

Abstract
Introduction
There is a strong female preponderance reported in many connective tissue diseases and in almost all systemic sclerosis (SSc) case series.
Methods
We compared gender differences in SSc patients in a large single-center cohort, including demographic features, disease subtype, environmental exposures, disease-specific serum autoantibodies, organ system involvement (frequency and severity) and survival. Adjustment for cutaneous subset (diffuse cutaneous [dc] and limited cutaneous [lc]) was performed.
Results
We identified key characteristics which distinguished female from male SSc patients. Females were more frequently younger at disease onset with a longer disease duration at the time of their first visit. Females more often had lcSSc and, if an overlap syndrome was present, it was most often systemic lupus erythematosus. In contrast, males more frequently had dcSSc and overlap with myositis. Females more frequently had peripheral vascular involvement but in males it was more often severe. Males were more often cigarette smokers and more frequently had environmental exposures. Males more frequently had interstitial lung disease (ILD or pulmonary fibrosis) which was more severe. Females had a significantly increased frequency of anti-centromere antibody and males anti-topoisomerase I and anti-U3RNP antibody. Males had significantly reduced survival (73% at 5 years and 45% at 10 years after onset of SSc). The most frequent causes of death were ILD in males and pulmonary hypertension in females.
Conclusions
Gender differences may be important clues to understanding the natural history and pathogenesis of SSc.
Keywords: Gender, Scleroderma, Systemic sclerosis
Introduction
Systemic sclerosis (SSc) is a multisystem autoimmune disease characterized by three key pathophysiologic features: vasculopathy with endothelial dysfunction; immune system activation and dysregulation; and collagen overproduction with fibrosis. Autoimmune diseases often have a female predominance and this is the case in SSc, which has an overall female to male ratio of 3:1 or greater. The incidence is highest among females of child bearing age (1). Thus, factors associated with gender are important in the occurrence of SSc.
Gender differences are also implicated in the severity of SSc. Several reports have shown that male SSc patients have more severe disease (2–4). Furthermore, the leading causes of death in SSc today, interstitial lung disease and pulmonary arterial hypertension (3, 4), have been reported to be more common and more severe in male SSc patients.
The objective of this study is to examine gender differences in a large single-center SSc cohort which has detailed clinical and laboratory data, serologic information, and longitudinal follow-up including the presence and severity of organ system involvement and survival status.
Methods
The University of Pittsburgh Scleroderma Database was used to identify consecutive new SSc patients first evaluated during 1985–2011. Adult (age >16) patients only were included. All had the diagnosis of SSc made by one of our three Scleroderma Clinic attending rheumatologists. Patient data examined included demographic features (gender, date/age at symptom onset, date/age at SSc diagnosis, date/age at first clinic visit, race), disease subtype (diffuse cutaneous [dc] vs. limited cutaneous [lc]), smoking status and occupational exposure, organ system involvement, organ system involvement severity (severe and/or end-stage) using the revised Medsger severity score for SSc (5), serum autoantibody profile and survival. Occupational exposures recorded were daily inhalation including silica dust (underground coal mining, sandblasting, foundry work), organic solvents and dust/fumes. Disease onset was defined as the time of the first symptom or finding attributable to SSc. First visit and all available follow-up visit data were examined.
SSc was classified as either dc (skin thickening proximal to the elbows or knees at any time during the illness) or lc (skin thickening only distal to the elbows or knees, face acceptable) (6). Patients with lc involvement at first visit who subsequently developed dc changes were classified as dcSSc. Those with a documented history of dcSSc whose skin thickness had regressed to a lc distribution were considered to have dcSSc. Overlap with another connective tissue disease was determined by the examining Scleroderma Center physician.
Organ system involvement
An organ system was considered to be involved if the following features were documented at any time during the course of the illness and not attributable to other diseases. Some of these definitions were modified from a prior publication (7).
peripheral blood vessels: Raynaud phenomenon, digital pitting scars, digital tip ulcers, digital gangrene, or abnormal nailfold capillaries
skin: any thickening according to the modified Rodnan skin scoring system (8)
joints or tendons: joint swelling or contractures, fingertip-to-palm distance >1.9 cm in full active flexion, or palpable tendon or bursal friction rubs
skeletal muscle: proximal muscle weakness on physical examination and any one of the following: elevated serum creatine kinase level, myopathic changes on electro-myogram, or inflammation on muscle biopsy
gastrointestinal tract: any of the following: esophageal hypomotility (by esophagram or manometry), esophageal stricture, radiographic evidence of hypomotility of the small intestine, wide-mouth colonic sacculations, or malabsorption syndrome (physician judgment)
lung: (a) interstitial lung disease (ILD): pulmonary fibrosis (PF) on chest radiograph or high resolution CT scan of the lungs; (b) “intrinsic” pulmonary arterial hypertension (PAH): mean PA pressure >25 mmHg on right heart catheterization or estimated PA systolic pressure >45 mmHg on echocardiogram and the clinical diagnosis of PAH made by a cardiologist.
heart: left-sided heart failure (clinical) or estimated ejection fraction <45%, pericarditis, or arrhythmia requiring treatment
kidney: clinical evidence of “renal crisis”
An organ system was considered to be adequately studied if at least some objective data were obtained. For example, the proportion of patients with organ involvement was determined by the number of patients with the organ involvement documented divided by the number of patients who had undergone objective evaluation of the organ’s function.
