

Abstract
Objective
Antiandrogen, aromatase inhibitor, and gonadotropin-releasing hormone analog (GnRHa) treatment normalizes growth rate and bone maturation and increases predicted adult height (AH) in boys with familial male-limited precocious puberty (FMPP). To evaluate the effect of long-term antiandrogen, aromatase inhibitor, and GnRHa on AH, boys with FMPP who were treated were followed to AH.
Study Design
Twenty-eight boys with FMPP, referred to the National Institutes of Health, were started on antiandrogen and aromatase inhibitor at 4.9 ± 1.5 years of age; GnRHa was added at 6.9 ± 1.5 years of age. Treatment was discontinued at 12.2 ± 0.5 years of age (bone age, 14.4 ± 1.3). AH was assessed at 16.4 ± 1.3 years of age (bone age, 18.5 ± 0.6).
Results
AH (mean ± standard deviation) for all treated subjects was 173.6 ± 6.8 cm (−0.4 ± 1.0 standard deviation relative to adult US males). For 25 subjects with pretreatment predicted AH, AH significantly exceeded predicted AH at treatment onset (173.8 ± 6.9 vs 164.9 ± 10.7 cm; P < .001), but fell short of predicted AH at treatment discontinuation (177.3 ± 9.0 cm; P < .001). For 11 subjects with maternal or sporadic inheritance, the mean AH was 3.1 cm (0.4 standard deviation score) below sex-adjusted midparental height (175.4 ± 5.8 vs 178.5 ± 3.1 cm [midparental height]; P = .10). For 16 subjects with affected and untreated fathers, AH was significantly greater than fathers’ AH (172.8 ± 7.4 vs 168.8 ± 7.2 cm; P < .05).
Conclusions
Long-term treatment with antiandrogen, aromatase inhibitor, and GnRHa in boys with FMPP results in AH modestly below sex-adjusted midparental height and within the range for adult males in the general population.
Familial male-limited precocious puberty (FMPP, also termed testotoxicosis) results from a luteinizing hormone (LH) receptor gene activating mutation.1–7 The mutation can occur de novo, but is usually inherited as an autosomal dominant. Affected males experience early pubertal development, usually by 3 years of age, with accelerated growth and bone maturation, premature epiphyseal fusion, and short adult stature.
Two therapeutic approaches, involving antiandrogens, aromatase inhibitors, and gonadotropin-releasing hormone analog (GnRHa; after the onset of central puberty), or steroid biosynthesis inhibitors, have resulted in reduced rate of linear growth rate, bone maturation, and virilization in boys with FMPP.8–15 Only limited data, however, are available on the adult height (AH) of patients after long-term treatment. For 5 boys treated for a median of 6.2 years with the steroidogenesis inhibitor ketoconazole, mean ± standard deviation (SD) AH (and height SD score [SDS]) were 173 ± 14 cm (−0.3 ± 1.4 SDS), which was significantly greater than the pretreatment predicted AH of 165 ± 12 cm and similar to the midparental height (MPH) of 175 ± 9 cm.16 By contrast, mean ± SD near-AH SDS for 7 boys with FMPP treated with cyproterone acetate (n = 4) or ketoconazole (n = 3), combined with GnRHa (after central puberty onset) in 4 boys and with medroxyprogesterone acetate or anastrozole in 1 subject each, was considerably less, at −1.5 ± 1.0 SDS.17
Since the mid-1980s, we have investigated whether antiandrogen (spironolactone) and aromatase inhibitor (testolactone or anastrozole), combined with GnRHa (daily deslorelin or depot leuprolide) after central puberty onset, can normalize growth, pubertal development, and AH in boys with FMPP.11 Previous interim reports have described the effects of this treatment regimen on linear growth, bone maturation, and predicted AH.10,11 The current report describes the AH outcome in 28 boys with FMPP who received long-term treatment with this regimen and were then followed until attainment of AH.
Methods
The Institutional Review Board of the National Institute of Child Health and Human Development approved the protocol, “Spironolactone and Testolactone Treatment of Boys with Familial Isosexual Preocious Puberty (National Institutes of Health 85-CH-0016; NCT00001202),” in 1985, and enrollment of subjects occurred between 1985 and 2001. Written informed consent was obtained from parents, and assent was obtained from children when appropriate. The study was in full compliance with the Health Insurance Portability and Accountability of 1996.
Subjects were assessed at the National Institutes of Health Clinical Center every 6 months while on study medications until the initiation of GnRHa, then yearly until AH was reached. At each visit, pubertal staging, routine laboratory measures (including complete blood count, electrolytes, blood urea nitrogen, creatinine, blood glucose, hepatic panel, mineral panel, total cholesterol, and thyroid function studies), reproductive hormone levels (including testosterone, and baseline and GnRH-stimulated LH and follicle-stimulating hormone [FSH] levels) were obtained as described.9–11 Also at each visit, the average of ≥3 stadiometer heights was recorded, and bone age (BA) films were interpreted by several radiologists from the National Institutes of Health (all with expertise in the interpretation of BA films), without knowledge of treatment status, using the method of Greulich and Pyle.18 The predicted AH was determined by the Bayley-Pinneau method.19 AH measurement was obtained when BA was ≥17 years of age (or up to 2 years later when additional follow-up visits were available).
