

Summary
The neural crest is a transient embryonic population that originates within the central nervous system, then migrates into the periphery and differentiates into multiple cell types. The mechanisms that govern neural crest stem-like characteristics and self-renewal ability are poorly understood. Here, we show that the proto-oncogene cMyc is a critical factor in the chick dorsal neural tube, where it regulates the size of the premigratory neural crest stem cell pool. Loss of cMyc dramatically decreases the number of emigrating neural crest cells due to reduced self-renewal capacity, increased cell death and shorter duration of the emigration process. Interestingly, rather than via E-Box binding, cMyc acts in the dorsal neural tube by interacting with another transcription factor, Miz1, to promote self-renewal. The finding that cMyc operates in a non-canonical manner in the premigratory neural crest highlights the importance of examining its role at specific timepoints and in an in vivo context.
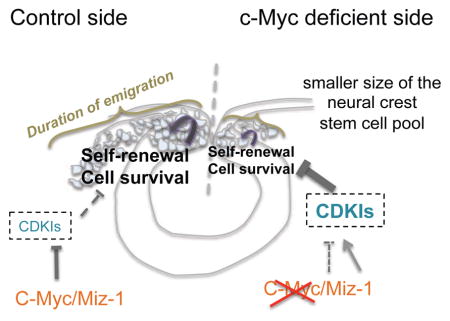
cMyc is a key regulator of the size of the neural crest stem cell pool
Our data suggest that cMyc binds to Miz1 to form a repressive complex that keeps factors such as Cyclin Dependent Kinase Inhibitors (CDKI) sufficiently low for adequate cell cycle progression and cell survival to take place in the self-renewing cells. The production of neural crest is regulated by: 1) the numbers of the neural crest cells generated in the dorsal neural tube and 2) the length of the emigration period as the newly produced neural crest cells delaminate and initiate migration towards multiple destinations in the developing embryo.
Introduction
Neural crest cells are multipotent cells that give rise to tens of different cell types in vertebrate embryos, ranging from craniofacial cartilage and bone to peripheral ganglia and melanocytes of the skin (Dupin and Coelho-Aguiar, 2013). Premigratory neural crest cells start as neuroepithelial cells in the dorsal neural tube that subsequently undergo an epithelial to mesenchymal transition (EMT), migrating from their site of origin in the central nervous system to diverse destinations within the developing embryo (Kerosuo and Bronner-Fraser, 2012). The premigratory phase lasts about 24 hours in the bird embryo. Due to its transient nature, the process of neural crest production must be tightly regulated. However, little is known about what regulates the size of the neural crest stem cell pool and/or whether it is comparable to other stem cell niches. Therefore, despite the high clinical relevance and need to understand how neural crest ‘stemness’ is maintained, the in vivo mechanisms that control the numbers of neural crest precursors and duration of their emigration process are poorly understood.
The transcription factor and proto-oncogene cMyc has been implicated in a broad range of cellular functions (Eilers and Eisenman, 2008), including cell proliferation and apoptosis. It has been estimated to regulate 15% of the genome, and has an established role in stem cell maintenance in both embryonic stem cells and tissue specific stem cell niches in adults and in the embryo (Chappell and Dalton, 2013; Dong et al., 2011; Kerosuo et al., 2008; Kwan et al., 2015; Varlakhanova et al., 2010; Wilson et al., 2004). Myc is often dysregulated in cancer cells, correlating with poor prognosis in many neural crest derived tumors such as neuroblastoma (Fredlund et al., 2008) and melanoma (Bosserhoff, 2006). In the chick, the Myc paralogs, cMyc and nMyc, exhibit complementary expression patterns during neural crest development with nMyc expressed early in the neural plate and its border and later in the middle and ventral neural tube (Khudyakov and Bronner-Fraser, 2009); in contrast, cMyc is expressed in premigratory neural crest cells within the dorsal neural tube. In the frog, the expression of these paralogs is switched such that cMyc is expressed early in the neural plate border, where it is required for induction of neural crest fate; loss of cMyc reverts cells to a neural fate, without affecting proliferation or cell death of the stem cell pool (Bellmeyer et al., 2003). In zebrafish, hMyc, which is genetically closer to nMyc than cMyc, is expressed in the neural plate during neural crest specification and its knockdown affects both neural induction and proper development of neural crest derived tissues such as the pharyngeal arches (Hong et al., 2008). Similarly, conditional knockdown of cMyc in mice using Wnt1-Cre results in defects in formation of pigment, skull and ear (Wei et al., 2007). Reprogramming of murine radial glial cells with Brn2 and cMyc induces characteristics of early neural crest cells (Bung et al., 2015). Finally, overexpression of avian nMyc after EMT in migratory neural crest cells results in differentiation of neurons at the expense of other neural crest derivatives (Wakamatsu et al., 1997). However, the relative roles of nMyc and cMyc in the early neural crest remain unclear.
Here, we tackle the role of the multifunctional protein cMyc (Eilers and Eisenman, 2008) in the premigratory neural crest of avian embryos. Initiation of cMyc expression begins concomitant with the onset of EMT, consistent with its proposed role as a late neural crest specifier gene (Sauka-Spengler and Bronner-Fraser, 2008). Moreover, we find that cMyc plays a critical role in regulating the size of the premigratory neural crest cell pool by promoting self-renewal and survival. This role is different than its canonical role in activating transcription together with its binding partner Max via E-Box binding. Instead, our results show that in the premigratory neural crest, cMyc/Max function depends upon interaction with another transcription factor Miz-1 to form a repressive complex. Our results suggest that the Myc/Miz1 complex transcriptionally regulates levels of factors including cyclin dependent kinase inhibitors, thus enabling cell cycle progression required for self-renewal. Although cMyc is well-known for its ability to induce proliferation, this does not appear to be the case in the neural crest, highlighting its tissue-specific roles. These results provide novel insights into the mechanisms that regulate self-renewal ability in the premigratory neural crest stem cell pool.
RESULTS
Given the existence of notable species specific differences in expression of cMyc in the early ectoderm, we performed a careful analysis of the expression pattern in the chick embryo by in situ hybridization (Figs 1 and S1). The results show that, unlike in the frog (Bellmeyer et al. 2003), cMyc expression is absent from the neural plate and neural plate border (Figs 1A–B, S1A–D) but is first detected in the cranial dorsal neural folds during neural tube closure at the 5–6 somite stage (Hamburger Hamilton, HH, stage 8+, Fig 1C). The expression overlaps with Pax7-immunopositive premigratory neural crest cells (Fig 1C′). Intense cMyc expression is maintained in neural crest cells throughout emigration and migratory stages (HH stages 9–12, Figs 1E–F S1E–F).
Figure 1. Onset Of cMyc Expression In The Premigratory Neural Crest.

