi: 10.1523/JNEUROSCI.20-21-07880.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campillo A, Cabanero D, Romero A, Garcia-Nogales P, Puig MM. Delayed postoperative latent pain sensitization revealed by the systemic administration of opioid antagonists in mice. Eur J Pharmacol. 2011;657:89–96. doi: 10.1016/j.ejphar.2011.01.059. [DOI] [PubMed] [Google Scholar]
- Chang P, Chandler KE, Williams RS, Walker MC. Inhibition of long-term potentiation by valproic acid through modulation of cyclic AMP. Epilepsia. 2010;51:1533–1542. doi: 10.1111/j.1528-1167.2009.02412.x. [DOI] [PubMed] [Google Scholar]
- Chen W, Marvizon JC. Acute inflammation induces segmental, bilateral, supraspinally mediated opioid release in the rat spinal cord, as measured by μ-opioid receptor internalization. Neuroscience. 2009;161:157–172. doi: 10.1016/j.neuroscience.2009.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Song B, Lao L, Perez OA, Kim W, Marvizon JCG. Comparing analgesia and μ-opioid receptor internalization produced by intrathecal enkephalin: Requirement for peptidase inhibition. Neuropharmacology. 2007;53:664–667. doi: 10.1016/j.neuropharm.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Walwyn W, Ennes H, Kim H, McRoberts JA, Marvizon JC. BDNF released during neuropathic pain potentiates NMDA receptors in primary afferent terminals. Eur J Neurosci. 2014;39:1439–1454. doi: 10.1111/ejn.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Zhang G, Marvizon JC. NMDA receptors in primary afferents require phosphorylation by Src family kinases to induce substance P release in the rat spinal cord. Neuroscience. 2010;166:924–934. doi: 10.1016/j.neuroscience.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers SR. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- Colquhoun D. Why the Schild method is better than Schild realised. Trends Pharmacol Sci. 2007;28:608–614. doi: 10.1016/j.tips.2007.09.011. [DOI] [PubMed] [Google Scholar]
- Connor M, Traynor J. Constitutively active mu-opioid receptors. Methods Enzymol. 2010;484:445–469. doi: 10.1016/B978-0-12-381298-8.00022-8. [DOI] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science. 2013;341:1394–1399. doi: 10.1126/science.1239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernfors P, Rosario CM, Merlio JP, Grant G, Aldskogius H, Persson H. Expression of mRNAs for neurotrophin receptors in the dorsal root ganglion and spinal cord during development and following peripheral or central axotomy. Brain Res Mol Brain Res. 1993;17:217–226. doi: 10.1016/0169-328x(93)90005-a. [DOI] [PubMed] [Google Scholar]
- Evans DI, Jones RS, Woodhall G. Differential actions of PKA and PKC in the regulation of glutamate release by group III mGluRs in the entorhinal cortex. J Neurophysiol. 2001;85:571–579. doi: 10.1152/jn.2001.85.2.571. [DOI] [PubMed] [Google Scholar]
- Fenner BM. Truncated TrkB: beyond a dominant negative receptor. Cytokine Growth Factor Rev. 2012;23:15–24. doi: 10.1016/j.cytogfr.2012.01.002. [DOI] [PubMed] [Google Scholar]
- Gao XF, Zhang HL, You ZD, Lu CL, He C. G protein-coupled inwardly rectifying potassium channels in dorsal root ganglion neurons. Acta Pharmacol Sin. 2007;28:185–190. doi: 10.1111/j.1745-7254.2007.00478.x. [DOI] [PubMed] [Google Scholar]
- Garzón J, Rodríguez-Muñoz M, Sánchez-Blázquez P. Direct association of Mu-opioid and NMDA glutamate receptors supports their cross-regulation: molecular implications for opioid tolerance. Curr Drug Abuse Rev. 2012;5:199–226. doi: 10.2174/1874473711205030199. [DOI] [PubMed] [Google Scholar]