Organ system severity
The severity of organ system involvement was defined as described previously (5). Severe involvement was defined as either grade 3 (severe) or grade 4 (end-stage) according to the revised Medsger severity index score for SSc (5). Grade 3 or 4 severity for each organ system was defined as follows:
peripheral blood vessels: presence of digital tip ulcers or gangrene
skin: modified Rodnan total skin thickness score (8) of >30
joints or tendons: fingertip-to-palm distance in full active flexion of ≥4.0 cm
skeletal muscle: severe proximal muscle weakness on physical examination or muscle weakness requiring assistance with ambulation aids (e.g., crutches or cane)
gastrointestinal tract: malabsorption syndrome (physician judgment), episodes of pseudo-obstruction, hyperalimentation required, or death due to intestinal involvement
lung: (a) pulmonary fibrosis detected on radiograph in addition to at least one of the following: FVC <50% predicted, requirement for oxygen in those with ILD, lung transplantation performed for ILD, or death due to SSc-related ILD, or (b) pulmonary arterial hypertension (PAH) (not secondary to pulmonary fibrosis) with estimated peak PA systolic pressure ≥65 mmHg on echocardiography or measured by right heart catheterization, requirement for oxygen due to SSc-related PAH, or death due to SSc-related PAH
heart: one of the following: left-sided congestive heart failure due to SSc (not secondary to renal crisis), arrhythmia requiring treatment, left ventricular ejection fraction of <40%, heart transplantation performed for SSc myocardial disease, or death due to SSc-related heart disease
kidney: scleroderma renal crisis or SRC (new malignant hypertension with a serum creatinine level of ≥3.0 mg/dL at any time), SRC with requirement for dialysis, renal transplantation performed for SSc-related renal disease, or death due to SSc-related renal disease.
Serum autoantibody detection
All patients with available serum samples had anti-nuclear antibody (ANA) testing performed in the University of Pittsburgh Scleroderma Research Laboratory using HEp-2 cells as substrate. SSc-associated antibodies, including anti-centromere, anti-topoisomerase I, anti-U1RNP, anti-RNA polymerase III, anti-PM-Scl, anti-U3RNP, anti-Ku, anti-Th/To, anti-U11/U12RNP and anti-RuvBL1/2 were detected as previously described (9–13).
Statistical analysis
The Pittsburgh Scleroderma Database uses the Medlog database management system. Quantitative variables were described with mean ± standard deviation (SD) or median and interquartile range for normally distributed or non-normally distributed continuous data, respectively. Qualitative variables were described with frequencies and percentages. Two-sample t test or Mann-Whitney U-test was performed to determine differences between female and male groups for normally distributed or non-normally distributed continuous data, respectively. Pearson’s chi-square or Fisher’s exact test, as appropriate, were used to compare the frequency distribution of categorical variables between female and male groups. Multiple linear, logistic or multinomial regression models were also fitted to adjust by cutaneous involvement subtype (dc, lc). If assumptions were not met (i.e., non-normally distributed continuous data) for multiple linear regression models, logarithm or square root transformations were used. Kaplan-Meier survival curves from first SSc symptom onset, first SSc physician diagnosis and first Pittsburgh visit were calculated for 5 and 10 years. Log-rank tests were performed to assess differences between survival rates. The proportional hazards assumption was tested for the variable of interest (sex) and was not violated. Survival curves adjusted for factors other than gender previously reported to be associated with increased mortality in SSc, including age, cutaneous subtype, smoking and involvement of the lung, heart or kidney, were generated from a Cox proportional hazards model.
Results
Comparison of all female versus all male SSc patients
Demographic and classification features and environmental exposures (Tab. I)
TABLE I.
Demographic features, classification and environmental exposures in female and male SSc patients
| Females (n = 2144) | Males (n = 542) | Significance (p value)
|
||
|---|---|---|---|---|
| n (%) | n (%) | Unadjusted | Adjusteda | |
| Race | ||||
| Caucasian (%) | 1929 (90%) | 488 (90%) | NS | NS |
| Age (mean years ± SD) | ||||
| Age at symptom onset | 43.8 ± 14.0 | 46.4 ± 13.7 | <0.001 | 0.0017 |
| Age at SSc diagnosis | 48.7 ± 13.6 | 49.8 ± 13.1 | NS | NS |
| Age at first Pittsburgh visit | 51.8 ± 13.4 | 51.8 ± 12.9 | NS | NS |
| Disease duration (years), median [interquartile range] | ||||
| Onset to first Pittsburgh visit | 3.9 [10.4] | 2.3 [5.0] | <0.0001 | 0.0005 |
| First Pittsburgh visit to last clinical follow-up | 7.7 [11.0] | 5.9 [9.4] | <0.0001 | <0.0001 |
| Residence [within 100 miles of Pittsburgh; referral area], n (%) | 1053 (49%) | 276 (51%) | NS | NS |
| Disease classification, n (%) | ||||
| Diffuse cutaneous | 794 (37%) | 262 (48%) | <0.0001 | <0.0001 |
| Limited cutaneous | 1142 (53%) | 232 (43%) | ||
| Overlap syndrome | 208 (10%) | 48 (9%) | 0.0008 | 0.0008 |
| Diffuse | 60 (29%) | 20 (42%) | ||
| Limited | 148 (71%) | 28 (58%) | ||
| Overlap types | ||||
| SSc, SLE | 56 (27%) | 2 (4%) | ||
| SSc, myositis | 119 (57%) | 42 (88%) | ||
| SSc, RA | 16 (8%) | 2 (4%) | ||
| SSc, SLE, myositis | 14 (7%) | 2 (4%) | ||
| SSc, RA, myositis | 3 (1%) | 0 (0%) | ||
| Environmental exposures, n (%) | ||||
| Cigarette smoking (ever) | 903 (42%) | 338 (63%) | <0.0001 | <0.0001 |
| Occupational | 48 (11%) | 58 (49%) | <0.0001 | <0.0001 |
Adjusted for cutaneous subtype (dc, lc).
