

Abstract
Among the 22 fibroblast growth factors (FGFs), FGF21 has now emerged as a key metabolic regulator. However, the mechanism whereby FGF21 mediates its metabolic actions per se remains largely unknown. Here, we show that FGF21 represses mammalian target of rapamycin complex 1 (mTORC1) and improves insulin sensitivity and glycogen storage in a hepatocyte-autonomous manner. Administration of FGF21 in mice inhibits mTORC1 in the liver, whereas FGF21-deficient mice display pronounced insulin-stimulated mTORC1 activation and exacerbated hepatic insulin resistance (IR). FGF21 inhibits insulin- or nutrient-stimulated activation of mTORC1 to enhance phosphorylation of Akt in HepG2 cells at both normal and IR condition. TSC1 deficiency abrogates FGF21-mediated inhibition of mTORC1 and augmentation of insulin signaling and glycogen synthesis. Strikingly, hepatic βKlotho knockdown or hepatic hyperactivation of mTORC1/ribosomal protein S6 kinase 1 abrogates hepatic insulin-sensitizing and glycemic-control effects of FGF21 in diet-induced insulin-resistant mice. Moreover, FGF21 improves methionine- and choline-deficient diet-induced steatohepatitis. Conclusions: FGF21 acts as an inhibitor of mTORC1 to control hepatic insulin action and maintain glucose homeostasis, and mTORC1 inhibition by FGF21 has the therapeutic potential for treating IR and type 2 diabetes.
Fibroblast growth factor 21(FGF21) has increasingly gained attention as a novel metabolic regulator. FGF21 is physiologically induced by prolonged fasting and feeding of a ketogenic diet(1) in rodents and functions as a hormone to regulate carbohydrate and lipid metabolism.(2) FGF21 binds to FGF receptor (FGFR) and the scaffold protein, βKlotho (KLB),(3) and activates FGFR substrate 2a and extracellular signal-regulated kinase 1 and 2 (ERK1/2).(4) Whereas FGFRs are widely expressed in most tissues, tissue specificity of FGF21 actions is determined by distribution of the coreceptor βKlotho.(5) The liver, where βKlotho is abundantly expressed,(6) is the major tissue mediating FGF21’s beneficial effects. FGF21 inhibits diet- and fasting-induced hepatic steatosis (HS),(1,7,8) induces ketogenesis,(1,9,10) enhances hepatic fatty acid oxidation and tricarboxylic acid cycle flux through peroxisome proliferator-activated receptor gamma coactivator 1 alpha,(3,7,11) and protects against methionine- and choline-deficient (MCD) diet-induced lipotoxicity.(11)
Pharmacological administration or transgenic overexpression of FGF21 has been shown to ameliorate hyperglycemia, hyperinsulinemia, and insulin resistance (IR) in obese rodents and monkeys.(3) Several lines of evidence support FGF21’s effects on improving insulin sensitivity in the liver. FGF21 gain of function protects against hepatic IR in diet-induced obese (DIO) mice.(8) Hyperinsulinemic clamp studies, the gold standard for assessing insulin action in vivo, have demonstrated that FGF21 improves hepatic insulin sensitivity and promotes glucose disposal in various rodent models, such as ob/ob mice, DIO mice, and Zucker fatty rats, in which FGF21 appears to have no effects on glucose uptake in adipose tissue or muscle.(12,13) Moreover, whole-body FGF21-deficient mice are prone to late onset of glucose intolerance,(14) and liver-specific FGF21 knockout mice show impaired insulin sensitivity.(15) However, it is currently unclear how FGF21 regulates hepatic and systemic glucose homeostasis.
Although adipose-produced hormone adiponectin has been previously shown to mediate some metabolic effects of FGF21, it is important to note that glycemic effects of acute administration of FGF21 remain largely intact in adiponectin−/− mice,(16) suggesting an adiponectin-independent mechanism that may contribute to glucose-lowering effects of FGF21. Recently, the mammalian target of rapamycin complex 1 (mTORC1) and its downstream effector, ribosomal protein S6 (S6) kinase 1 (S6K1) have been revealed as critical regulators for nutrient overload-induced pathogenesis of IR and type 2 diabetes (T2D).(17) S6K1 phosphorylates insulin receptor substrate 1 (IRS-1) at Ser1101(18) and Ser302.(19) Phosphorylation of IRS-1 on serine residues causes a disruption of phosphatidylinositol 3-kinase and has emerged as a key molecular basis underlying induction of IR. However, whether mTORC1 mediates effects of FGF21 on insulin sensitivity remains unknown.