Parental and fraternal AHs were measured at the Clinical Center when possible, and reported AHs were used for family members not available for measurement. Sex-adjusted MPH for subjects with unaffected fathers (n = 11) was calculated as follows:
Corrected, sex-adjusted MPH for subjects with untreated affected fathers (n = 16) was calculated as follows, to correct for the 7.3-cm mean height decrement of affected fathers relative to US adult males20:
Spironolactone was administered daily, every 12 hours, in 2 equally divided oral doses. The dose was increased weekly from 1.5 to 3.0 mg/kg per day and then 5.7 mg/kg per day. Because spironolactone can deplete sodium and retain potassium, liberal intake of salt and moderate intake of high-potassium foods were recommended. In addition, subjects and parents were instructed to withhold spironolactone during diarrhea, vomiting, or other illness involving increased fluid loss or decreased fluid intake. For all 28 subjects, mean ± SD treatment duration was 6.9 ± 1.8 years (range, 2.7–10.3) for spironolactone.
Testolactone was administered daily in 4 equally divided oral doses (every 6 hours) from 1985 to 1994 and then, to enhance convenience, in 3 equally divided oral doses (every 8 hours). Testolactone therapy was increased weekly from 20 to 30 mg/kg per day and then 40 mg/kg per day. When testolactone became unavailable in 2005, the 5 subjects remaining on study drug transitioned to anastrozole, 1 mg daily (oral) at bedtime. For all 28 subjects, mean ± SD treatment duration was 6.9 ± 1.8 years (range, 2.7–10.3) for testolactone and 0.3 ± 0.8 years (range, 0.0–3.0) for anastrozole.
The decision to start GnRHa was based on both clinical evidence of central puberty (an acute increase in the signs and symptoms of puberty) and a GnRH stimulation test that was either frankly pubertal (peak LH > 20 mIU/mL and/or peak LH greater than peak FSH) or approaching a pubertal response, as described.9–11 Initially, all subjects in secondary central puberty were treated with deslorelin 4 g/kg per day subcutaneously each evening. When deslorelin became unavailable in 2003, 4 subjects who were still on the study drug were transitioned to 1-month depot leuprolide, 7.5–15 mg (based on weight and response) every 3 weeks. Also, 3 subjects who had not yet been treated with GnRHa were later treated with depot leuprolide. Once GnRHa was initiated, GnRH stimulation testing was performed every 12 months to ensure suppression of secondary central puberty (suppressed basal and peak LH and FSH levels). For the 27 AH subjects treated with GnRHa, the mean ± SD treatment duration was 5.5 ± 1.6 years (range, 2.1–8.3) for deslorelin and 2.8 ± 0.9 years (range, 1.5–4.3) for leuprolide.
All weight-based medication dosages were adjusted at 6-month intervals.
Statistical Analyses
Among the 39 total study subjects, 8 patients who dropped out of the study before attaining AH were excluded from this completer analysis. Study discontinuation resulted from parent decision owing to divorce (n = 2), desire to receive care closer to home (n = 1), and inconvenience of study visits (n = 5). No subjects discontinued owing to adverse effects or perceived lack of efficacy. Three additional subjects were excluded because they had not yet attained AH, leaving 28 subjects with measured AHs as the subject of this report.
All data were expressed as mean ± SD. AH measurements were compared with predicted AH at start of treatment, predicted AH at end of treatment, sex-adjusted MPH, paternal height, and fraternal height using the 2-tailed paired Student t test. AH comparison between paternally versus maternally inherited (or sporadic) cases was made with the 2-tailed Student t test.
Results
All 28 subjects had a documented heterozygous activating mutation of the LH receptor (23 Asp578Gly, 2 Asp564Gly, 1 Asp572Val, 1 Ile575Leu, 1 Met398Thr; Tables I and II). Sixteen inherited the mutation from their affected, untreated father, nine from their mother, two had de novo mutations, and one had a mutation of unknown source (mother did not have the mutation; father unavailable for analysis). There was no known consanguinity between study subjects, although approximately one-half of the subjects with the most common mutation (Asp578Gly) reported having ancestors from Dothan, Alabama, suggesting that some of may have been distant relatives. Mean chronological age and BA at start of therapy were 4.9 ± 1.5 and 9.7 ± 3.5 years of age, respectively (mean BA advancement 4.8 ± 2.6 years). GnRHa was started at a mean chronological age of 6.9 ± 1.5 years (mean BA, 12.1 ± 1.6).
Table I.