SSc = systemic sclerosis; dc = diffuse cutaneous; lc = limited cutaneous; SLE = systemic lupus erythematosus; SD = standard deviation; RA = rheumatoid arthritis; NS = non-significant.
A total of 2686 consecutive new SSc patients were included (2144 females and 542 males; female to male ratio 4:1). The female to male ratio at the time of first-SSc symptom for patients aged 16–45 (child bearing years) was 5:1. There were no significant differences in race, age at SSc diagnosis, or age at first evaluation between female and male SSc patients. Females were significantly younger at symptom onset (mean 43.8 vs. 46.4 years, p = 0.0017) but not at first Pittsburgh visit. Thus females had longer disease duration at first visit (median 3.9 vs. 2.3 years, p = 0.0005). There were no differences in gender regarding residence location (<100 vs. 100+ miles from Pittsburgh) and thus referral bias was minimal. Consequently, for analysis purposes, data were combined for both of these residence groups.
Two thousand and sixty-four (2064) or 77% of patients satisfied the 1980 classification criteria for definite SSc (14). Of the remaining 23%, 464 (17%) satisfied the criteria for early SSc proposed by LeRoy and Medsger in 2001 (15). Only 158 (6%) did not satisfy either of these criteria, primarily because no serum was available for autoantibody testing, they had a negative ANA or the ANA was positive but no SSc-associated autoantibody was identified. Our database does not contain adequate information to examine the proportion of patients satisfying the recently published 2013 classification criteria (16).
Females significantly more frequently had lcSSc without overlap compared to males (53% vs. 43%, p<0.0001). Patients with SSc sine scleroderma (ssSSc) were included in the lcSSc category (17). The proportion of all SSc patients with ssSSc (8%) was somewhat higher in females (11%) than in males (7%). Overlap syndromes were equally frequent (9%–10%), but the distribution was different. Overlap with lcSSc was more frequent in females (71%) compared with males (58%). Females had a higher frequency of overlap with SLE (27% vs. 4%), and males with myositis (88% vs. 57%).
Male SSc patients were significantly more likely to have ever smoked cigarettes (p<0.0001) and to have had potentially contributing occupational exposures (p<0.0001).
Organ system involvement and severity (Tab. II)
TABLE II.
Organ system involvement and severity in female and male SSc patients
| Females (n = 2144) | Males (n = 542) | Significance (p value)
|
||
|---|---|---|---|---|
| Unadjusted | Adjustedd | |||
| Organ system involvement | ||||
| Peripheral blood vessels, n (%) | 2102 (98%) | 521 (96%) | 0.0085 | 0.0188 |
| Skin score, median maximum [interquartile range] | 8 [21] | 15 [25] | <0.0001 | NS |
| Joints or tendons, n (%) | ||||
| Joint tenderness | 1667 (78%) | 443 (82%) | 0.0313 | NS |
| Tendon or bursal rubs | 528 (25%) | 178 (33%) | <0.0001 | NS |
| Skeletal muscle, n (%) | 265 (20%) | 83 (24%) | NS | NS |
| Gastrointestinal tract, n (%)a | 1237 (78%) [1584] | 319 (81%) [395] | NS | NS |
| Interstitial lung disease, n (%)a | 699 (39%) [1809] | 257 (52%) [492] | <0.0001 | <0.0001 |
| Pulmonary arterial hypertension, n (%)a | 238 (19%) [1229] | 56 (17%) [328] | NS | NS |
| Heart, n (%)a | 192 (11%) [1728] | 79 (18%) [445] | 0.0002 | 0.0017 |
| Kidney, n (%) | 199 (9%) | 57 (11%) | NS | NS |
| Organ system severityb | ||||
| Peripheral blood vessels, n (%) | 804 (38%) | 231 (44%) | 0.0099 | 0.0140 |
| Skin score (mean maximum) | 384 (18%) | 137 (25%) | <0.0001 | NS |
| Joints or tendons, n (%) | 374 (20%) | 132 (28%) | 0.0002 | NS |
| Skeletal muscle, n (%) | 20 (8%) | 5 (6%) | NS | NS |
| Gastrointestinal tract, n (%)c | 161 (13%) [1237] | 41 (13%) [319] | NS | NS |
| Interstitial lung disease, n (%)c | 278 (42%) [660] | 125 (53%) [237] | <0.0048 | 0.0013 |
| Pulmonary arterial hypertension, n (%)c | 192 (81%) [238] | 47 (84%) [56] | NS | NS |
| Heart, n (%)c | 114 (59%) [192] | 52 (66%) [79] | NS | NS |
| Kidney, n (%)c | 122 (61%) [199] | 37 (65%) [57] | NS | NS |
Denominators in brackets reflect the number who had objective testing performed.
Frequencies are the number of patients with severe or end-stage disease.
Denominators in brackets reflect the number who had objective testing performed and had organ system involvement.