Our recent studies demonstrate that sirtuin 1 (SIRT1) and retinoic acid receptor beta act as potent upstream regulators of FGF21.(7,10) In this study, we utilized pharmacological and genetic approaches to characterize that the mTORC1 complex serves as a novel downstream target of FGF21. These in vivo and in vitro studies illustrate that (1) FGF21 inhibits nutrient- and hormonal-induced hepatic mTORC1 activity; (2) hepatic βKlotho is necessary for FGF21 to inhibit mTORC1 and enhance insulin sensitivity; (3) FGF21 down-regulates mTORC1 in a tuberous sclerosis complex (TSC)-dependent manner; and (4) FGF21 enhances hepatic insulin sensitivity through mTORC1 inhibition.
Materials and Methods
ANIMALS
FGF21 knockout mice were described.(16) Tamoxifen-inducible TSC1 knockout mice were generated by crossing Cre-ER recombinase transgenic mice with floxed TSC1 mice(20) (The Jackson Laboratory, Bar Harbor, ME). Male C57BL/6 mice at 8 weeks of age were purchased from Shanghai Laboratory Animal Co. (Shanghai, China). All animal experimental protocols were approved by the institutional animal care and use committee at Institute for Nutritional Sciences, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China).
LIVER HISTOLOGICAL ANALYSIS
Livers were fixed in 10% phosphate-buffered formalin acetate at 48C overnight and embedded in paraffin wax. Paraffin sections (5 μm) were cut and mounted on glass slides for hematoxylin and eosin (H&E) staining as described(21) and for Periodic Acid-Schiff (PAS) staining and Sirius Red staining according to the manufacturer’s instructions (Maixin Biotech, Fujan, China).
SHORT-HAIRPIN RNA KNOCKDOWN
Adenoviruses (Ad) expressing short-hairpin RNAs (shRNAs) targeting βKlotho (shKLB) and negative control (shNC) were purchased from GenePharma (Shanghai, China). The following shRNA targeting sequences were used: shKLB-17, GCGACTACCCTGAGTTCATGA; shKLB-18, GCAATCTGTCCAAAGTTAACA; shKLB-19, GCTCTGGATCACCATCAATGA; shKLB-20, GGAATACGATGACCCTCAAAT; shNC, TTCTCCGAACGTGTCACGT.
IN VIVO ADENOVIRAL GENE TRANSFER
Adenovirus-mediated gene transfer in livers of C57/ BL/6 mice was accomplished as described.(7,10) Adenoviruses (5 × 109 plaque-forming units [PFU] ~ 1 × 1010 PFU/mouse) were delivered into mice by tail vein injection. Two weeks postinjection, animals were killed and tissues were rapidly collected.
STATISTICAL ANALYSIS
Data are expressed as mean ± SEM. Statistical significance was evaluated using the unpaired two-tailed Student t test and among more than two groups by analysis of one-way analysis of variance. Differences were considered significant at a P value <0.05.
Additional materials and methods are detailed in the Supporting Information.