(A–D) In situ hybridization shows that cMyc expression initiates in the premigratory cranial neural crest within the dorsal neural tube at Hamburger Hamilton (HH) stage 8 prior to EMT and remains on during emigration from the neural tube at stage 9. (C′) cMyc expression overlaps with that of the neural crest marker Pax7, observed in a transverse section by immunostaining. (E–F, E′) cMyc expression persists in neural crest cells throughout migration. Scale bar for whole embryos 150 μm; sections 20μm. See also figure S1.
We next examined the effects of cMyc loss of function in the neural crest by electroporating a translation blocking morpholino onto one side of the embryo at gastrula stages. As an internal control, a control morpholino was electroporated on the contralateral side of the same embryo, which was cultured and analyzed the following day (Fig 2A). 1.2mM morpholino had no effect on the induction of neural crest cells at the neural plate border prior to the time of initiation of cMyc expression in the neural folds, which also demonstrates that the morpholino treatment did not cause any non-specific effects (Figs S2A–C). In contrast, at premigratory stages (HH8+/9−), cMyc knockdown reduced the numbers of FoxD3 and Pax7 expressing neural crest cells compared to control embryos. As a result, the neural crest cell domain in the tip of the dorsal neural tube was significantly smaller on the cMyc-morphant side (Fig 2B–D). However, expression of the neural marker Sox2 was not affected (Fig 2D). The same phenotype persisted throughout the emigration and migration period (Figs 2E–H) with on average 85% (for stage 8–10 combined) of the embryos resulting in a decrease in size of the neural crest population on the cMyc morpholino-treated side compared with 10% in control embryos (Fig 2G). During migratory stages (stage 9), this resulted in fewer Sox10 positive neural crest cells on the cMycMO treated side. In addition, emigration was delayed, such that none of the cMyc morpholino electroporated cells had delaminated at the time when the first neural crest cells on the control morpholino side already were migrating (Figs 2E–F). As emigration continued, neural crest cells migrated further laterally on the control side as viewed in whole embryos. In contrast, a Sox10-negative area was visible adjacent to the midline in cMyc morpholino-treated embryos. Transverse sections revealed that emigration had ended on the cMyc morpholino-treated side such that no more neural crest cells were emerging from the dorsal neural tube. In contrast, production of Sox10-positive cells was still observed on the control side (Figs 2H, h and hh). Higher dosage of the cMycMO (1.5mM and up) resulted in almost a complete loss in expression of neural crest markers, FoxD3 and Sox10, and caused severe degradation of the dorsal neural tube (Fig. S2D–G). Because cMyc and nMyc have non-overlapping patterns in the neural tube, we analyzed whether cMyc knockdown caused a compensatory increase in nMyc levels in the neural crest cells. However, no changes in nMyc expression were observed after loss of cMyc (Fig S2H).
Figure 2. cMyc Regulates The Size Of The Neural Crest Stem Cell Pool.

(A) Translation blocking cMyc morpholino was electroporated into gastrula stage (HH stage 4) embryos on one side, while the contralateral side was injected with control morpholino as an internal control and analyzed the next day. (B–C) cMyc knockdown reduces the numbers of premigratory neural crest cells in the dorsal neural tube as shown by in situ hybridization for FoxD3 whereas control embryos show similar levels of FoxD3 expression on both sides. (D) The size of the neural crest cell domain is smaller as shown by Pax7 immunostaining but Sox2 protein levels are similar on both sides (n=9/9 for Sox2/Pax7 immunos). (E) At HH stage 9, fewer Sox10-positive neural crest cells (black arrow) are noted on the cMyc morphant side and their emigration has not yet started whereas neural crest cells have emigrated on the control side (blue arrow); (F) control embryos are similar on both sides. (G) 85% of embryos have a reduced number of neural crest cells and thus a smaller neural crest stem cell pool on the cMyc morphant side compared with only 10% of control embryos (n=58 for stage 8–10 combined, n=20 for control embryos). (H) The phenotype persists during the migration phase: emigration starts later and ends sooner on the cMyc morphant side leaving a Sox10-negative region next to the midline (magnified in h and marked by black arrow in hh) whereas new Sox10-positive cells are still produced in the dorsal neural tube on the contralateral side (phenotype seen in 83% of embryos, n=30). Overall, this leads to a smaller size of the neural crest cell pool as visualized by the brackets with different length. * marks the treated side. Scale bar 20μm. See also figure S2.
The apparent decrease in size of the neural crest cell pool prompted us to test whether loss of cMyc affects the ability of the cells to self-renew. To this end, we performed a colony forming assay using chick crestospheres that we previously showed could be kept in vitro as primary epithelial neural crest stem cells (Kerosuo et al., 2015) and thus maintained high expression levels of cMyc similar to that of premigratory neural crest cells (Figs 3A,B). After cMyc knockdown achieved by using two different siRNAs, we found that the self-renewal capacity was reduced by half (Fig. 3C) after a ~ 30–40% decrease in cMyc expression levels assayed by QPCR (Fig. 3D). In addition, many of the newly formed cMyc deficient spheres were smaller in size (~15 cells/sphere as compared to ~30–50 cells on average in untreated spheres). The capacity of secondary sphere formation was also reduced after cMyc knockdown, further demonstrating an effect on self-renewal rather than proliferation (Fig. 3E).
Figure 3. cMyc Regulates Self-Renewal But Not Multipotency Of Neural Crest Cells.

(A) Self-renewal was measured in vitro by using the colony forming assay on crestospheres (B) that express high levels of cMyc mRNA as shown by in situ hybridization. (C) siRNA knockdown of cMyc significantly reduces neural crest cell self-renewal capacity (siRNA1: average=0.41 fold, SEM= 0.076; n=6; siRNA2: average= 0.54 fold, SEM 0.13, n=6; CosiRNA average = 1 fold, SEM=0.152, n=12, ttest siRNA1/Co p=0.00358; siRNA2/Co p=0.0355). (D) Reduced cMyc expression levels after siRNA knockdown (average for siRNA1 = 0.577, SEM 0.121, n=3; siRNA2= 0.748, SEM 0.022: n=3). (E) cMyc knockdown also reduces secondary sphere formation capacity (cMyc siRNA = 0.456208929, sem 0.092996221 n=6; control siRNA = 1, sem= 0.237695836 n=6; p=0.04310364). (F–G). Differentiation of neural tube explants electroporated with cMycMO. cMyc deficient neural crest cells were able to differentiate into all major types of neural crest derivatives: melanoblasts (Mitf), Smooth muscle (SMA), neurons (TUJ1) and glial cells (BLBP) as a proof of multipotency (n=24 embryos). Scale bar 50 μm.
We also examined the differentiation capacity of cMyc morpholino-treated neural tube explants. After a week of in vitro differentiation, we detected several types of neural crest derivatives similar to those seen in control cultures (not shown, n=12 embryos), as indicated by staining of neurons with Tuj1 antibody, melanoblasts by Mitf protein expression, smooth muscle cells by SMA antibody and glial cells with BLBP antibody (Figs 3F–G).