- Grady EF, Baluk P, Bohm S, Gamp PD, Wong H, Payan DG, Ansel J, Portbury AL, Furness JB, McDonald DM, Bunnett NW. Characterization of antisera specific to NK1, NK2, and NK3 neurokinin receptors and their utilization to localize receptors in the rat gastrointestinal tract. J Neurosci. 1996;16:6975–6986. doi: 10.1523/JNEUROSCI.16-21-06975.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood S, Richards P, Murray F, Harrison N, Wingrove PB, Hutson PH. Characterisation of N-methyl-D-aspartate receptor-specific [3H]Ifenprodil binding to recombinant human NR1a/NR2B receptors compared with native receptors in rodent brain membranes. J Neurochem. 2000;75:2455–2463. doi: 10.1046/j.1471-4159.2000.0752455.x. [DOI] [PubMed] [Google Scholar]
- Hannon HE, Atchison WD. Omega-conotoxins as experimental tools and therapeutics in pain management. Mar Drugs. 2013;11:680–699. doi: 10.3390/md11030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinke B, Gingl E, Sandkuhler J. Multiple targets of μ-opioid receptor-mediated presynaptic inhibition at primary afferent Aδ- and C-Fibers. J Neurosci. 2011;31:1313–1322. doi: 10.1523/JNEUROSCI.4060-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore P, Menning PM, Rogers SD, Nichols ML, Basbaum AI, Besson JM, Mantyh PW. Spinal cord substance P receptor expression and internalization in acute, short-term, and long-term inflammatory pain states. J Neurosci. 1999;19:7670–7678. doi: 10.1523/JNEUROSCI.19-17-07670.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DI, Scott DT, Riddell JS, Todd AJ. Upregulation of substance P in low-threshold myelinated afferents is not required for tactile allodynia in the chronic constriction injury and spinal nerve ligation models. J Neurosci. 2007;27:2035–2044. doi: 10.1523/JNEUROSCI.5401-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TS, Yaksh TL. Spinal monoamine and opiate systems partly mediate the antinociceptive effects produced by glutamate at brainstem sites. Brain Res. 1984;321:287–297. doi: 10.1016/0006-8993(84)90181-1. [DOI] [PubMed] [Google Scholar]
- Jin W, Klem AM, Lewis JH, Lu Z. Mechanisms of inward-rectifier K+ channel inhibition by tertiapin-Q. Biochemistry. 1999;38:14294–14301. doi: 10.1021/bi991206j. [DOI] [PubMed] [Google Scholar]
- Keith DE, Anton B, Murray SR, Zaki PA, Chu PC, Lissin DV, Monteillet-Agius G, Stewart PL, Evans CJ, von Zastrow M. mu-Opioid receptor internalization: opiate drugs have differential effects on a conserved endocytic mechanism in vitro and in the mammalian brain. Mol Pharmacol. 1998;53:377–384. [PubMed] [Google Scholar]
- Keith RA, Mangano TJ, Salama AI. Inhibition of N-methyl-D-aspartate- and kainic acid-induced neurotransmitter release by omega-conotoxin GVIA. Br J Pharmacol. 1989;98:767–772. doi: 10.1111/j.1476-5381.1989.tb14604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Kim Y, Cho H-y, Ahn YJ, Kim J, Yoon YW. Effect of NMDA NR2B antagonist on neuropathic pain in two spinal cord injury models. Pain. 2012;153:1022–1029. doi: 10.1016/j.pain.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Kondo I, Marvizon JC, Song B, Salgado F, Codeluppi S, Hua XY, Yaksh TL. Inhibition by spinal mu- and delta-opioid agonists of afferent-evoked substance P release. J Neurosci. 2005;25:3651–3660. doi: 10.1523/JNEUROSCI.0252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer TH, Shook JE, Kazmierski W, Ayres EA, Wire WS, Hruby VJ, Burks TF. Novel peptidic mu-opioid antagonists: pharmacologic characterization in vitro and in vivo. J Pharmacol Exp Ther. 1989;249:544–551. [PubMed] [Google Scholar]