Results
HEPATIC KNOCKDOWN OF βKLOTHO ABROGATES FGF21’S EFFECTS ON IMPROVING INSULIN SENSITIVITY AND GLYCOGEN STORAGE IN LIVERS OF HIGH-FAT, HIGH-SUCROSE DIET-FED MICE
To investigate the relative contribution of FGF21 to insulin signaling and glucose homeostasis, hepatic knockdown of βKlotho using adenovirus-mediated shRNA (Ad-shKLB) was performed. shKLB-18, which produced the greatest knockdown efficiency, was chosen for the following in vivo experiments (Supporting Fig. S1A,B). We next administrated FGF21 to Ad-shKLB-injected mice fed on a type 2 diabetogenic diet composed of high fat, high sucrose (HFHS).(21) Strikingly, as shown in Fig. 1A, Ad-shKLB largely abrogated insulin-sensitizing effects of FGF21, including fasting glucose, plasma insulin levels, and the calculated value for the homeostasis model assessment of IR (HOMA-IR). Importantly, the stimulatory effect of FGF21 on hepatic glycogen, as determined by either biochemical measurements or PAS staining, was diminished by Ad-shKLB (Fig. 1B,C), which is consistent with increased glycogen content by FGF21 in ob/ob mice.(13) These results suggest a critical role of hepatic βKlotho in mediating insulin-sensitizing effects of FGF21. Notably, hepatic knockdown efficiency of βKlotho was evidenced (Fig. 1E and Supporting Fig. S1C,D). Consistent with the effects of FGF21 in DIO mice,(8) FGF21 treatment caused a significant reduction of body weight, likely through increased energy expenditure,(7,22) which was not obviously affected by Ad-KLB. Similar to previous studies,(16) FGF21 treatment stimulated messenger RNA (mRNA) levels of adiponectin in the white adipose tissue (Supporting Fig. S1F), suggesting a potential role of adiponectin in mediating some of FGF21’s hepatic actions, such as partial reduction of liver triglyceride levels as shown in (Supporting Fig. S1E). Moreover, FGF21’s beneficial effects on glucose tolerance and insulin sensitivity in HFHS diet-fed mice as measured by glucose tolerance tests (GTTs) and insulin tolerance tests (ITTs) were largely abrogated by Ad-shKLB (Fig. 1F), further supporting the essential role of hepatic βKlotho in mediating FGF21 effects on insulin sensitivity.
FIG. 1.
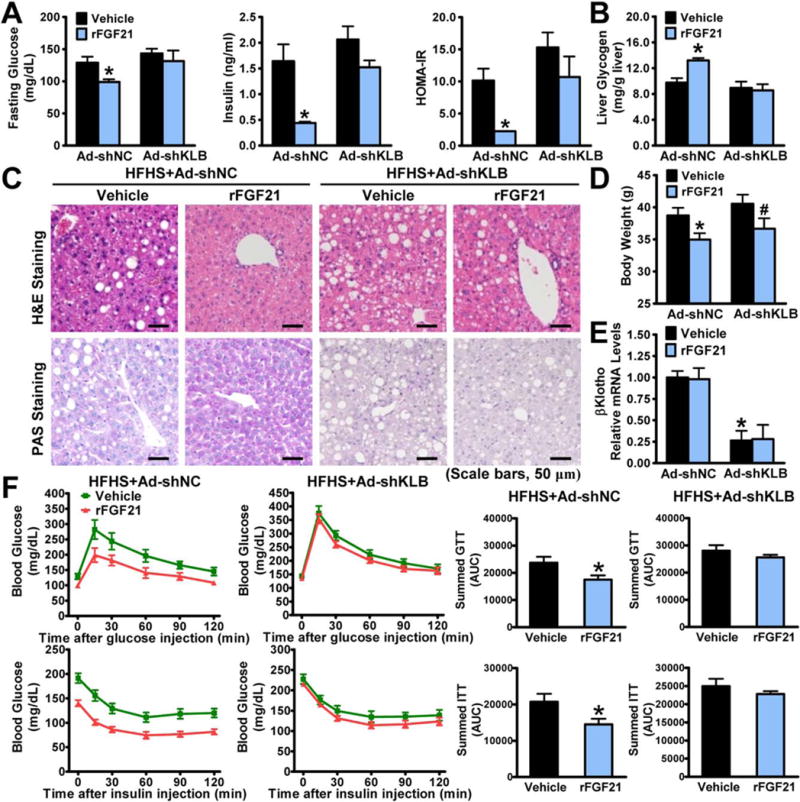
Beneficial effects of FGF21 on insulin sensitivity and hepatic glycogen storage are compromised by hepatic knockdown of βKlotho in HFHS diet-fed mice. Eight-week-old male C57BL/6 mice were fed on a HFHS diet for 14 weeks, followed by treatment with adenoviruses encoding shRNAs targeting βKlotho (shKLB) or control shNC by tail vein injection, and then treated with recombinant FGF21 (0.4 mg/kg/day) or vehicle (phosphate-buffered saline) by subcutaneous injection once-daily for 10 days. (A) Blood glucose, plasma insulin levels, and calculated HOMA-IR were assessed. (B) Liver glycogen content was determined. (C) Representative H&E staining and PAS staining of liver sections (scale bars, 50 μm). (D) FGF21 reduced body weight in both shNC- and shKLB-treated mice. (E) mRNA amounts of βKlotho were decreased by shKLB in livers. (F) GTTs (1 g/kg) or ITTs (1 U/kg) were performed. Data are presented as the mean ± SEM; n = 5–7. *P < 0.05 versus shNC and vehicle; #P < 0.05 versus shKLB and vehicle.