Patient characteristics at baseline, treatment initiation, treatment discontinuation, and AH
| Characteristics | All Subjects (N = 28) | Paternal Inheritance (n = 16) | Maternal (n = 9) or Sporadic (N = 2) Inheritance | Unknown Inheritance (n = 1) |
|---|---|---|---|---|
| Initiation of antiandrogen/aromatase inhibitor (baseline) | ||||
| CA (y) | 4.9 ± 1.5 | 4.9 ± 1.5 | 4.8 ± 1.7 | 6.2 |
| BA (y) | 9.7 ± 3.5 | 9.7 ± 3.4 | 9.3 ± 3.8 | 12.5 |
| BA minus CA (y) | 4.8 ± 2.6 | 4.8 ± 2.6 | 4.6 ± 2.7 | 6.3 |
| Predicted AH (cm) | 164.9 ± 10.7§ | 163.7 ± 10.7§ | 166.5 ± 11.7§ | 165.8 |
| Predicted AH (SDS) | −1.7 ± 1.5§ | −1.8 ± 1.5§ | −1.4 ± 1.6§ | −1.5 |
| MPH (cm)* | 177.0 ± 5.3¶ | 175.9 ± 6.3 | 178.5 ± 3.1 | N/A |
| MPH (SDS)* | 0.0 ± 0.7¶ | −0.1 ± 0.9 | 0.2 ± 0.4 | N/A |
| Initiation of GnRHa** | ||||
| CA (y) | 6.9 ± 1.5 | 7.0 ± 1.5 | 6.9 ± 1.6 | 6.6 |
| BA (y) | 12.1 ± 1.6 | 12.2 ± 1.6 | 11.9 ± 1.7 | 13.5 |
| BA minus CA (y) | 5.2 ± 1.9 | 5.3 ± 1.6 | 5.0 ± 2.2 | 6.9 |
| Discontinuation of treatment | ||||
| CA (y) | 12.2 ± 0.5 | 12.2 ± 0.6 | 12.2 ± 0.6 | 12.1 |
| BA (y) | 14.4 ± 1.3 | 14.7 ± 1.5 | 14.0 ± 0.9 | 14.5 |
| BA minus CA (y) | 2.2 ± 1.5 | 2.4 ± 1.7 | 1.8 ± 1.2 | 2.5 |
| Antiandrogen/aromatase inhibitor treatment duration (y) | 7.3 ± 1.8 | 7.3 ± 1.7 | 7.5 ± 2.1 | 5.9 |
| GnRHa treatment duration (y) | 5.3 ± 1.6 | 5.3 ± 1.4 | 5.3 ± 2.0 | 5.5 |
| Predicted AH (cm) | 177.2 ± 9.0 | 176.8 ± 9.9 | 178.5 ± 7.9 | 170.1 |
| Predicted AH (SDS) | 0.1 ± 1.3 | 0.0 ± 1.4 | 0.2 ± 1.1 | −1.0 |
| AH (study end) | ||||
| CA (y) | 16.4 ± 1.3 | 16.6 ± 1.5 | 16.1 ± 0.9 | 15.1 |
| BA (y) | 18.5 ± 0.6 | 18.5 ± 0.4 | 18.5 ± 0.7 | 19.0 |
| AH (cm) | 173.6 ± 6.8 | 172.8 ± 7.4 | 175.4 ± 5.8 | 166.8 |
| AH (SDS) | −0.4 ± 1.0 | −0.5 ± 1.0 | −0.1 ± 0.8 | −1.4 |
CA, Chronological age.
MPH calculation depends on mode of inheritance:
Subjects with maternal or sporadic inheritance: MPH = (father’s height [cm] + mother’s height [cm] + 13 cm)/2
Subjects with paternal inheritance: MPH = (father’s height [cm] + [176.1 cm† − 168.8 cm‡] + mother’s height [cm] + 13 cm)/2
Mean adult male height according to the 2000 National Health Examination Survey.21
Mean paternal height (affected fathers).
Three subjects started treatment at BA too low to determine predicted height (inheritance—2 paternal, 1 maternal).
One subject with unknown inheritance could not be included in these calculations.
One subject (with paternal inheritance) declined treatment with GnRHa.
Table II.
Patient characteristics during interval between previously reported analysis (Leschek et al11) and AH measurement of patients in current analysis
| CAs* (y) | Patient No. | Height Velocity (cm/y) | BA (y) | Testicular Volume (mL) | Peak LH† (IU/L) | Peak FSH† (IU/L) | Testosterone (ng/dL) |
|---|---|---|---|---|---|---|---|
| 6 | 6 | 6.4 ± 2.0 | 11.0 ± 2.0 | 6.5 ± 1.4 | 4.8 ± 3.3 | 2.8 ± 2.5 | 209 ± 100 |
| 7 | 9 | 6.3 ± 1.8 | 11.4 ± 1.3 | 7.1 ± 1.2 | 6.6 ± 4.3 | 3.5 ± 2.7 | 195 ± 138 |
| 8 | 8 | 6.6 ± 1.4 | 11.7 ± 0.9 | 7.9 ± 0.9 | 7.2 ± 10.2 | 1.8 ± 1.7 | 204 ± 113 |
| 9 | 9 | 5.4 ± 1.3 | 12.0 ± 1.1 | 8.3 ± 1.4 | 1.7 ± 2.1 | 1.4 ± 1.8 | 210 ± 141 |
| 10 | 10 | 5.6 ± 1.8 | 12.7 ± 0.9 | 8.5 ± 1.6 | 2.1 ± 1.0 | 1.9 ± 1.3 | 214 ± 170 |
| 11 | 12 | 4.6 ± 1.3 | 13.2 ± 0.6 | 8.5 ± 2.3 | 2.7 ± 2.0 | 2.1 ± 1.9 | 235 ± 167 |
| 12 | 12 | 4.7 ± 2.3 | 13.8 ± 0.6 | 10.7 ± 5.7 | 3.0 ± 3.3 | 1.8 ± 1.8 | 232 ± 221 |
| 13 | 14 | 6.3 ± 6.0 | 15.3 ± 1.2 | 14.9 ± 5.2 | No data | No data | 414 ± 162 |
| 14 | 17 | 3.7 ± 2.8 | 16.6 ± 0.9 | 16.7 ± 4.6 | 18.5 ± 7.0 | 11.7 ± 7.3 | 443 ± 186 |
| 15 | 16 | 2.1 ± 1.7 | 17.3 ± 0.9 | 17.0 ± 4.3 | 25.3 ± 17.5 | 11.2 ± 7.4 | 507 ± 217 |
| 16 | 13 | 0.8 ± 0.6 | 18.2 ± 0.6 | 17.2 ± 2.9 | 24.9 ± 22.0 | 12.3 ± 11.1 | 499 ± 234 |
| 17 | 5 | 0.6 ± 0.7 | 18.8 ± 0.3 | 16.5 ± 5.2 | No data | No data | 432 ± 238 |
CA, Chronologic age.