Adjusted for cutaneous subtype (dc, lc).
SSc = systemic sclerosis; dc = diffuse cutaneous; lc = limited cutaneous; SD = standard deviation; NS = non-significant.
After adjustment for cutaneous subtype, females more often had peripheral vascular involvement (98% vs. 96%, p = 0.0188). However, males more often had severe or end-stage peripheral vascular involvement (44% vs. 38%, p = 0.014). Males more often had interstitial lung disease (52% vs. 39%, p<0.0001) which was also more frequently severe or end-stage (53% vs. 42%, p = 0.0013). Cardiac involvement was more common in males (p = 0.0017).
In addition, gender differences were found for severe lung fibrosis before (odds ratio [OR] = 1.53, 95% confidence interval [CI] 1.14, 2.07, p = 0.0049) and after adjustment by smoking status and disease subtype classification (OR − 1.62, 95% CI 1.18, 2.20, p = 0.0025). Thus, males have higher odds of severe lung fibrosis when compared to females before and after adjustment for smoking status and disease subtype classification.
Scleroderma-related serum autoantibodies (Tab. III)
TABLE III.
Scleroderma-associated serum autoantibodies in female and male SSc patients
| Autoantibody | Females (n = 2144) | Males (n = 542) | Significance (p values)
|
||
|---|---|---|---|---|---|
| Unadjusted | Adjusteda | ||||
| Single autoantibody | Anti-RNA polymerase III | 455 (21%) | 124 (23%) | NS | NS |
| Anti-centromere | 451 (21%) | 52 (10%) | <0.0001 | <0.0001 | |
| Anti-topoisomerase I | 353 (16%) | 121 (22%) | 0.0008 | 0.0151 | |
| Anti-U1RNP | 109 (5%) | 27 (5%) | NS | NS | |
| Anti-Th/To | 147 (7%) | 36 (7%) | NS | NS | |
| Anti-U3RNP | 56 (3%) | 25 (5%) | 0.0138 | 0.0213 | |
| Anti-U11/U12 | 23 (1%) | 10 (2%) | NS | NS | |
| Anti-PM-Scl | 67 (3%) | 17 (3%) | NS | NS | |
| Anti-Ku | 13 (1%) | 5 (1%) | NS | NS | |
| Anti-RuvBL1/2 | 10 (<1%) | 7 (1%) | 0.0296 | NS | |
| More than one of above | Antibody combinationsb | 57 (3%) | 7 (1%) | NS | 0.0336 |
| None of the above | ANA negative | 47 (2%) | 22 (4%) | NS | NS |
| ANA positive | 252 (12%) | 74 (14%) | |||
| Serum not available | 104 (5%) | 15 (3%) | 0.0352 | 0.0305 | |
Adjusted for cutaneous subtype (dc, lc).
More than one SSc-associated autoantibody.
SSc = systemic sclerosis; dc = diffuse cutaneous; lc = limited cutaneous; NS = non-significant; ANA = anti-nuclear antibody.
Serum was obtained on over 95% of patients and antibody testing was performed in our laboratory on over 85% of all patients. As expected, 98% of the patients tested had a positive ANA. Overall, three serum autoantibodies (anti-RNA polymerase III, anti-topoisomerase I, and anti-centromere) accounted for over 70% of all single antibody specificities detected. After adjustment for cutaneous subtype, female SSc patients were significantly more likely to have anti-centromere antibody (p<0.0001). In contrast, male SSc patients were significantly more likely to have anti-topoisomerase I (p = 0.0151) and anti-U3RNP antibody (p = 0.0213). There were no significant gender differences in the frequency of other SSc-associated serum autoantibodies. More than one antibody was detected in 3% of females and 1% of males (p = 0.0336). Two SSc-related antibodies, which we have recently described (anti-U11/U12 RNP and anti-RuvBL1/2) (12, 13), were not tested for in all patients, and thus may account for some of the patients who are ANA positive with none of the other eight listed antibodies detected.
Cumulative survival and causes of death (Tab. IV, Fig. 1)
TABLE IV.
Cumulative survival and causes of death in female and male SSc patients
| CSR (%) | Females (n = 2144) | Males (n = 542) | Significance (p value)
|
|
|---|---|---|---|---|
| Unadjusted | Adjusteda | |||
| 5-year CSR | ||||
| From symptom onset | 82% | 73% | <0.0006 | 0.0109 |
| From SSc diagnosis | 66% | 54% | <0.0327 | 0.0409 |
| From first visit | 54% | 50% | <0.0001 | NS |
| 10-year CSR | ||||
| From symptom onset | 62% | 45% | <0.0001 | 0.0002 |
| From SSc diagnosis | 42% | 31% | <0.0011 | 0.0038 |
| From first visit | 63% | 22% | NS | NS |
|
| ||||
| Causes of death | Females | Males | ||
| (n = 955) | (n = 295) | |||
|
| ||||
| Unknown | 278 (29%) | 85 (29%) | ||
| Non-CTD related | 274 (29%) | 66 (22%) | NS | NS |
| CTD related | 403 (42%) | 144 (49%) | ||
| CTD related | ||||
| “Intrinsic” PAH | 126 (31%) | 28 (20%) | 0.0006 | 0.0006 |
| Pulmonary fibrosis | 93 (23%) | 61 (42%) | ||
| Cardiac | 34 (8%) | 11 (8%) | ||
| Renal | 60 (15%) | 18 (13%) | ||
| Gastrointestinal | 42 (10%) | 16 (11%) | ||
| Other | 48 (12%) | 10 (7%) | ||
Adjusted for cutaneous subtype (dc, lc).