Since cMyc is known for its role in cell proliferation and survival (Eilers and Eisenman, 2008), we examined whether cell proliferation and/or survival were affected after cMyc morpholino knockdown. The results show that the proliferation rate in the neural crest was not changed in a statistically significant manner, though we did detect a slight decrease in the numbers of proliferating cells by using Phospho-Histone 3 staining (Figs 4A,S3A). Instead, we consistently detected fewer cells in the dorsal neural tube (Fig 4B). The tip of the neural tube also appeared thinner and improperly folded on the cMyc morpholino-treated side (Figs 4D,F). Since proliferation was not noticeably affected and higher cMycMO concentrations caused degradation of the dorsal neural tube (Fig S2), we next examined whether lack of cMyc caused apoptosis in late stage 8 neural folds examined by Caspase3 immunostaining. As compared to the control morpholino injected side, cMycMO increased apoptosis 4 fold (Figs 4C,E).
Figure 4. Neural Crest Cell Survival Requires cMyc.

(A) cMyc deficiency does not affect proliferation in a statistically significant way (cMycMO average= 0.702, SEM 0.12; n=7 embryos; coMO=1, SEM 0.105; n=7 embryos), (B, D) although the dorsal neural tube has fewer cells on the Myc deficient side (on average 47 cells in the tip of the dorsal neural tube, SEM 1.499, as compared to 55 cells on the tips on the coMo side, SEM 1.51; n= 26 slides in 6 embryos, ttest p= 0.00020924); (C, E–F) likely due to a massive decrease in cell survival as shown by Caspase 3 immunostaining (average = 4.13, SEM cMycMo 0.2039, n=6 embryos and CoMo 0.1700912, ttest p= 4.534E-07, n=6 embryos) and (D, D′) incorrect folding of the dorsal neural folds. (G) QPCR data shows no change in known targets of cMyc involved in cell proliferation (CyclinE: cMycMo/CoMo average 0.997, SEM 0.098, n=8; CoMo/CoMo average 1.1, SEM 0.22, n=4; ttest cMycMo/CoMo p=0.63) or survival (BCL2: cMycMo/CoMo average 0.977, SEM 0.082, n=8; CoMo/CoMo average 1.09, SEM 0.26, n=4; ttest cMycMo/CoMo p=0.62), even though neural crest genes (FoxD3, Sox10) are downregulated (FoxD3 cMycMo/CoMo average 0.41, SEM0.07, n=6; Sox10 cMycMo/CoMo average 0.50, SEM 0.08, n=6, FoxD3 CoMo/CoMo average = 1.13, SEM 0.13, n=7; Sox10 CoMo/CoMo average 1.03, SEM0.06, n=7; ttest cMycMO/CoMo FoxD3 p=0.00111, Sox10 p= 0.00036). Instead, the CDKI p27 is increased in cMyc morphants (cMycMO/CoMo average 1.39, SEM 0.148, n=7; CoMo/CoMo average 0.93, SEM 0.082, n=4; ttest p=0.0215). This suggests an alternative mechanism for cMyc function other than E-Box binding. (H) Immunostaining for proliferation (PH3) and apoptosis (Caspase 3) in crestospheres transfected with cMycSiRNA. (I–J) Knockdown of cMyc does not alter proliferation or cell survival in crestospheres (PH3 cMycSiRNA average=1.13, SEM 0.17, n= 10; CosiRNA = 1, SEM 0.17; ttest p= 0.672; Caspase (cMycSiRNA average= 1.05, SEM= 0.11, n=10; control = 1, SEM 0.23, n=7, ttest p= 0.68). (K) Knockdown of cMyc increses p27 mRNA levels in crestospheres (cMycsiRNA average=1.21; SEM= 0.06; n=5; CoSiRNA=1, n=6, p=0.025). * marks the treated side. Scale bar 50 μm. See also figure S3A,B.
To gain insight into possible downstream effectors of cMyc, we performed QPCR to examine changes in expression of known cMyc targets associated with proliferation and cell survival (Fig 4G). Surprisingly, neither the cell cycle activator CyclinE or the anti-apoptotic marker BCL2 were affected by the loss of cMyc as compared to the contralateral side treated with control morpholino. Expression values were similar to control embryos treated with control morpholino on both sides. However, in the same embryos, neural crest markers FoxD3 and Sox10 were significantly decreased on the cMyc morpholino treated side, consistent with loss of neural crest cells whereas control embryos had no phenotype. Interestingly, we also noted upregulation of p27 on the cMycMO treated side (Fig 4G), a Cyclin Dependent Kinase Inhibitor (CDKI) associated with cMyc activity (Yang et al., 2001). In light of this, we analyzed the expression of p27 in wild type embryos. The results reveal p27 transcripts throughout the neural tube, albeit with much reduced expression in the premigratory and early migrating neural crest cells (Fig S3B).
To gain mechanistic insight, we repeated these experiments in the crestospheres. Similar to the embryo, the results show that proliferation was unaffected in crestospheres by loss of cMyc (Fig. 4H–I). However, unlike the situation in the embryo, cMyc knockdown in crestospheres did not trigger apoptosis, suggesting that the cell death in the embryo may be a secondary consequence of loss of self-renewal ability in vivo. Despite the rather low transfection efficiency in crestospheres (~30%), we also detected a significant increase in the levels of p27 transcript expression (Fig. 4K).
The QPCR results raised the possibility that, in the neural crest, cMyc did not affect known “canonical” downstream targets whereby cMyc with its binding partner Max activates transcription via E-Box binding. In search of an alternative mechanism for the role of cMyc in cell self-renewal and survival, we investigated the possible involvement of Miz1, a transcription factor that works as a transcriptional repressor when bound to cMyc/Max. Miz1/Myc targets include CDKIs (Adhikary and Eilers, 2005). Moreover, we previously found that Miz1 is required for proper induction of neural crest cells at the neural plate border (Fig. 5A; Kerosuo and Bronner, 2014).
Figure 5. cMyc Requires Miz1 For Its Function In The Neural Crest Cells At Premigratory Stage.