Data are mean ± SD of all study measurements during interval between previously reported and current analysis.
Chronological age (y) of patient at previously reported analysis (Leschek et al11).
GnRH stimulation testing was not performed when GnRHa was unavailable owing to discontinuation by the manufacturer.
AH was significantly greater than pretreatment predicted AH (173.8 ± 6.9 cm [−0.3 ± 0.9 SDS] vs 164.9 ± 10.7 cm [−1.5 ± 1.5 SDS]; n = 25; P < .001; Figure). Three subjects could not be included in this analysis because their pretreatment BAs were too immature for a Bayley-Pinneau height prediction. Analysis of subjects by parental inheritance showed similar, statistically significant increases in AH compared with pretreatment predicted AH for individuals with affected fathers (172.4 ± 7.8 cm vs 163.7 ± 10.7 cm; n = 14; P < .001; Figure) and for those with maternal or sporadic inheritance (176.5 ± 4.8 cm vs 166.5 ± 11.7 cm; n = 10; P < .05; Figure). One subject with unknown inheritance could not be analyzed according to parental origin.
Figure.
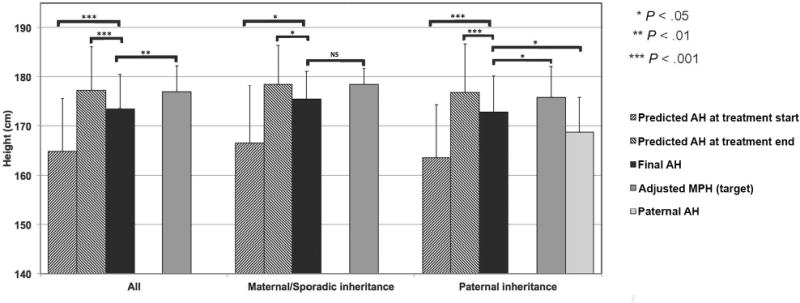
Predicted AH at treatment start and end, final AH, and adjusted midparental (target) height in all treated FMPP patients (left-hand bars, n = 28). Center and right-hand bars indicate results for patients with maternal or sporadic inheritance (center bars, n = 11) and those with paternal inheritance (right-hand bars, n = 16). Data for boys with paternal inheritance include a bar (far right) indicating paternal AH for their affected untreated fathers. One patient had unknown inheritance. Values are expressed as mean ± SD.
AH decreased significantly below the estimated predicted AH at treatment discontinuation (173.6 ± 6.8 cm [−0.4 ± 0.9 SDS] vs 177.2 ± 9.0 cm [0.1 ± 1.2 SDS]; n = 28; P < .001; Figure). Analysis of subjects by parental inheritance demonstrated similar, statistically significant shortfalls in AH versus end-of-treatment predicted AH for individuals with affected fathers (172.8 ± 7.4 cm vs 176.8 ± 9.9 cm; n = 16; P < .001; Figure) and for those with maternal or sporadic inheritance (175.4 ± 5.8 cm vs 178.5 ± 7.9 cm; n = 11; P < .05; Figure).
In 11 subjects with either maternal inheritance or sporadic FMPP, whose fathers were unaffected by FMPP and for whom sex-adjusted MPH provided a meaningful estimate of genetic height potential, AH did not differ significantly from MPH (175.4 ± 5.8 cm [−0.1 ± 0.8 SDS] vs 178.5 ± 3.1 cm [0.3 ± 0.4 SDS], respectively; P = .10; Figure). Of note, AH for unaffected fathers was 181.0 ± 0.3 cm (range, 175.3–185.4), which is 4.9 cm greater than the mean normal US adult male height at age 19 years.20
In the 16 boys with affected fathers, a similar comparison between AH and paternally corrected MPH (calculated after correcting paternal heights for the mean 7.3-cm deficit in affected father height versus mean National Health Examination Survey male height data21) did reach statistical significance (172.8 ± 7.4 cm [AH] vs 175.9 ± 6.3 cm; P < .05; Figure), although the mean decrease in AH versus MPH (sex-adjusted and/or paternally corrected) was identical (3.1 cm [0.4 SDS]).