SSc = systemic sclerosis; dc = diffuse cutaneous; lc = limited cutaneous; PAH = pulmonary arterial hypertension; CTD = connective tissue disease; CSR = cumulative survival rate; NS = non-significant.
Fig. 1.
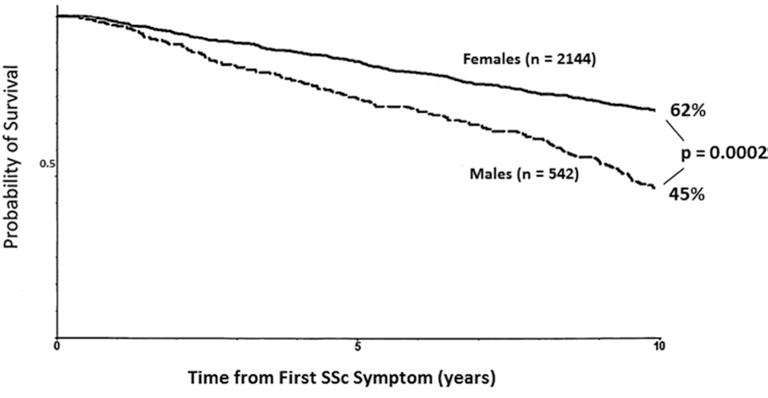
Adjusted cumulative survival rate at 10 years from the time of first symptom in all females and males with systemic sclerosis.
Female SSc patients had significantly better unadjusted cumulative survival rates at 5 and 10 years after first symptom attributable to SSc (p = 0.0006), first SSc diagnosis (p<0.0327), and first Pittsburgh visit (p<0.0001). Adjustment for age, cutaneous subtype, smoking status, and involvement of the lung (pulmonary fibrosis), heart and kidney did not change this conclusion for 5 and 10 years after SSc onset or diagnosis.
The distribution of causes of death was significantly different (Tab. IV, p = 0.0006), primarily because pulmonary fibrosis is a relatively more frequent cause in males (42% vs. 23%) and pulmonary hypertension in females (31% vs. 20%).
Summary of female and male SSc patient profiles (Tab. V)
TABLE V.
Summary of female and male SSc patient profiles
| Females | Males | |
|---|---|---|
| Demographic features | Younger age at onset; longer duration at first visit | Older age at onset; shorter duration at first visit |
| Subset classification | lcSSc; lupus overlap | dcSSc; myositis overlap |
| Exposures | Cigarette smoking; occupational | |
| Organ involvement | ||
| (a) Frequency | Peripheral vascular | ILD; heart |
| (b) Severity | ILD | |
| Serum autoantibodies Outcomes | Centromere | Topoisomerase I |
| (a) Survival | Better | Worse |
| (b) Most frequent cause of death | PAH | ILD |
PAH = pulmonary arterial hypertension; ILD = interstitial lung disease; SSc = systemic sclerosis; dc = diffuse cutaneous; lc = limited cutaneous.
In essence, the disease profile in females is that of lcSSc with younger age of onset, longer disease duration at first Pittsburgh visit, more frequent but less severe peripheral vascular involvement, anti-centromere antibody, and improved survival, with the most frequent cause of death being pulmonary arterial hypertension. In contrast, males are older at onset, present earlier in their disease, are more frequently cigarette smokers with potentially contributing occupational exposures, have dcSSc, more severe peripheral vascular disease, more frequent and more severe involvement the lungs (ILD), more frequent heart involvement, anti-topoisomerase I and anti-U3RNP antibody and reduced survival with the most common cause of death being ILD. Overlap with lupus is more frequent in females and with myositis in males. Other distinctive female- and male-associated SSc disease characteristics may exist, but we were unable to identify them in this study.
Discussion
Autoimmune diseases are more prevalent in women, as is the case with SSc. The objective of this study was to describe gender differences in SSc in a large single-center cohort. Our comparisons were comprehensive, analyzing demographic information, disease subtype, environmental exposures, organ system involvement and severity, serum autoantibody profile, survival and causes of death. A recent abstract including over 1500 SSc patients concluded that dc-SSc, cigarette smoking, ILD and reduced survival were significantly more frequent among males (18). While there were many similarities when comparing the Pittsburgh female with male SSc patients, several key differences emerged, as illustrated in Table V.
An environmental factor, i.e., cigarette smoking, is implicated in the pathogenesis of rheumatoid arthritis (19). Our finding of similar concordance for SSc in monozygotic and dizygotic twins favors a participation of the environment in disease development (20). Cigarette smoking and occupational exposures may contribute to the frequency and severity of ILD in our male SSc patients and to their reduced survival.
Our findings are consistent with other publications concluding that in SSc and related autoimmune diseases, male gender and dcSSc are poor prognostic factors (21–23). Several studies have suggested that males were more likely to have dcSSc and to have pulmonary involvement, including interstitial lung disease (ILD) and pulmonary hypertension (PH) (21, 24–26). In systemic lupus erythematosus (SLE), male patients have more severe disease and worse survival (27, 28). In SLE patients who have renal disease, males have more severe lupus nephritis as well (29). These previous reports in combination with our results raise the question of whether gender differences are integral to the pathogenesis of SSc and other autoimmune diseases. Interestingly, reduced survival in males has also been observed in another fibrosing disorder, idiopathic pulmonary fibrosis (30).