(A) Miz1 knockdown causes defects in neural crest formation in the dorsal neural tube similar to those seen with cMyc. (B) Consistent with this, endogenous cMyc is detected by Western blot after co-immunoprecipitation with a Flag-tagged chicken Miz1 protein (electroporated in low levels 1 μg/μl) from HH stage 8/9 chick neural tubes by using an anti-Flag antibody but not with control IgG (n=3). (C) The requirement for Miz1 binding was tested by performing a rescue experiment by using the wild type full length (FL cMyc) human cMyc and a mutated form (V394Myc) unable to bind to Miz1; (D) both FL cMyc and V394DMyc are expressed at similar levels in the chick embryo after electroporation as verified by a Western blot and compared to a chick control lysate (n=3). (E–G) The FL Myc but not mutated V394DMyc, rescues the cMycMo phenotype assayed by QPCR (average cMycMO alone 0.44, SEM 0.061, n=6; cMycMo rescued with FL cMYC average 0.88, SEM 0.156, n=7; average CoMo on both sides 0.92, SEM 0.032, n=7; cMycMO rescued with V394DMyc average 0.48, SEM 0.988, n=4, ttest MycMO/MycMO + FLcMyc p= 0.0183; ttest CoMo/MycMO p=0.00042; ttest CoMo/cMycMO + V394DMyc p=0.0080) and in situ hybridization showing requirement for Miz1 binding for the function of cMyc in the neural crest cells at premigratory stage. (H–I) The pRetroX-G1-red construct was electroporated to monitor cell cycle. An almost three-fold increase in cells in G1 phase was seen on the cMycMO side as compared to the control side. FITC-labeled morpholinos are shown as a control of equal electroporation on both sides of the embryo (cMycMO 2.9 fold, SEM 0.30; CoMo 1 fold, SEM 0.12; p=6.184E-06; n=12 embryos). * marks the treated side. Scale bar 20 μm. See also figures S3C.
Based on the elevated levels of p27 after loss of cMyc, we hypothesized that cMyc may bind to Miz1 in the premigratory neural crest to form a repressive complex. To test this possibility, we performed co-immunoprecipitation in vivo of neural tubes at the HH stage 8–9. The results show that an antibody to a flag-tagged chicken Miz1 expressed in moderate levels (1μg/μl) precipitates endogenous cMyc, demonstrating that the two transcription factors indeed form a complex in the neural crest cells at the correct stage (Fig 5B).
To examine the importance of the Miz1/cMyc interaction, we tested the ability of either full length cMyc (FL cMyc) or cMyc with a single point mutation that renders it incapable of binding to Miz1 (V394DMyc, Herold et al.; 2002) to rescue the loss of cMyc phenotype. To this end, we simultaneously knocked down cMyc in the premigratory neural crest cells in vivo and added back either full length human cMyc or the mutated V394D version that is unable to bind to Miz1 (Fig 5C). Using the human version of the protein has the additional advantage of being unaffected by the morpholino. Moreover, there is high conservation of the Myc binding domains (Peukert et al., 1997) between human and chicken Miz1 proteins (Fig S3C). We used Western Blots (WB) to verify efficient expression of the human constructs in the HH stage 9 embryos after electroporation at gastrula stages (Fig 5D). As confirmation of the specificity of the morpholino, FL cMyc was able to rescue the morpholino knock-down phenotype and restore normal levels of Sox10 expression by QPCR and in situ hybridization (Figs 5E,F). In contrast, the mutated V394DMyc that is unable to bind to Miz1 was unable to rescue the cMyc loss-of-function phenotype (Figs 5E,G). Furthermore, we used a Fucci construct to analyze possible differences in the cell cycle phases and indeed detected more cells arrested in G1 phase on the cMyc morphant side, thus further supporting the involvement of Myc/Miz1 in cell cycle progression and self-renewal (Figs. 5H–I).
Next, we asked whether overexpression of cMyc in the premigratory/early migrating neural crest cells affects neural crest development. We found that overexpression (2 μg/μl) of FL cMyc caused an increase in neural crest cell production, yielding more premigratory neural crest cells and a prolonged emigration time (Figs. 6A,C–E). The cells emigrated prematurely in ~60% of the embryos (Figs 6A,C), and in roughly half of them, the formation of Sox10 positive cells also continued in the dorsal neural tube on the cMyc over-expressing side at a time when Sox10 positive cells were no longer observed on the control side (Figs. 6D black arrow and S3D). While this phenotype was predominant, a minority of embryos lacked neural crest cells (Fig. 6C), perhaps due to variability in levels of expression, as high expression levels also caused degradation and failure of neural tube closure in the majority of embryos (Fig S3E). At lower levels (0.5 μg/μl), neural crest cells also emigrated from the neural tube earlier than on the control side (Fig S3F), similar to effects observed with higher levels. In contrast to FL cMyc, embryos over-expressing the V394DMyc mutant (2μg/μl) had no phenotype and only a small percentage (11%) had neural tube closure defects (Figs 6B,C,F and S3D,E). These results suggest that the ability to bind to Miz1 is required for the effect of cMyc on premigratory neural crest cells.
Figure 6. cMyc Overexpression Increases Self-Renewal But Miz1 Binding Mutant V394dmyc Causes No Phenotype In The Neural Crest Cells.

(A–C) Overexpression of FL Myc causes premature emigration and an increased number of neural crest cells in roughly ~60% (n=25 for stages 9–11 combined) of the embryos whereas V394DMyc doesn’t induce a phenotype (n=27 for stages 9–11 combined). (D) We also observed prolongation of neural crest production by the dorsal neural tube on the Myc overexpressing side (black arrow) at HH stage 10, (E) which results in increased numbers of neural crest cells that have migrated further laterally (white arrow) than on the control side at stage 11. (F) V394DMyc overexpression does not induce a phenotype at a later stage either. * marks the overexpression side. (G) Overexpression of FL cMyc, but not V394DMyc, increases self-renewal capacity in crestospheres (FL= 1.6 fold, SEM= 0.08, n=6; V394D= 1.1, SEM=0.05, n=6; control=1, SEM= 0.05, n=6; ttest FL/Co p= 0.0002; FL/V394D p=0.0007; V394D/Co p= 0.17). (H–I) Proliferation (PH3) or cell survival (Caspase 3) are not affected after overexpression of cMyc FL or the mutated V394DMyc that has lost Miz1 binding ability (PH3: FL cMyc= 0.9 fold, SEM=0.06, n=14 spheres; V394D= 1.25, SEM=0.16, n=14 spheres; Control= 1, SEM=0.26, n=17 spheres;ttest for all p>0.5); and for Caspase 3: cMyc FL=0.96; SEM=0.06; n=14 spheres; V394D= 1.29, SEM=0.13, n=14 spheres; control= 1, SEM=0.08, n=17 spheres; ttest for all p>0.5). Scale bar 20 μm. See also Figures S3D–G.
Finally, we investigated whether overexpression of either FL cMyc or V394DMyc mutant had an impact on self-renewal, cell proliferation or cell survival in crestospheres. Similar to observations in the embryo, overexpression of FL cMyc clearly showed a significant increase in self-renewal capacity with the FL but not V394D Myc. Neither proliferation, as assayed by PH3 staining, or cell survival were affected. These results suggest that self-renewal and proliferation may be regulated via different mechanisms.