- Lao L, Marvizon JCG. GABAA receptor facilitation of neurokinin release from primary afferent terminals in the rat spinal cord. Neuroscience. 2005;130:1013–1027. doi: 10.1016/j.neuroscience.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Lao L, Song B, Chen W, Marvizon JC. Noxious mechanical stimulation evokes the segmental release of opioid peptides that induce μ-opioid receptor internalization in the presence of peptidase inhibitors. Brain Res. 2008;1197:85–93. doi: 10.1016/j.brainres.2007.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao L, Song B, Marvizon JCG. Neurokinin release produced by capsaicin acting on the central terminals and axons of primary afferents: relationship with NMDA and GABAB receptors. Neuroscience. 2003;121:667–680. doi: 10.1016/s0306-4522(03)00501-3. [DOI] [PubMed] [Google Scholar]
- Lau CG, Takeuchi K, Rodenas-Ruano A, Takayasu Y, Murphy J, Bennett MV, Zukin RS. Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem Soc Trans. 2009;37:1369–1374. doi: 10.1042/BST0371369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laulin JP, Maurette P, Corcuff JB, Rivat C, Chauvin M, Simonnet G. The role of ketamine in preventing fentanyl-induced hyperalgesia and subsequent acute morphine tolerance. Anesth Analg. 2002;94:1263–1269. doi: 10.1097/00000539-200205000-00040. [DOI] [PubMed] [Google Scholar]
- Lee SL, Kim JK, Kim DS, Cho HJ. Expression of mRNAs encoding full-length and truncated TrkB receptors in rat dorsal root ganglia and spinal cord following peripheral inflammation. Neuroreport. 1999;10:2847–2851. doi: 10.1097/00001756-199909090-00027. [DOI] [PubMed] [Google Scholar]
- Li J, McRoberts JA, Ennes HS, Trevisani M, Nicoletti P, Mittal Y, Mayer EA. Experimental colitis modulates the functional properties of NMDA receptors in dorsal root ganglia neurons. Am J Physiol Gastrointest Liver Physiol. 2006;291:G219–228. doi: 10.1152/ajpgi.00097.2006. [DOI] [PubMed] [Google Scholar]
- Li J, McRoberts JA, Nie J, Ennes HS, Mayer EA. Electrophysiological characterization of N-methyl-d-aspartate receptors in rat dorsal root ganglia neurons. Pain. 2004;109:443–452. doi: 10.1016/j.pain.2004.02.021. [DOI] [PubMed] [Google Scholar]
- Liljebris C, Baranczewski P, Bjorkstrand E, Bystrom S, Lundgren B, Tjernberg A, Warolen M, James SR. Oxidation of protein tyrosine phosphatases as a pharmaceutical mechanism of action: a study using 4-hydroxy-3,3-dimethyl-2H-benzo[g]indole-2,5(3H)-dione. J Pharmacol Exp Ther. 2004;309:711–719. doi: 10.1124/jpet.103.062745. [DOI] [PubMed] [Google Scholar]
- Liu H, Mantyh PW, Basbaum AI. NMDA-receptor regulation of substance P release from primary afferent nociceptors. Nature. 1997;386:721–724. doi: 10.1038/386721a0. [DOI] [PubMed] [Google Scholar]
- Liu H, Wang H, Sheng M, Jan LY, Jan YN, Basbaum AI. Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the spinal cord dorsal horn. Proc Natl Acad Sci U S A. 1994;91:8383–8387. doi: 10.1073/pnas.91.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Weight FF. Glutamate induces a depolarization of adult rat dorsal root ganglion neurons that is mediated predominantly by NMDA receptors. Neurosci Lett. 1988;94:314–320. doi: 10.1016/0304-3940(88)90037-7. [DOI] [PubMed] [Google Scholar]
- Ma QP, Hargreaves RJ. Localization of N-methyl-D-aspartate NR2B subunits on primary sensory neurons that give rise to small-caliber sciatic nerve fibers in rats. Neuroscience. 2000;101:699–707. doi: 10.1016/s0306-4522(00)00419-x. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Fernandes K, Tomlinson DR. NMDA receptor activation modulates evoked release of substance P from rat spinal cord. Br J Pharmacol. 1998;125:1625–1626. doi: 10.1038/sj.bjp.0702260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE. Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science. 1995;268:1629–1632. doi: 10.1126/science.7539937. [DOI] [PubMed] [Google Scholar]