Analysis of all 27 subjects with known inheritance (paternal, maternal, or sporadic) similarly demonstrated a significant shortfall between AH and sex-adjusted, paternally corrected MPH (173.8 ± 6.8 cm [AH] vs 177.0 ± 5.3 cm; n = 27; P < .01; Figure).
AH was greater than paternal height for the 16 subjects with FMPP-affected, untreated fathers (172.8 ± 7.4 cm vs 168.8 ± 7.2 cm; P < .05; Figure). Similarly, the mean AH was greater than fraternal height for the 2 subjects with affected, untreated brothers (168.6 cm vs 157.5 cm [−1.1 vs −2.6 SDS] and 185.9 cm vs 170.2 cm [1.3 vs −0.9 SDS]).
The AH in subjects with paternal inheritance did not differ significantly from AH in those with maternal or sporadic inheritance (172.8 ± 7.4 cm [−0.5 ± 1.0 SDS] vs 175.4 ± 5.8 cm [−0.1 ± 0.8 SDS]; P < .4).
The first-generation aromatase inhibitor testolactone was changed, late in the study, to anastrozole, and daily deslorelin was changed to depot leuprolide. There was no acute effect of these changes on growth rate or bone maturation (data not shown). The transition from deslorelin to depot leuprolide did not affect the baseline and GnRH-stimulated LH and FSH levels. The shift from testolactone to anastrozole did not alter serum testosterone levels (data not shown). Serum estradiol levels were not assessed because testolactone (and/or its metabolites) cross-reacted in the available estradiol assay.
Subjects were highly compliant with medication and evaluation regimens as assessed by visit history, return of unused study medication, parental and caregiver report, and pubertal suppression as measured by height velocity, rate of bone maturation, and baseline and stimulated LH and FSH levels during GnRH-stimulation testing (once GnRHa was initiated for secondary central puberty). Subjects and families were highly motivated to comply with treatment and follow-up regimens because the androgen blockade achieved while on the study drug resulted in significant and immediate improvement in the signs and symptoms of precocious puberty.
No deaths or treatment-related severe adverse events occurred during the study. No hepatic, renal, hematologic, or lipid abnormalities were attributed to study medications. Transient, mild abdominal pain, nausea, and/or vomiting occurred occasionally after starting testolactone treatment, and resolved quickly in all cases without dose reduction. One episode of hyponatremia (sodium 125 mEq/L [125 mmol/L]) was observed during poor fluid intake, fever, and pneumonia in a subject in whom spironolactone had been continued despite the protocol provision and instructions to discontinue spironolactone during acute illness involving fluid loss or reduced intake. The hyponatremia resolved spontaneously after temporarily discontinuing spironolactone. The potassium level remained normal.
Discussion
The current study of 28 boys with FMPP, treated from early childhood with a combination of antiandrogen, aromatase inhibitor, and GnRHa (after premature secondary gonadotropin activation), represents the largest study to date of AH after long-term antiandrogen and aromatase inhibitor treatment in FMPP. Mean achieved AH was only modestly (−0.4 SD units) below that of the general US male population, or the child’s own MPH (−0.4 SD units), and for boys with affected, untreated fathers, it was significantly greater than the mean AH of their fathers, which was −1.0 SDS (16th percentile) relative to US adult males. Bayley-Pinneau predicted AH increased throughout treatment, but height prediction after mean treatment duration of 7.3 years significantly overestimated actual AH (mean overprediction, 0.5 SD units). This overestimation of AH at treatment end, which has been observed in other precocious puberty studies,20,22 may be attributable to the underlying disease process or prediction method inaccuracies, and has implications for physician, patient, and parental expectation. Despite the regimen’s complexity, treatment was well-tolerated with only minor adverse events.
Several factors may explain the 0.4 SD-unit AH decrement compared with mean US male AH and sex-adjusted MPH. First, mean BA was advanced 4.8 years at treatment onset, and whether delayed treatment can normalize AH completely at this point in development is unclear. Second, because estrogen levels of normal boys are undetectable in all but the most sensitive research assays, it was not possible to confirm whether testolactone or anastrozole reduced estrogen levels continuously to the normal childhood range. Third, compliance with study medication administration was not assessed quantitatively, and nonadherence to study medication must be considered a potential factor in any long-term study involving children.
Compared with previous reports, the current AH data are consistent with those of Sorianoo-Guillén et al,16 who observed a mean AH SDS of −0.3 in 5 boys with FMPP after long-term treatment with ketoconazole. By contrast, the current AH values seem to be greater than the mean near-final height SDS of −1.5 observed by Almeida et al17 in 7 boys treated with ketoconazole or cyproterone acetate. Because of multiple study design differences, the reasons for this apparent difference are unclear.