DISCUSSION
Neural crest cells are a population of multipotent cells with broad developmental potential in vivo, exceeded only by the inner cell mass cells of the blastocyst in their ability to contribute to multiple cell types characteristic of “all three germ layers”. Understanding neural crest cell function is highly clinically relevant given the large spectrum of neural crest derived disorders, including craniofacial, cardiovascular, peripheral nervous system and enteric syndromes. Aggressive cancers like neuroblastoma and melanoma also originate from this cell population (Takahashi et al., 2013). Furthermore, due to its broad range of derivatives, neural crest stem cells hold great promise for regenerative medicine. Multiple in vitro approaches have been used to understand cues required for their development and stem cell maintenance (Kerosuo et al., 2015; Lee et al., 2010; Lee et al., 2007). Neural crest cells can be derived from human embryonic stem (ES) cells or postnatal fibroblast cells in vitro; however, these do not resemble premigratory neural crest cells within the neural tube. Rather, these cultures exhibit various features of late, committed neural crest cells intermixed with multipotent cells (Kim YJ, 2014). Despite the importance of understanding these initial regulatory events, the regulation of the premigratory neural crest stem cell pool is poorly understood. To address this question, we have used an in vivo approach in the chick embryo combined with in vitro premigratory ‘crestospheres’ (Kerosuo et al., 2015). Here, we show that cMyc is involved in regulating the size of the premigratory neural crest stem cell pool as well as the duration of the neural crest cell emigration period. Our results thus provide new in vivo insight into the timing and maintenance of neural crest production.
We show that cMyc is expressed in neural crest cells in the dorsal neural tube at stages correlating with initiation of EMT (Figs 1 and S1). This contrasts with observations in the frog where there appears to have been a paralog switch between cMyc and nMyc compared with the chick. In Xenopus embryos, cMyc is expressed much earlier at the neural plate border where it appears to play a role in neural crest induction, but not in later in maintenance of the stem cell pool (Bellmeyer et al., 2003). In chick, we find that knockdown of cMyc using a translation blocking morpholino (cMycMO) results in fewer neural crest cells and a reduction in neural crest markers, Sox10, FoxD3 and Pax7. While the expression levels of these markers in individual cells was unchanged (Fig 2), indicating that neural crest induction occurred normally, the numbers of neural crest cells appeared decreased. Importantly, cMyc knockdown resulted in premature cessation of the neural crest cell production by the dorsal neural tube (Fig 2), likely reflecting a reduced self-renewal capacity. Conversely, we observed prolonged neural crest production and emigration after cMyc overexpression (Fig 6). Using a complementary in vitro approach of crestospheres (Kerosuo et al., 2015), we noted a reduction in the self-renewal ability of premigratory neural crest cells due to cMyc knockdown (Fig 3). Reciprocally, self-renewal was increased upon over-expression of cMyc (Fig 6). This is consistent with the strong connection previously noted between cMyc and maintenance of self-renewal in several other types of stem cells (Chappell and Dalton, 2013; Kerosuo et al., 2008; Murphy et al., 2005; Takahashi and Yamanaka, 2006). Loss of cMyc seemed to affect the numbers of the multipotent stem cells, rather than affecting capacity to maintain a certain lineage, as shown by the fact that cMyc deficient neural crest cells are still able to form all major types of neural crest derivatives (Fig 3). Previous studies point to cMyc’s pivotal role in adhesion related changes during exit from the hematopoietic and epidermal stem cell niches (Frye et al., 2003; Varlakhanova et al., 2010; Waikel et al., 2001; Wilson et al., 2004). Consistent with this, the timing of cMyc expression correlates with onset of neural crest EMT that is accompanied by adhesion changes in which Miz1 has been implicated (Kerosuo and Bronner, 2014). Moreover, in keratinocytes, Miz1 binding is required for these changes (Gebhardt et al., 2006) in line with the results shown here. However, a direct link between cMyc-regulated self-renewal and cell adhesion changes during neural crest EMT has yet to be established.
We detected an increase in apoptotic cells on the cMyc morpholino-electroporated side, resulting in a thinner dorsal neural tube. On the other hand, cell proliferation, perhaps representing the most commonly known function of cMyc, was not significantly altered. Moreover, levels of known cMyc downstream targets that promote proliferation or cell survival were unaffected (Fig 4). Therefore, we investigated alternative mechanisms whereby cMyc might affect target gene expression by interacting with Miz1 or RAREs (Adhikary and Eilers, 2005; Uribesalgo et al., 2012). Indeed, we detected upregulated levels of p27, a cyclin dependent kinase inhibitor that suggested a possible connection with Miz1. Down-regulation of p27 has been associated previously with cMyc albeit not been linked to Miz1 (Yang et al., 2001; Zindy et al., 2006), and highlights the possibility that additional CDKIs may be involved in maintenance of the neural crest stem cell pool. To test a possible interaction between cMyc and Miz1, we performed rescue experiments by co-electroporating cMyc morpholino with either wild type or a mutated version of cMyc incapable of binding to Miz1. Whereas the former gave efficient rescue, demonstrating the specificity of the phenotype, the latter was unable to rescue the cMyc deficient phenotype (Fig 5), consistent with cMyc binding to Miz1 and repressing its activating function. Moreover, co-immunoprecipitation showed that cMyc and Miz1 indeed bind to one another in the chick neural tube during premigratory neural crest stages. We also detected an increase of neural crest cells in G1 cell cycle arrest after loss of cMyc (Fig 5), which we speculate triggers apoptosis of these cells as shown in Figure 4. Although a similar increase in apoptosis was not observed in crestospheres (Fig. 4), this may be due to the relatively low transfection efficiency in vitro compared with much higher penetration of morpholino in the embryo. In addition to being important for maintenance of the stem cell pool shown here, Miz1 also plays an earlier role in neural crest specification that is independent of cMyc and occurs prior to expression of the latter (Kerosuo and Bronner, 2014). Consistent with these results, Miz1 is known to form repressive complexes with transcription factors other than cMyc (Basu et al., 2009; Phan et al., 2005). Interestingly, overexpression of the mutated cMycV394D form that is incapable of binding to Miz1 had no effect on neural crest development or self-renewal (Figs 5 and 6), further strengthening the hypothesis that the action of cMyc in the premigratory neural crest cells is specific to its interaction with Miz1.
It has been shown that heterogeneous endogenous levels of cMyc expression are key for creating cell competition for fitness in mouse epiblast cells, resulting in apoptotic elimination of those cells with lowest cMyc levels (Claveria et al., 2013). This phenomenon also is seen in the Drosophila wing after ectopic expression (Johnston, 2009) suggesting a widely utilized mechanism for stem cell niche homeostasis during development. Similarly, we show that transient knockdown or overexpression of cMyc via electroporation, which produces unequal levels of cMyc in neighboring cells, alters the size of the neural crest precursor pool. Our results implicate cMyc in regulating the overall amount of neural crest production in the embryo. We show that cMyc acts in the neural crest via binding to Miz1, thus changing it from an activator to a repressor.
In summary, our results reveal a novel mechanism of self-renewal regulation in the neural crest population at premigratory stages. This involves a non-canonical function for the proto-oncoprotein cMyc: despite the fact that cMyc plays a variety of roles in development and cancer, its primary and perhaps sole function in the premigratory neural crest seems to be in maintenance of self-renewal. This is distinct from its role at later stages, for example where cMyc has been show to promote neuronal differentiation in the neural tube in vivo (Zinin et al., 2014). The neural crest is often characterized as a transient cell population that emigrates from the neural tube and differentiates into multiple derivatives. Despite its ephemeral nature, our results reveal the importance of the self-renewal capability of premigratory cells, regulated by cMyc/Miz, in determining the size of the neural crest stem cell pool.