In a previous study, we demonstrated that estradiol (E2) promotes the development of a fibrotic phenotype, both in vitro and ex vivo in human skin (31). In addition, we have shown that serum E2 levels are significantly elevated in postmenopausal female patients with early diffuse cutaneous SSc compared to healthy postmenopausal female controls, with neither group having received any hormone replacement therapy (HRT) (31). These findings suggest the possibility that estradiol participates in the pathogenesis of skin thickening and possibly other organ fibrosis in SSc. Elevated estradiol may also underlie worse prognosis in males. As men age, their estrogen levels increase, most commonly due to conversion of testosterone to estrogen via aromatase (32). Increased estradiol was associated with increased risk of all-cause mortality in a study of 5350 men (33). Since the men in our study are older at disease onset, they likely have elevated estrogen levels which contribute to the increased severity of disease. Current research in our laboratory is focused on measuring estrogen levels in SSc males.
In addition to hormonal status, the female preponderance in SSc is proposed to be due to genetic/epigenetic differences and X chromosome gene reactivation or skewed X chromosome inactivation (reviewed in D’Amico et al) (34). Skewed X chromosome inactivation in various peripheral blood cells in women with SSc has been described in several reports (35–38). Differential methylation of genes on the X chromosome was identified in peripheral blood mononuclear cells (PBMC) of monozygotic twins discordant for SSc (39). The majority of the differentially methylated genes were linked in a network involving IL-6, a cytokine implicated in the pathogenesis of SSc whose production is increased in dermal fibroblasts and serum of patients with the disease (40, 41).
Other genetic factors could also be implicated in SSc gender differences. SLE is the connective tissue disease (CTD) most frequently associated with Klinefelter syndrome (chromosome complement 47, XXY) (42). The second most common CTD reported with Klinefelter syndrome is SSc, but this coexistence is rare, with fewer than 10 cases found in the medical literature (43–45). Other patients with “mixed connective tissue disease”, some of whom had SSc features, have also been reported (42). The Klinefelter-SLE association and the presence of SLE in females with 47, XXX support a possible role of the X chromosome in the pathogenesis of lupus. An intriguing case report of a phenotypic male SSc patient with hypogonadism having an XX sex chromosome complement was published (46). By fluorescent in situ hybridization (FISH) karyotype analysis, it was shown that this patient had a 46, XX, Xp22.3 (SRY+) gene translocation. We recently identified a similar male SSc patient also with hypogonadism 46, XX. We showed by microarray analysis that both the Velasco patient and ours have a nearly identical short arm X chromosome deletion with the SRY region attached to the X (Yatsenko et al, unpublished observations).
Our study is primarily descriptive. In the future we intend to further examine this patient population by analyzing the relationships between gender and autoantibodies, between gender, cigarette smoking and pulmonary fibrosis and in various patient subsets including early and late dcSSc and lcSSc.
Conclusions
In females, systemic sclerosis (SSc) occurs at a younger age and is more likely to be of the limited cutaneous (lc) subtype with anti-centromere antibody and improved survival. In contrast, male SSc patients present at an older age, are more likely to be cigarette smokers, have diffuse cutaneous (dc) involvement, anti-topoisomerase I and anti-U3RNP antibody, pulmonary fibrosis and reduced survival. The reasons for these gender differences in occurrence, clinical features, autoantibody profiles and mortality remain to be explained.
Acknowledgments
Financial support: This work was supported in part by the NIH through grants K24AR060297 to CAF-B,d UL1TR000005 (pilot project funding to CAF-B) and UL1 RR024153.
Footnotes
Disclosures
Conflict of interest: None of the authors has financial interest related to this study to disclose.
References
- 1.Steen VD, Oddis CV, Conte CG, Janoski J, Casterline GZ, Medsger TA., Jr Incidence of systemic sclerosis in Allegheny County, Pennsylvania. A twenty-year study of hospital-diagnosed cases, 1963–1982. Arthritis Rheum. 1997;40(3):441–445. doi: 10.1002/art.1780400309. [DOI] [PubMed] [Google Scholar]
- 2.Medsger TA. Classification, prognosis. In: Clements PJ, Furst DE, editors. Systemic sclerosis. 2nd. Philadelphia: Lippincott, Williams & Wilkins; 2004. pp. 17–28. [Google Scholar]