During the 3 decades over which the current study was conducted, aromatase inhibitors improved in potency and convenience, depot versus daily GnRHa became established in pediatric patients, and study drugs were changed when 2 of the 3 original study drugs became unavailable. The study design does not allow determination of possible differences in effectiveness between initial and subsequent choices of aromatase inhibitor or GnRHa. In addition, the study did not permit comparison of spironolactone with bicalutamide, a more potent androgen receptor blocker. Thus, the results should be viewed as a proof of concept for combined antiandrogen and aromatase inhibitor treatment to normalize growth rate, bone maturation, and pubertal progression and, when combined with GnRHa after central puberty onset, to achieve a near-normal AH outcome in boys with FMPP. Further long-term studies, such as those that are ongoing,12–15 will be required to determine whether newer, more convenient antiandrogen and aromatase inhibitor combinations can achieve similar or better results, and to provide more evidence on the benefits and risks of antiandrogen and aromatase inhibitor versus ketoconazole-induced blockade of steroidogenesis. We recommend that physicians support such ongoing studies so that information in this rare disorder can be accrued more rapidly.
Initially, only an antiandrogen and an aromatase inhibitor were administered to boys with FMPP, with the goal of blocking androgen effects on masculinization and behavior, and estrogen effects on growth rate and bone maturation.9 We added GnRHa to our treatment regimen because of unequivocal height velocity and/or BA acceleration coincident with pubertal gonadotropin secretion and testicular enlargement.10 However, the need for GnRH treatment may be specific to the early generation aromatase inhibitor and antiandrogen that were used during the majority of this study. With more potent aromatase inhibitors and antiandrogens now available, it may be possible to block the effects of central puberty with the combined aromatase inhibitor and antiandrogen regimen alone, in which case the cost and burden of GnRHa treatment may be avoidable. Because all but one subject received all 3 components of this regimen, the relative contributions of the different components, and in particular of GnRHa after central puberty had begun, are unknown.
Bone mineral density was not included as an outcome measure when this study was designed because methods of bone density measurement were still evolving, normative data for children were not available, and estrogen’s role in male bone density was less well-appreciated before the discovery of men with osteoporosis owing to estrogen receptor or aromatase deficiency.23,24 Although this lack of bone density data represents a knowledge gap for the current treatment, the age at onset and duration of estrogen suppression in our subjects was similar to that of boys with central precocious puberty treated with GnRHa—a setting in which bone density after treatment has been shown to be normal.25
The discovery that estrogen mediates epiphyseal fusion has prompted interest in aromatase inhibitor treatment as a potential means to increase AH in other pediatric conditions associated with short stature, such as constitutional delay of growth and adolescence,26 growth hormone deficiency,27 and idiopathic short stature.28–32 Although the improved AH outcome in the current study of boys with FMPP is consistent with this potential, we agree with the recommendation to complete ongoing long-term clinical trials before expanding pediatric aromatase inhibitor treatment to a much wider patient group.33,34
In conclusion, the combined regimen of an antiandrogen, an aromatase inhibitor, and GnRHa substantially achieved the study goals of normalizing growth, development, and AH in boys with FMPP. The regimen, although complex, was well-tolerated with only minor adverse events. Until more specific therapy is developed for this rare disorder, we recommend antiandrogen and aromatase inhibitor treatment, with added GnRHa where clinically appropriate, as a safe and effective approach based on results observed over the past 30 years.
Data Analysis
Data were analyzed by Ellen W. Leschek. ■
Acknowledgments
We gratefully acknowledge (1) the study subjects and their families for their ongoing participation in this research study, (2) the National Institutes of Health Clinical Center nurses and staff for their assistance in conducting this research and for the devoted care they provided to the subjects and their families, and (3) The Eunice Kennedy Shriver National Institute of Child Health and Human Development and National Institute of Diabetes and Digestive and Kidney Diseases pediatric and adult endocrinology fellows for their assistance in caring for these subjects during their National Institutes of Health Clinical Center admissions and clinic visits. We especially acknowledge the late Louisa Laue, MD (The Eunice Kennedy Shriver National Institute of Child Health and Human Development), who led this project during its early years.
Funded by The Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health.
Abbreviations
- AH
Adult height
- BA
Bone age
- FMPP
Familial male-limited precocious puberty
- FSH
Follicle-stimulating hormone
- GnRHa
Gonadotropin-releasing hormone analog
- LH
Luteinizing hormone
- MPF
Midparental height
- SD
Standard deviation
- SDS
SD score
Footnotes
The authors declare no conflicts of interest.