Experimental Procedures
In situ hybridization
Embryos were fixed with 4% paraformaldehyde, washed with PBS/0.1% Tween, dehydrated in MeOH, and stored at −20°C. The avian cMyc probe was made by using cEST191o11 (www.chick.manchester.ac.uk) and the Sox10, FoxD3, Dlx-5 and Sox2 probes were made by cloning respective genes to DNA vectors from RT-PCR products made by using chicken whole embryo cDNA as template. Whole-mount in situ hybridization was performed as described (Acloque et al., 2008). The digoxigenin-conjugated RNA probes were visualized by using anti dig-AP antibody (11093274910, Roche Diagnostics GmbH in 1/2000) and NCB/BCIP (11383213001 and 11383221001, Roche Diagnostics GmbH). Embryos were sectioned at 12– 18 μm.
Morpholino knockdown and HH4 electroporation of the chicken embryos
FITC-conjugated morpholinos were purchased from Gene Tools LLC (www.gene-tools.com). The cMyc (CTGCGAGACGGAGCGAGGCAATAAT), and Miz-1 (AACTGGGACAGCTGCTGCAAGCCAC) translation blocking morpholinos were targeted to the respective 5’ UTR in close proximity of the ATG and a control morpholino was designed to assure lack of non-specific effects from electroporation (CTGCGATGAAAAACACGGGAGCACA). The MOs were diluted to a 1–2mM concentration and electroporated together with an empty pGAG vector as carrier DNA (1 μg/μl). The morpholino was injected as two-sided injections with control morpholino on the contralateral side. The electroporation was carried out as previously described (Sauka-Spengler T, 2008). Briefly, the chicken embryos were collected on Whatman filter papers and electroporated at Hamburger and Hamilton (HH) stage 4 by using 5.3V and 5 pulses (50mA/100mA) and incubated on individual petri dishes (Falcon 1008 35×10mm) in thin albumin until they reached the desired stage.
Q-PCR
mRNA was isolated from stage 8–9 embryos by using the Ambion® RNAqueous-Micro Kit by individually collecting neural tube halves from the cMycMo treated and the contralateral CoMo treated sides, respectively. The control embryos were treated with CoMo on both sides. The results are shown as the relative expression fold of the treated side versus the control side, or treated crestospheres versus respective controls, and were analyzed using the ΔΔCT method (Livak and Schmittgen, 2001). Results from 3–8 individual embryos were pooled and are shown as average values. The error bars represent the standard error of mean (SEM). Primers were designed to target an exon-exon boundary and their amplification rate was verified as linear within the margin of a +/− 10% amplification rate change between points in the logarithmically diluted cDNA standard curve. The following primers were used: Sox10fwd AGCCAGCAATTGAGAAGAAGG; Sox10rev GAGGTGCGAAGAGTTGTCC; FoxD3fwd TCTGCGAGTTCATCAGCAAC; FoxD3rev TTCACGAAGCAGTCGTTGAG; cMycfwd GCAGCGACTCGGAAGAAGAACAAGAA cMycrev TGCTGGATTCAGACTCGTTCGCTT; BCL2Fwd CGGCAACAGTATGAGGCCTTTGTT BCL2rev ATAAGCGCCAAGAGTGATGCAAGC and P27fwd AGCCCGAGACGACATCAAACGTAA; P27rev TTTATATCTTCCTGGCTTCACCGCCC with GaphdHfwd1 ATCACTATCTTCCAGGAGCGTG; GapdHrev1 AGCACCACCCTTCAGATGAG; as well as CyclinEfwd TACCGTGCCTGTTTGTCTCTGGAA and CyclinErev AGAGGCTTTGAAATGTCGCCTTGC with GapdHfwd2 CAGAGGACCAGGTTGTCTCC and GapdHrev2 CAGGGTTGCTGTATCCAAAC.
Immunostaining, Western Blot and Immunoprecipitation
The following primary antibodies were used for immunofluorescence: Caspase 3 (1/200, AF835 R&D Systems) and Pax7 (1/10; Developmental Studies Hybridoma Bank, University of Iowa), Sox2 (rb), Mitf (1/500, Abcam ab122982), Tuj-1 (1/400, Covance MMS-435P), SMA (1/1000, Sigma A5228), BLBP (1/200, Millipore ABN14), Cherry (1/150, Clontech Living colors 632543), Histone H3 Phospho S10 (1/2000, Abcam 14955). Antibodies were incubated in PBS/0.2% tween, 1% DMSO and 10% goat serum with whole mount embryos for 2 days in +4°C, or alternatively on 12μm cryosections over night, and by using the respective secondary antibodies by Alexa (1/1000; www.bdbiosciences.com). The cells were imaged using fluorescence microscopy (Zeiss Axioscope 2 and Zeiss ApoTome.2)
For Western blot, SDS-PAGE was performed by using Bolt™ 4–12% or 8%, respectively, Bis-Tris Plus gels (NW04120Box, NW0080Box, Invitrogen) with the Novex electrophoresis chamber system (Life Technologies) followed by a transfer onto the nitrocellulose filter by using the Criterion Blotter (Bio-Rad). The filters were blocked with 4% milk/PBS-0.2% Tween for 1h RT and stained with primary Ab O/N +4*C, washed 5x with PBS-0.2% Tween RT and incubated with the secondary Ab in block 1.5h RT, washed 5x and illuminated by using ECL™ (RPN2232 Amersham). The following primary antibodies were used for Western Blot: Flag (1/4000; M2 F1804 Sigma), cMyc (c-8, 1/50, Santa Cruz SC-41), cMyc (N262, 1/3000 Santa Cruz SC-764), β-Tubulin (1/10000 Sigma T5168) ribosomal S6 (1/5000 cell Signalling 5G10). The following secondary antibodies were used for Western Blot: peroxidase labeled Gt anti-Rb IgG (KPL 074-1806, 1/20 000), Gt anti-Ms IgG (KPL074-1506; 1/20 000). For the IP, Pre-swollen protein G-agarose beads (Sigma P7700) were coated with 20 μg of Flag antibody (M2 F1804 Sigma) or Ms IgG1 (Santa Cruz SC-3877) o/n +4*C, washed and added onto lysed stage 8–9 (5–7 som) chick embryo neural tubes (from anterior tip up to somite 2-level in 900 μl lysis buffer consisting of 50mM Tris-HCL pH7.4, 150mM NaCl, 1% NP-40 with Protease Inhibitor Cocktail (Complete EDTA-free, Roche 11873580001)) and incubated RT*C for 3h. The beads were then washed 6x with lysis buffer and beads were detached by using 50 μl 8M Urea/2.5% SDS (30min 65*C), and the supernatant was run on a SDS-Page gel as described above and blotted with the anti-Myc ab.