- Marker CL, Lujan R, Loh HH, Wickman K. Spinal G-protein-gated potassium channels contribute in a dose-dependent manner to the analgesic effect of mu- and delta- but not kappa-opioids. J Neurosci. 2005;25:3551–3559. doi: 10.1523/JNEUROSCI.4899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizon JC, Grady EF, Waszak-McGee J, Mayer EA. Internalization of μ-opioid receptors in rat spinal cord slices. Neuroreport. 1999;10:2329–2334. doi: 10.1097/00001756-199908020-00020. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Martinez V, Grady EF, Bunnett NW, Mayer EA. Neurokinin 1 receptor internalization in spinal cord slices induced by dorsal root stimulation is mediated by NMDA receptors. J Neurosci. 1997;17:8129–8136. doi: 10.1523/JNEUROSCI.17-21-08129.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizon JC, McRoberts JA, Ennes HS, Song B, Wang X, Jinton L, Corneliussen B, Mayer EA. Two N-methyl-D-aspartate receptors in rat dorsal root ganglia with different subunit composition and localization. J Comp Neurol. 2002;446:325–341. doi: 10.1002/cne.10202. [DOI] [PubMed] [Google Scholar]
- Marvizon JC, Walwyn W, Minasyan A, Chen W, Taylor BK. Latent sensitization: a model for stress-sensitive chronic pain. Curr Protoc Neurosci. 2015;71:9 50 51–59 50 14. doi: 10.1002/0471142301.ns0950s71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizon JC, Wang X, Matsuka Y, Neubert JK, Spigelman I. Relationship between capsaicin-evoked substance P release and neurokinin 1 receptor internalization in the rat spinal cord. Neuroscience. 2003a;118:535–545. doi: 10.1016/s0306-4522(02)00977-6. [DOI] [PubMed] [Google Scholar]
- Marvizon JCG, Wang X, Lao L, Song B. Effect of peptidases on the ability of exogenous and endogenous neurokinins to produce neurokinin 1 receptor internalization in the rat spinal cord. Br J Pharmacol. 2003b;140:1389–1398. doi: 10.1038/sj.bjp.0705578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP. Inhibition of calcium channels in rat central and peripheral neurons by omega-conotoxin MVIIC. J Neurosci. 1996;16:2612–2623. doi: 10.1523/JNEUROSCI.16-08-02612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRoberts JA, Coutinho SV, Marvizon JC, Grady EF, Tognetto M, Sengupta JN, Ennes HS, Chaban VV, Amadesi S, Creminon C, Lanthorn T, Geppetti P, Bunnett NW, Mayer EA. Role of peripheral N-methyl-D-aspartate (NMDA) receptors in visceral nociception in rats. Gastroenterology. 2001;120:1737–1748. doi: 10.1053/gast.2001.24848. [DOI] [PubMed] [Google Scholar]
- McRoberts JA, Li J, Ennes HS, Mayer EA. Sex-dependent differences in the activity and modulation of N-methyl-d-aspartic acid receptors in rat dorsal root ganglia neurons. Neuroscience. 2007;148:1015–1020. doi: 10.1016/j.neuroscience.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moises HC, Rusin KI, Macdonald RL. Mu- and kappa-opioid receptors selectively reduce the same transient components of high-threshold calcium current in rat dorsal root ganglion sensory neurons. J Neurosci. 1994;14:5903–5916. doi: 10.1523/JNEUROSCI.14-10-05903.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JA, Stein IS, Lau CG, Peixoto RT, Aman TK, Kaneko N, Aromolaran K, Saulnier JL, Popescu GK, Sabatini BL, Hell JW, Zukin RS. Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. J Neurosci. 2014;34:869–879. doi: 10.1523/JNEUROSCI.4538-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian A, Gu G, Gracias NG, Wilkinson K, Hua XY, Vasko MR, Yaksh TL. Spinal NMDA receptors and nociception-evoked release of primary afferent substance P. Neuroscience. 2008;152:119–127. doi: 10.1016/j.neuroscience.2007.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nockemann D, Rouault M, Labuz D, Hublitz P, McKnelly K, Reis FC, Stein C, Heppenstall PA. The K+ channel GIRK2 is both necessary and sufficient for peripheral opioid-mediated analgesia. EMBO Mol Med. 2013;5:1263–1277. doi: 10.1002/emmm.201201980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piros ET, Hales TG, Evans CJ. Functional analysis of cloned opioid receptors in transfected cell lines. Neurochem Res. 1996;21:1277–1285. doi: 10.1007/BF02532368. [DOI] [PubMed] [Google Scholar]
- Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci. 2007;10:285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randic M, Jiang MC, Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci. 1993;13:5228–5241. doi: 10.1523/JNEUROSCI.13-12-05228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richebe P, Rivat C, Laulin JP, Maurette P, Simonnet G. Ketamine improves the management of exaggerated postoperative pain observed in perioperative fentanyl-treated rats. Anesthesiology. 2005;102:421–428. doi: 10.1097/00000542-200502000-00028. [DOI] [PubMed] [Google Scholar]
- Rivat C, Vera-Portocarrero LP, Ibrahim MM, Mata HP, Stagg NJ, De Felice M, Porreca F, Malan TP. Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci. 2009;29:727–737. doi: 10.1111/j.1460-9568.2009.06616.x. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, de la Torre-Madrid E, Sánchez-Blázquez P, Wang JB, Garzón J. NMDAR-nNOS generated zinc recruits PKCgamma to the HINT1-RGS17 complex bound to the C terminus of Mu-opioid receptors. Cell Signal. 2008;20:1855–1864. doi: 10.1016/j.cellsig.2008.06.015. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, Sánchez-Blázquez P, Vicente-Sánchez A, Bailón C, Martín-Aznar B, Garzón J. The histidine triad nucleotide-binding protein 1 supports mu-opioid receptor-glutamate NMDA receptor cross-regulation. Cell Mol Life Sci. 2011;68:2933–2949. doi: 10.1007/s00018-010-0598-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Muñoz M, Sánchez-Blázquez P, Vicente-Sánchez A, Berrocoso E, Garzón J. The mu-opioid receptor and the NMDA receptor associate in PAG neurons: implications in pain control. Neuropsychopharmacology. 2012;37:338–349. doi: 10.1038/npp.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusin KI, Moises HC. μ-Opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. J Neurosci. 1995;15:4315–4327. doi: 10.1523/JNEUROSCI.15-06-04315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusin KI, Moises HC. Mu-opioid and GABAB receptors modulate different types of Ca2+ currents in rat nodose ganglion neurons. Neuroscience. 1998;85:939–956. doi: 10.1016/s0306-4522(97)00674-x. [DOI] [PubMed] [Google Scholar]
- Schneider SP, Eckert WA, III, Light AR. Opioid-activated postsynaptic, inward rectifying potassium currents in whole cell recordings in substantia gelatinosa neurons. J Neurophysiol. 1998;80:2954–2962. doi: 10.1152/jn.1998.80.6.2954. [DOI] [PubMed] [Google Scholar]
- Shi WX, Bunney BS. Roles of intracellular cAMP and protein kinase A in the actions of dopamine and neurotensin on midbrain dopamine neurons. J Neurosci. 1992;12:2433–2438. doi: 10.1523/JNEUROSCI.12-06-02433.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AB, Cunnane TC. Omega-conotoxin GVIA-resistant neurotransmitter release from postganglionic sympathetic nerves in the guinea-pig vas deferens and its modulation by presynaptic receptors. Br J Pharmacol. 1998;123:167–172. doi: 10.1038/sj.bjp.0701577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone LS, Broberger C, Vulchanova L, Wilcox GL, Hokfelt T, Riedl MS, Elde R. Differential distribution of α2A and α2C adrenergic receptor immunoreactivity in the rat spinal cord. J Neurosci. 1998;18:5928–5937. doi: 10.1523/JNEUROSCI.18-15-05928.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storkson RV, Kjorsvik A, Tjolsen A, Hole K. Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods. 1996;65:167–172. doi: 10.1016/0165-0270(95)00164-6. [DOI] [PubMed] [Google Scholar]