- 3.Gaultier JB, Hot A, Cathébras P, Grange C, Ninet J, Rousset H. Systemic sclerosis in men. Rev Med Interne. 2008;29(3):181–186. doi: 10.1016/j.revmed.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66(7):940–944. doi: 10.1136/ard.2006.066068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bombardieri S, Medsger TA, Jr, Silman AJ, Valentini G. The assessment of the patient with systemic sclerosis. Introduction Clin Exp Rheumatol. 2003;21(3 Suppl 29):S2–S4. [PubMed] [Google Scholar]
- 6.Clements PJ, Feghali CA. Cutaneous involvement in systemic sclerosis. Philadelphia: Lippincott Williams & Wilkins; 2004. [Google Scholar]
- 7.Koschik RW, II, Fertig N, Lucas MR, Domsic RT, Medsger TA., Jr Anti-PM-Scl antibody in patients with systemic sclerosis. Clin Exp Rheumatol. 2012;30(2 Suppl 71):S12–S16. [PubMed] [Google Scholar]
- 8.Clements P, Lachenbruch P, Siebold J, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol. 1995;22(7):1281–1285. [PubMed] [Google Scholar]
- 9.Okano Y, Medsger TA., Jr Autoantibody to Th ribonucleoprotein (nucleolar 7-2 RNA protein particle) in patients with systemic sclerosis. Arthritis Rheum. 1990;33(12):1822–1828. doi: 10.1002/art.1780331210. [DOI] [PubMed] [Google Scholar]
- 10.Okano Y, Steen VD, Medsger TA., Jr Autoantibody to U3 nucleolar ribonucleoprotein (fibrillarin) in patients with systemic sclerosis. Arthritis Rheum. 1992;35(1):95–100. doi: 10.1002/art.1780350114. [DOI] [PubMed] [Google Scholar]
- 11.Okano Y, Steen VD, Medsger TA., Jr Autoantibody reactive with RNA polymerase III in systemic sclerosis. Ann Intern Med. 1993;119(10):1005–1013. doi: 10.7326/0003-4819-119-10-199311150-00007. [DOI] [PubMed] [Google Scholar]
- 12.Fertig N, Domsic RT, Rodriguez-Reyna T, et al. Anti-U11/U12 RNP antibodies in systemic sclerosis: a new serologic marker associated with pulmonary fibrosis. Arthritis Rheum. 2009;61(7):958–965. doi: 10.1002/art.24586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaji K, Fertig N, Medsger TA, Jr, et al. Autoantibodies to RuvBL1 and RuvBL2: a novel systemic sclerosis-related antibody associated with diffuse cutaneous and skeletal muscle involvement. Arthritis Care Res (Hoboken) 2014;66(4):575–584. doi: 10.1002/acr.22163. [DOI] [PubMed] [Google Scholar]
- 14.Masi AT, Rodnan GP, Medsger TA, Jr, et al. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum. 1980;23(5):581–590. doi: 10.1002/art.1780230510. [DOI] [PubMed] [Google Scholar]
- 15.LeRoy EC, Medsger TA., Jr Criteria for the classification of early systemic sclerosis. J Rheumatol. 2001;28(7):1573–1576. [PubMed] [Google Scholar]
- 16.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72(11):1747–1755. doi: 10.1136/annrheumdis-2013-204424. [DOI] [PubMed] [Google Scholar]
- 17.Poormoghim H, Lucas M, Fertig N, Medsger TA., Jr Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum. 2000;43(2):444–451. doi: 10.1002/1529-0131(200002)43:2<444::AID-ANR27>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 18.Freire M, Simeon-Aznar CP, Rivera Gallego A, et al. Clinic and mortality differences between sclerodermic men and women of the Rescle Cohort. Journal of Scleroderma and Related Disorders. 2016;1(1):151. [Google Scholar]
- 19.Sparks JA, Chang SC, Deane KD, et al. Associations of smoking and age with inflammatory joint signs among first-degree relatives without rheumatoid arthritis: Results from the Studies of the Etiology of RA. Arthritis Rheumatol. 2016 Feb 11; doi: 10.1002/art.39630. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feghali-Bostwick C, Medsger TA, Jr, Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003;48(7):1956–1963. doi: 10.1002/art.11173. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen C, Bérezné A, Baubet T, et al. Groupe Français de Recherche sur la Sclérodermie Association of gender with clinical expression, quality of life, disability, and depression and anxiety in patients with systemic sclerosis. PLoS ONE. 2011;6(3):e17551. doi: 10.1371/journal.pone.0017551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters-Golden M, Wise RA, Schneider P, Hochberg M, Stevens MB, Wigley F. Clinical and demographic predictors of loss of pulmonary function in systemic sclerosis. Medicine (Baltimore) 1984;63(4):221–231. doi: 10.1097/00005792-198407000-00004. [DOI] [PubMed] [Google Scholar]
- 23.Wynn J, Fineberg N, Matzer L, et al. Prediction of survival in progressive systemic sclerosis by multivariate analysis of clinical features. Am Heart J. 1985;110(1 Pt 1):123–127. doi: 10.1016/0002-8703(85)90525-3. [DOI] [PubMed] [Google Scholar]
- 24.Al-Dhaher FF, Pope JE, Ouimet JM. Determinants of morbidity and mortality of systemic sclerosis in Canada. Semin Arthritis Rheum. 2010;39(4):269–277. doi: 10.1016/j.semarthrit.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Ferri C, Valentini G, Cozzi F, et al. Systemic Sclerosis Study Group of the Italian Society of Rheumatology (SIR-GSSSc) Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002;81(2):139–153. doi: 10.1097/00005792-200203000-00004. [DOI] [PubMed] [Google Scholar]