Cell survival and proliferation
Cell survival and proliferation were measured by counting the percentage of Caspase3 and PH3 immunopositive cells on cryosections or z-stack images on whole crestospheres, respectively, from all Dapi-stained nuclei in the dorsal neural tube at premigratory neural crest stage. MycMo electroporated side was compared to CoMo side. The results from 10–20 sections from each embryo were pooled to represent each embryo (n). For the crestosphere experiments, 2–4 z-stack slices per sphere (at least 5μm apart from each other) were counted.
Expression constructs
The coding sequence of the human cMyc as well as the mutated cMycV394D (Herold et al., 2002), a kind gift from Martin Eilers, were subcloned, and the chicken Miz1 gene was cloned into the pCIG-RFP chicken expression vector. For Miz-1, the 5′ end was tagged with a 1x Flag sequence that was inserted into the PCR primers and chicken cDNA was used as a template (Miz1gallusFwd with Flag/Bstb1 TTTTTCGAAGCCACCATGGACTACAAGGACGACGACGACAAGGCAGCAatggatttcccccagcacag, Miz1gallusRev with NheI TTTT GCTAGC ttttacaccgtttccaagca). The G1 cell cycle arrest was monitored by expression of pRetroX-G1-red (Clontech 631463) by electroporation (1 μg/μl) together with either cMycMO or CoMO, respectively, the expression was enhanced by using an anti-Cherry antibody, and the percentage of positive cells was measured as compared to Dapi-staining on sections as described above.
Self-renewal assay, siRNA transfection and DNA transfection into crestospheres
Self-renewal was measured in crestospheres by counting the ability of single cells to form new spheres after a week in crestosphere stem cell culture as described in figure 3A (Kerosuo et al., 2015). Accumax (innovative cell technologies) was used (15min RT*C accompanied with trituration with a p1000 pipette) to dissociate the crestospheres into single cells. The custom designed siRNA for avian cMyc (Ambion, designed to target the following sequences #1 GCAUCAGAGGAGCACUGUA and #2 GCAGGGUCCUCAAACAGAU) as well as the control (Silencer Select Negative Control #1, Ambion 4390843) was transfected by using either the HiPerfect (Quiagen 301704) or Lipofectamine 3000 (L3000008 Invitrogen) kit, respectively, by using 750ng siRNA/200 000 cells incubated for 3 days before performing the colony forming self-renewal assay. DNA was transfected according to instructions by using the Lipofectamine 3000 (L3000008 Invitrogen) kit with 1.5μl Lipofectamine and 500ng of DNA per each 24-well plate well, and the self-renewal assay was performed on day 3 after the transfection.
Explant cultures
Embryos were electroporated at gastrula (HH4) stage with either cMycMo or CoMo on one side of the embryo and grown on filter papers until they reached premigratory (4–5 som) stage. Halves of cranial neural tubes were dissected out and placed on fibronectin coated (5μg/ml in PBS 2h RT) cover glasses and cultured in a DMEM with 1% FBS for 1 week (for smooth muscle and melanocyte differentiation); or alternatively laminin coated (50μg/ml 30min 37°C), cover glasses with DMEM complemented with N2 supplement (for neural and glial differentiation), fixed 20min in 4% PFA at RT and immunostained.
Statistical methods
All averages represent the data pooled from at least 3 biological samples (n), either individual embryos or individually prepared batches of crestospheres. Error bars represent the standard mean of error (SEM) and the p-values were calculated by using student’s ttest; * represents <0.05 and **<0.01.
Supplementary Material
Acknowledgments
This work was funded by NIH grants HD037105 and DE024157 (to M.E.B.) and by fellowships from The Sigrid Juselius Foundation, Finnish Cultural Foundation, Jane and Aatos Erkko Foundation, Väre Foundation, and Ella and Georg Ehrnrooth Foundation (to L.K.). We thank Drs. Crystal Rogers, Stephen Green and Michael Piacentino and the entire Bronner lab members for technical advice.
Footnotes
Author contributions
L.K. and M.E.B conceived and designed the experimental approach. L.K. performed the experiments and analyzed the data. L.K. and M.E.B. wrote the manuscript.
References
- Acloque H, Wilkinson DG, Nieto MA. Chapter 9 In Situ Hybridization Analysis of Chick Embryos in Whole-Mount and Tissue Sections. In: Marianne B-F, editor. Methods Cell Biol. Academic Press; 2008. pp. 169–185. [DOI] [PubMed] [Google Scholar]
- Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nature reviews Molecular cell biology. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- Basu S, Liu Q, Qiu Y, Dong F. Gfi-1 represses CDKN2B encoding p15INK4B through interaction with Miz-1. Proc Natl Acad Sci U S A. 2009;106:1433–1438. doi: 10.1073/pnas.0804863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmeyer A, Krase J, Lindgren J, LaBonne C. The protooncogene c-myc is an essential regulator of neural crest formation in xenopus. Dev Cell. 2003;4:827–839. doi: 10.1016/s1534-5807(03)00160-6. [DOI] [PubMed] [Google Scholar]
- Bosserhoff AK. Novel biomarkers in malignant melanoma. Clin Chim Acta. 2006;367:28–35. doi: 10.1016/j.cca.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Bung R, Worsdorfer P, Thier MC, Vogt K, Gebhardt M, Edenhofer F. Partial dedifferentiation of murine radial glia type neural stem cells by Brn2 and c-Myc yields early neuroepithelial progenitors. J Mol Biol. 2015 doi: 10.1016/j.jmb.2015.10.028. [DOI] [PubMed] [Google Scholar]
- Chappell J, Dalton S. Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb Perspect Med. 2013;3:a014381. doi: 10.1101/cshperspect.a014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveria C, Giovinazzo G, Sierra R, Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39–U53. doi: 10.1038/nature12389. [DOI] [PubMed] [Google Scholar]
- Dong J, Sutor S, Jiang G, Cao Y, Asmann YW, Wigle DA. c-Myc regulates self-renewal in bronchoalveolar stem cells. PLoS One. 2011;6:e23707. doi: 10.1371/journal.pone.0023707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin E, Coelho-Aguiar J. Isolation and differentiation properties of neural crest stem cells. Cytometry Part A. 2013;83:38–47. doi: 10.1002/cyto.a.22098. [DOI] [PubMed] [Google Scholar]
- Eilers M, Eisenman R. Myc’s broad reach. Genes Dev. 2008;22:2755–2766. doi: 10.1101/gad.1712408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredlund E, Ringner M, Maris J, Påhlman S. High Myc pathway activity and low stage of neuronal differentiation associate with poor outcome in neuroblastoma. Proc Natl Acad Sci U S A. 2008;105:14094–14099. doi: 10.1073/pnas.0804455105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Gardner C, Li ER, Arnold I, Watt FM. Evidence that Myc activation depletes the epidermal stem cell compartment by modulating adhesive interactions with the local microenvironment. Development. 2003;130:2793–2808. doi: 10.1242/dev.00462. [DOI] [PubMed] [Google Scholar]