References
- 1.Schedewie HK, Reiter EO, Beitins IZ, Seyed S, Wooten VD, Jimenez JF, et al. Testicular Leydig cell hyperplasia as a cause of familial sexual precocity. J Clin Endocrinol Metab. 1981;52:271–8. doi: 10.1210/jcem-52-2-271. [DOI] [PubMed] [Google Scholar]
- 2.Rosenthal SM, Grumbach MM, Kaplan SL. Gonadotropin-independent familial sexual precocity with premature Leydig and germinal cell maturation (familial testotoxicosis): effects of a potent luteinizing hormone-releasing factor agonist and medroxyprogesterone acetate therapy in four cases. J Clin Endocrinol Metab. 1983;57:571–9. doi: 10.1210/jcem-57-3-571. [DOI] [PubMed] [Google Scholar]
- 3.Reiter EO, Brown RS, Longcope C, Beitins IZ. Male-limited familial precocious puberty in three generations apparent Leydig-cell autonomy and elevated glycoprotein hormone alpha subunit. N Engl J Med. 1984;311:515–9. doi: 10.1056/NEJM198408233110807. [DOI] [PubMed] [Google Scholar]
- 4.Egli CA, Rosenthal SM, Grumbach MM, Motalvo JM, Gondos B. Pituitary gonadotropin-independent male-limited autosomal dominant sexual precocity in 4 generations: “familial testotoxicosis”. J Pediatr. 1985;106:33–40. doi: 10.1016/s0022-3476(85)80460-1. [DOI] [PubMed] [Google Scholar]
- 5.Beas F, Zurbrugg RP, Leibow SG, Patton RG, Gardner LI. Familial male sexual precocity: report of the eleventh kindred found with observations on blood group linkage and urinary C19-steroid excretion. J Clin Endocrinol Metab. 1962;22:1095–102. doi: 10.1210/jcem-22-11-1095. [DOI] [PubMed] [Google Scholar]
- 6.Shenker A, Laue L, Kosugi S, Merendino JJ, Jr, Minegishi T, Cutler GB., Jr A constitutively activating mutation of the luteinizing hormone receptor in familial male precocious puberty. Nature. 1993;365:652–4. doi: 10.1038/365652a0. [DOI] [PubMed] [Google Scholar]
- 7.Wierman ME, Beardsworth DE, Mansfield MJ, Badger TM, Crawford JD, Crigler JF, Jr, et al. Puberty without gonadotropins: a unique mechanism of sexual development. N Engl J Med. 1985;312:65–72. doi: 10.1056/NEJM198501103120201. [DOI] [PubMed] [Google Scholar]
- 8.Holland FJ, Fishman L, Bailey JD, Fazekas AT. Ketoconazole in the management of precocious puberty not responsive to LHRH-analogue therapy. N Engl J Med. 1985;312:1023–8. doi: 10.1056/NEJM198504183121604. [DOI] [PubMed] [Google Scholar]
- 9.Laue L, Kenigsberg D, Pescovitz OH, Hench KD, Barnes KM, Loriaux DL, et al. Treatment of familial male precocious puberty with spironolactone and testolactone. N Engl J Med. 1989;320:496–502. doi: 10.1056/NEJM198902233200805. [DOI] [PubMed] [Google Scholar]
- 10.Laue L, Jones J, Barnes KM, Cutler GB., Jr Treatment of familial male precocious puberty with spironolactone, testolactone, and deslorelin. J Clin Endocrinol Metab. 1993;76:151–5. doi: 10.1210/jcem.76.1.8421081. [DOI] [PubMed] [Google Scholar]
- 11.Leschek EW, Jones J, Barnes KM, Hill SC, Cutler GB., Jr Six-year results of spironolactone and testolactone treatment of familial male-limited precocious puberty with addition of deslorelin after central puberty onset. J Clin Endocrinol Metab. 1999;8:175–8. doi: 10.1210/jcem.84.1.5413. [DOI] [PubMed] [Google Scholar]
- 12.Kreher NC, Pescovitz OH, Delameter P, Tiulpakov A, Hochberg Z. Treatment of familial male-limited precocious puberty with bicalutamide and anastrazole. J Pediatr. 2006;149:416–20. doi: 10.1016/j.jpeds.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 13.Eysette-Guerreau S, Pinto G, Sultan A, Le Merrer M, Sultan C, Polak M. Effectiveness of anastrozole and cyproterone acetate in two brothers with familial male precocious puberty. J Pediatr Endocrinol Metab. 2008;21:995–1002. [Google Scholar]
- 14.Reiter EO, Mauras N, McCormick K, Kulshreshtha B, Amrhein J, De Luca F, et al. Bicalutamide plus anastrozole for the treatment of gonadotropin-independent precocious puberty in boys with testostoxicosis: a phase II, open-label pilot study (BATT) J Pediatr Endocrinol Metab. 2010;23:999–1009. doi: 10.1515/jpem.2010.161. [DOI] [PubMed] [Google Scholar]
- 15.Lenz AM, Shulman D, Eugster EA, Rahhal S, Fuqua JS, Pescovitz OH, et al. Bicalutamide and third-generation aromatase inhibitors in testotoxicosis. Pediatrics. 2010;126:e728–33. doi: 10.1542/peds.2010-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sorianoo-Guillén L, Lahlou N, Chauvet G, Roger M, Chaussain JL, Carel JC. Adult height after ketoconazole treatment in patients with familial male-limited precocious puberty. J Clin Endocrinol Metab. 2005;90:147–51. doi: 10.1210/jc.2004-1438. [DOI] [PubMed] [Google Scholar]