- 26.Hesselstrand R, Scheja A, Akesson A. Mortality and causes of death in a Swedish series of systemic sclerosis patients. Ann Rheum Dis. 1998;57(11):682–686. doi: 10.1136/ard.57.11.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crosslin KL, Wiginton KL. Sex differences in disease severity among patients with systemic lupus erythematosus. Gend Med. 2011;8(6):365–371. doi: 10.1016/j.genm.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 28.Schwartzman-Morris J, Putterman C. Gender differences in the pathogenesis and outcome of lupus and of lupus nephritis. Clin Dev Immunol. 2012;2012:604892. doi: 10.1155/2012/604892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Carvalho JF, do Nascimento AP, Testagrossa LA, Barros RT, Bonfá E. Male gender results in more severe lupus nephritis. Rheumatol Int. 2010;30(10):1311–1315. doi: 10.1007/s00296-009-1151-9. [DOI] [PubMed] [Google Scholar]
- 30.Gribbin J, Hubbard RB, Le Jeune I, Smith CJP, West J, Tata LJ. Incidence and mortality of idiopathic pulmonary fibrosis and sarcoidosis in the UK. Thorax. 2006;61(11):980–985. doi: 10.1136/thx.2006.062836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aida-Yasuoka K, Peoples C, Yasuoka H, et al. Estradiol promotes the development of a fibrotic phenotype and is increased in the serum of patients with systemic sclerosis. Arthritis Res Ther. 2013;15(1):R10. doi: 10.1186/ar4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vermeulen A, Kaufman JM, Goemaere S, van Pottelberg I. Estradiol in elderly men. Aging Male. 2002;5(2):98–102. [PubMed] [Google Scholar]
- 33.Holmboe SA, Vradi E, Jensen TK, et al. The association of reproductive hormone levels and all-cause, cancer, and cardiovascular disease mortality in men. J Clin Endocrinol Metab. 2015;100(12):4472–4480. doi: 10.1210/jc.2015-2460. [DOI] [PubMed] [Google Scholar]
- 34.D’Amico F, Skarmoutsou E, Mazzarino MC. The sex bias in systemic sclerosis: on the possible mechanisms underlying the female disease preponderance. Clin Rev Allergy Immunol. 2014;47(3):334–343. doi: 10.1007/s12016-013-8392-9. [DOI] [PubMed] [Google Scholar]
- 35.Ozbalkan Z, Bagişlar S, Kiraz S, et al. Skewed X chromosome inactivation in blood cells of women with scleroderma. Arthritis Rheum. 2005;52(5):1564–1570. doi: 10.1002/art.21026. [DOI] [PubMed] [Google Scholar]
- 36.Uz E, Loubiere LS, Gadi VK, et al. Skewed X-chromosome inactivation in scleroderma. Clin Rev Allergy Immunol. 2008;34(3):352–355. doi: 10.1007/s12016-007-8044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Broen JC, Wolvers-Tettero IL, Geurts-van Bon L, et al. Skewed X chromosomal inactivation impacts T regulatory cell function in systemic sclerosis. Ann Rheum Dis. 2010;69(12):2213–2216. doi: 10.1136/ard.2010.129999. [DOI] [PubMed] [Google Scholar]
- 38.Lian X, Xiao R, Hu X, et al. DNA demethylation of CD40l in CD4+ T cells from women with systemic sclerosis: a possible explanation for female susceptibility. Arthritis Rheum. 2012;64(7):2338–2345. doi: 10.1002/art.34376. [DOI] [PubMed] [Google Scholar]
- 39.Selmi C, Feghali-Bostwick CA, Lleo A, et al. X chromosome gene methylation in peripheral lymphocytes from monozygotic twins discordant for scleroderma. Clin Exp Immunol. 2012;169(3):253–262. doi: 10.1111/j.1365-2249.2012.04621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasegawa M, Sato S, Ihn H, Takehara K. Enhanced production of interleukin-6 (IL-6), oncostatin M and soluble IL-6 receptor by cultured peripheral blood mononuclear cells from patients with systemic sclerosis. Rheumatology (Oxford) 1999;38(7):612–617. doi: 10.1093/rheumatology/38.7.612. [DOI] [PubMed] [Google Scholar]
- 41.Feghali CA, Bost KL, Boulware DW, Levy LS. Mechanisms of pathogenesis in scleroderma. I. Overproduction of interleukin 6 by fibroblasts cultured from affected skin sites of patients with scleroderma. J Rheumatol. 1992;19(8):1207–1211. [PubMed] [Google Scholar]
- 42.Rovenský J, Imrich R, Lazúrová I, Payer J. Rheumatic diseases and Klinefelter’s syndrome. Ann N Y Acad Sci. 2010;1193(1193):1–9. doi: 10.1111/j.1749-6632.2009.05292.x. [DOI] [PubMed] [Google Scholar]
- 43.Kobayashi S, Shimamoto T, Taniguchi O, Hashimoto H, Hirose S. Klinefelter’s syndrome associated with progressive systemic sclerosis: report of a case and review of the literature. Clin Rheumatol. 1991;10(1):84–86. doi: 10.1007/BF02208039. [DOI] [PubMed] [Google Scholar]
- 44.Bargagli E, Bartalini G, Galeazzi M, Maggiorelli C, Anichini C, Rottoli P. Chromosome 5 inversion in two siblings, one with Klinefelter syndrome and systemic sclerosis, the other with rheumatoid arthritis. Rheumatol Int. 2008;28(7):725–726. doi: 10.1007/s00296-007-0501-8. [DOI] [PubMed] [Google Scholar]
- 45.De Keyser F, Mielantes H, Veys EM. Klinefelter’s syndrome and scleroderma. J Rheumatol. 1989;16(12):1613–1614. [PubMed] [Google Scholar]
- 46.Velasco G, Savarese V, Sandorfi N, Jimenez SA, Jabbour S. 46, XX SRY-positive male syndrome presenting with primary hypogonadism in the setting of scleroderma. Endocr Pract. 2011;17(1):95–98. doi: 10.4158/EP10184.CR. [DOI] [PubMed] [Google Scholar]