- Gebhardt A, Frye M, Herold S, Benitah S, Braun K, Samans B, Watt F, Elsässer H-P, Eilers M. Myc regulates keratinocyte adhesion and differentiation via complex formation with Miz1. The Journal of cell biology. 2006;172:139–149. doi: 10.1083/jcb.200506057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, Syvaoja J, Saluz H-P, Haenel F, Eilers M. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell. 2002;10:509–521. doi: 10.1016/s1097-2765(02)00633-0. [DOI] [PubMed] [Google Scholar]
- Hong S-K, Tsang M, Dawid I. The mych gene is required for neural crest survival during zebrafish development. PLoS ONE. 2008;3:e2029–e2029. doi: 10.1371/journal.pone.0002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA. Competitive interactions between cells: death, growth, and geography. Science. 2009;324:1679–1682. doi: 10.1126/science.1163862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerosuo L, Bronner M. Biphasic influence of Miz1 on neural crest development by regulating cell survival and apical adhesion complex formation in the developing neural tube. Mol Biol Cell. 2014;25:347–355. doi: 10.1091/mbc.E13-06-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerosuo L, Bronner-Fraser M. What is bad in cancer is good in the embryo: importance of EMT in neural crest development. Semin Cell Dev Biol. 2012;23:320–332. doi: 10.1016/j.semcdb.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerosuo L, Nie S, Bajpai R, Bronner ME. Crestospheres: Long-Term Maintenance of Multipotent, Premigratory Neural Crest Stem Cells. Stem Cell Reports. 2015;5:499–507. doi: 10.1016/j.stemcr.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerosuo L, Piltti K, Fox H, Angers-Loustau A, Häyry V, Eilers M, Sariola H, Wartiovaara K. Myc increases self-renewal in neural progenitor cells through Miz-1. J Cell Sci. 2008;121:3941–3950. doi: 10.1242/jcs.024802. [DOI] [PubMed] [Google Scholar]
- Khan FH, Pandian V, Ramraj S, Aravindan S, Herman TS, Aravindan N. Reorganization of metastamiRs in the evolution of metastatic aggressive neuroblastoma cells. BMC Genomics. 2015;16:501. doi: 10.1186/s12864-015-1642-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khudyakov J, Bronner-Fraser M. Comprehensive spatiotemporal analysis of early chick neural crest network genes. Dev Dyn. 2009;238:716–723. doi: 10.1002/dvdy.21881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, LH, Li Z, Oh Y, Kovlyagina I, Choi IY, Dong X, Lee G. Generation of Multipotent Induced Neural Crest by Direct Reprogramming of Human Postnatal Fibroblasts with a Single Transcription Factor. Cell Stem Cell. 2014;15:1–10. doi: 10.1016/j.stem.2014.07.013. [DOI] [PubMed] [Google Scholar]
- Kwan KY, Shen J, Corey DP. C-MYC transcriptionally amplifies SOX2 target genes to regulate self-renewal in multipotent otic progenitor cells. Stem Cell Reports. 2015;4:47–60. doi: 10.1016/j.stemcr.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Chambers SM, Tomishima MJ, Studer L. Derivation of neural crest cells from human pluripotent stem cells. Nat Protoc. 2010;5:688–701. doi: 10.1038/nprot.2010.35. [DOI] [PubMed] [Google Scholar]
- Lee G, Kim H, Elkabetz Y, Al Shamy G, Panagiotakos G, Barberi T, Tabar V, Studer L. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat Biotechnol. 2007;25:1468–1475. doi: 10.1038/nbt1365. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Murphy M, Wilson A, Trumpp A. More than just proliferation: Myc function in stem cells. Trends Cell Biol. 2005;15:128–137. doi: 10.1016/j.tcb.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Peukert K, Staller P, Schneider A, Carmichael G, Hänel F, Eilers M. An alternative pathway for gene regulation by Myc. EMBO J. 1997;16:5672–5686. doi: 10.1093/emboj/16.18.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan R, Saito M, Basso K, Niu H, Dalla Favera R. BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6:1054–1060. doi: 10.1038/ni1245. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, BM Gain-and loss-of-function approaches in the chick embryo. Methods Cell Biol. 2008:237–256. doi: 10.1016/S0091-679X(08)00212-4. [DOI] [PubMed] [Google Scholar]
- Sauka-Spengler T, Bronner-Fraser M. A gene regulatory network orchestrates neural crest formation. Nature reviews Molecular cell biology. 2008;9:557–568. doi: 10.1038/nrm2428. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Sipp D, Enomoto H. Tissue interactions in neural crest cell development and disease. Science. 2013;341:860–863. doi: 10.1126/science.1230717. [DOI] [PubMed] [Google Scholar]
- Uribesalgo I, Benitah SA, Di Croce L. From oncogene to tumor suppressor: the dual role of Myc in leukemia. Cell Cycle. 2012;11:1757–1764. doi: 10.4161/cc.19883. [DOI] [PubMed] [Google Scholar]
- Varlakhanova NV, Cotterman RF, deVries WN, Morgan J, Donahue LR, Murray S, Knowles BB, Knoepfler PS. myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation. 2010;80:9–19. doi: 10.1016/j.diff.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waikel RL, Kawachi Y, Waikel PA, Wang XJ, Roop DR. Deregulated expression of c-Myc depletes epidermal stem cells. Nat Genet. 2001;28:165–168. doi: 10.1038/88889. [DOI] [PubMed] [Google Scholar]
- Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by N-myc: promotion of ventral migration and neuronal differentiation. Development. 1997;124:1953–1962. doi: 10.1242/dev.124.10.1953. [DOI] [PubMed] [Google Scholar]
- Wei K, Chen J, Akrami K, Galbraith G, Lopez I, Chen F. Neural crest cell deficiency of c-myc causes skull and hearing defects. Genesis. 2007;45:382–390. doi: 10.1002/dvg.20304. [DOI] [PubMed] [Google Scholar]
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, MacDonald HR, Trumpp A. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 2004;18:2747–2763. doi: 10.1101/gad.313104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu Mourez R, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene. 2001;20:1688–1702. doi: 10.1038/sj.onc.1204245. [DOI] [PubMed] [Google Scholar]
- Zindy F, Knoepfler PS, Xie S, Sherr CJ, Eisenman RN, Roussel MF. N-Myc and the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 coordinately regulate cerebellar development. Proc Natl Acad Sci U S A. 2006;103:11579–11583. doi: 10.1073/pnas.0604727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinin N, Adameyko I, Wilhelm M, Fritz N, Uhlen P, Ernfors P, Henriksson MA. MYC proteins promote neuronal differentiation by controlling the mode of progenitor cell division. Embo Rep. 2014;15:383–391. doi: 10.1002/embr.201337424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.