- 17.Almeida MQ, Brito VN, Lins TS, Guerra-Junior G, de Castro M, Antonini SR, et al. Long-term treatment of familial male-limited precocious puberty (testotoxicosis) with cyproterone acetate or ketoconazole. Clin Endocrinol (Oxf) 2008;69:93–8. doi: 10.1111/j.1365-2265.2007.03160.x. [DOI] [PubMed] [Google Scholar]
- 18.Greulich WW, Pyle SI. Radiographic skeletal development of the hand and wrist. Stanford: Stanford University Press; 1985. [Google Scholar]
- 19.Zachmann M, Sabradillo B, Frank M, Frish H, Prader A. Bayley-Pinneau Roche-Weiner-Thissen and Tanner height predictions in normal children and in patients with various pathologic conditions. J Pediatr. 1978;93:749–55. doi: 10.1016/s0022-3476(78)81071-3. [DOI] [PubMed] [Google Scholar]
- 20.Carel J-C, Roger M, Ispas S, Tondu F, Lahlou N, Blumberg J, et al. Final height after long-term treatment with Triptorelin slow release for central precocious puberty: importance of statural growth after interruption of treatment. J Clin Endocrinol Metab. 1999;84:1973–8. doi: 10.1210/jcem.84.6.5647. [DOI] [PubMed] [Google Scholar]
- 21.Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, Flegal KM, Mei Z, et al. 2000 CDC growth charts for the United States: methods and development. National Center for Health Statistics. Vital Health Stat 11. 2002;246:1–190. [PubMed] [Google Scholar]
- 22.Klein KO, Barnes KM, Jones JV, Feuillan PP, Cutler GB., Jr Increased final height in precocious puberty after long-term treatment with LHRH agonists: the National Institutes of Health Experience. J Clin Endocrinol Metab. 2001;86:4711–6. doi: 10.1210/jcem.86.10.7915. [DOI] [PubMed] [Google Scholar]
- 23.Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in man. N Engl J Med. 1994;331:1056. doi: 10.1056/NEJM199410203311604. [DOI] [PubMed] [Google Scholar]
- 24.Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80:3689–98. doi: 10.1210/jcem.80.12.8530621. [DOI] [PubMed] [Google Scholar]
- 25.Bertelloni S, Baroncelli GI, Ferdeghini M, Menchini-Fabris F, Saggese G. Final height, gonadal function and bone mineral density of adolescent males with central precocious puberty after therapy with gonadotropin-releasing hormone analogues. Eur J Pediatr. 2000;159:369–74. doi: 10.1007/s004310051289. [DOI] [PubMed] [Google Scholar]
- 26.Wickman S, Sipilä I, Ankarberg-Lindgren C, Norjavaara E, Dunkel L. A specific aromatase inhibitor and potential increase in adult height in boys with delayed puberty: a randomized controller trial. Lancet. 2001;357:1743–8. doi: 10.1016/S0140-6736(00)04895-9. [DOI] [PubMed] [Google Scholar]
- 27.Zhou P, Shah B, Prasad K, David R. Letrozole significantly improves growth potential in a pubertal boy with growth hormone deficiency. Pediatrics. 2005;115:e245–8. doi: 10.1542/peds.2004-1536. [DOI] [PubMed] [Google Scholar]
- 28.Shams K, Cameo T, Fennoy I, Hassoun AA, Lerner SE, Aranoff GS. Outcome analysis of aromatase inhibitor therapy to increase adult height in males with predicted adult short stature and/or rapid pubertal progress: a retrospective chart review. J Pediatr Endocrinol Metab. 2014;27:725–30. doi: 10.1515/jpem-2013-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neely EK, Kumar RB, Payne SL, Ranadive SA, Suchet DI. Letrozole vs anastrozole for height augmentation in short pubertal males: first year data. J Clin Endocrinol Metab. 2014;99:4086–93. doi: 10.1210/jc.2014-2432. [DOI] [PubMed] [Google Scholar]
- 30.Rothenbuhler A, Linglart A, Bougnères P. A randomized pilot trial of growth hormone with anastrozole versus growth hormone alone, starting at the very end of puberty with idiopathic short stature. Int J Pediatr Endocrinol. 2015;2015:4. doi: 10.1186/1687-9856-2015-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mauras N, Ross JL, Gagliardi P, Yu YM, Hossain J, Permuy J, et al. Randomized trial of aromatase inhibitors, growth hormone, or combination in pubertal boys with idiopathic short stature. J Clin Endocrinol. 2016;101:4984–93. doi: 10.1210/jc.2016-2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wit JM, Hero M, Nunez SB. Aromatase inhibitors in pediatrics. Nature Rev Endocrinol. 2012;8:135–47. doi: 10.1038/nrendo.2011.161. [DOI] [PubMed] [Google Scholar]
- 33.Shulman DI, Francis GL, Palmert MR, Eugster EA. Use of aromatase inhibitors in children and adolescents with disorders of growth and adolescent development. Pediatrics. 2008;121:e975–83. doi: 10.1542/peds.2007-2081. [DOI] [PubMed] [Google Scholar]
- 34.Geffner ME. Aromatase inhibitors to augment height: continued caution and study required. J Clin Res Pediatr Endocrinol. 2009;1:256–61. doi: 10.4274/jcrpe.v1i6.256. [DOI] [PMC free article] [PubMed] [Google Scholar]