- Strock J, Diverse-Pierluissi MA. Ca2+ channels as integrators of G protein-mediated signaling in neurons. Mol Pharmacol. 2004;66:1071–1076. doi: 10.1124/mol.104.002261. [DOI] [PubMed] [Google Scholar]
- Tan PH, Yang LC, Shih HC, Lan KC, Cheng JT. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther. 2005;12:59–66. doi: 10.1038/sj.gt.3302376. [DOI] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marchand S, Mantyh PW, Basbaum AI. Spinal opioid analgesia: how critical is the regulation of substance P signaling? J Neurosci. 1999;19:9642–9653. doi: 10.1523/JNEUROSCI.19-21-09642.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walwyn W, Chen W, Kim H, Minasyan A, Ennes H, McRoberts JA, Marvizon JC. Sustained suppression of hyperalgesia during latent sensitization by μ, δ and κ opioid receptors and α2A adrenergic receptors - role of constitutive activity. J Neurosci. 2016;36:204–221. doi: 10.1523/JNEUROSCI.1751-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DA, Fay FS. Intracellular calibration of the fluorescent calcium indicator Fura-2. Cell Calcium. 1990;11:75–83. doi: 10.1016/0143-4160(90)90061-x. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Woolf CJ, Thompson SW. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44:293–299. doi: 10.1016/0304-3959(91)90100-C. [DOI] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Chen H, Pan HL. Bortezomib induces neuropathic pain through protein kinase C-mediated activation of presynaptic NMDA receptors in the spinal cord. Neuropharmacology. 2017;123:477–487. doi: 10.1016/j.neuropharm.2017.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabaluri N, Medzihradsky F. Down-regulation of mu-opioid receptor by full but not partial agonists is independent of G protein coupling. Mol Pharmacol. 1997;52:896–902. doi: 10.1124/mol.52.5.896. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Jessell TM, Gamse R, Mudge AW, Leeman SE. Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature. 1980;286:155–157. doi: 10.1038/286155a0. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Yan X, Yan E, Gao M, Weng HR. Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J Physiol. 2013;591:2001–2019. doi: 10.1113/jphysiol.2012.250522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Zhou LJ, Hu NW, Xin WJ, Liu XG. Activation of spinal δ1/δ5 receptors induces late-phase LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2005;94:961–967. doi: 10.1152/jn.01324.2004. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Goldstein BD. Delta-Opioid receptor modulation of the release of substance P-like immunoreactivity in the dorsal horn of the rat following mechanical or thermal noxious stimulation. Brain Res. 1996a;736:305–314. doi: 10.1016/0006-8993(96)00718-4. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Goldstein BD. Kappa-opioid receptor modulation of the release of substance P in the dorsal horn. Brain Res. 1996b;706:80–88. doi: 10.1016/0006-8993(95)01182-x. [DOI] [PubMed] [Google Scholar]
- Zachariou V, Goldstein BD. Dynorphin-(1–8) inhibits the release of substance P-like immunoreactivity in the spinal cord of rats following a noxious mechanical stimulus. Eur J Pharmacol. 1997;323:159–165. doi: 10.1016/s0014-2999(97)00038-1. [DOI] [PubMed] [Google Scholar]
- Zeng J, Thomson LM, Aicher SA, Terman GW. Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J Neurosci. 2006;26:12033–12042. doi: 10.1523/JNEUROSCI.2530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Chen W, Marvizon JC. Src family kinases mediate the inhibition of substance P release in the rat spinal cord by μ-opioid receptors and GABAB receptors, but not α2 adrenergic receptors. Eur J Neurosci. 2010;32:963–973. doi: 10.1111/j.1460-9568.2010.07335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]