HEPATIC βKLOTHO IS NECESSARY FOR FGF21 TO INHIBIT mTORC1 ACTIVITY TO IMPROVE HEPATIC INSULIN SENSITIVITY IN HFHS DIET-FED MICE
Our recent study demonstrated that FGF21 is up-regulated by the nutrient sensor, SIRT1,(7) and SIRT1 ameliorates hepatic IR in diabetic obese mice through suppression of mTORC1.(23) These studies suggest a potential link between FGF21 and mTORC1. To investigate the causal association between FGF21-mediated mTORC1 inhibition and insulin-sensitizing effects, immunoblotting analysis was performed using mouse livers. Administration of FGF21 significantly decreased mTORC1 kinase activity toward S6K, S6, and 4E-BP1, resulting in decreased phosphorylation of IRS-1 at Ser1101 that is critical for development of hepatic IR,(18) whereas phosphorylation of Akt (protein kinase B) was increased, suggesting improved hepatic insulin sensitivity (Fig. 2A,B). The effect of FGF21 on hepatic glycogen synthesis was further determined. Glycogen synthesis is controlled by glycogen synthase (GS). Phosphorylation of glycogen synthase kinase 3 (GSK-3) inhibits its own kinase activity, which prevents its inhibition of GS and leads to increased glycogen synthesis.(24) Notably, FGF21 increased phosphorylation of GSK-3β and reduced phosphorylation of GS, suggesting enhanced hepatic glycogen synthesis. Strikingly, the ability of FGF21 to attenuate mTORC1 activity and promote hepatic insulin sensitivity and glycogen synthesis was abrogated by Ad-shKLB.
FIG. 2.
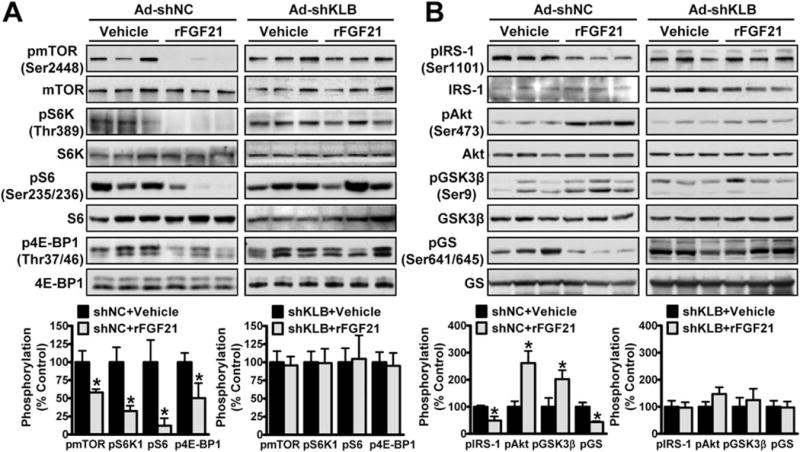
Administration of FGF21 inhibits mTORC1 activity to improve hepatic insulin sensitivity in a βKlotho-dependent manner in HFHS diet-fed mice. (A,B) FGF21 inhibited mTORC1 activity (A) and improved insulin sensitivity (B) in liver, and the effects were eliminated by Ad-KLB. Data are presented as the mean ± SEM; n = 4–6. *P < 0.05 versus Ad-shNC and vehicle.
FGF21 IS SUFFICIENT TO INHIBIT mTORC1 ACTIVATION AND ENHANCE HEPATIC INSULIN SIGNALING IN MICE
To determine the effects of FGF21 on insulin-stimulated mTOR/S6K activity in vivo, adenovirus-mediated overexpression of FGF21 (Ad-FGF21) was performed in mice injected without or with insulin. Overexpression of FGF21 was confirmed by increased hepatic FGF21 mRNAs and elevated plasma levels of FGF21 and β-hydroxybutyrate (Fig. 3A and Supporting Fig. S2A,B). No obvious changes in body weight and plasma lipid levels were evident (Supporting Fig. S2C–E). Strikingly, FGF21 caused a robust reduction in mTORC1 activity and serine phosphorylation of IRS-1 in livers of insulin-injected mice, resulting in increased phosphorylation of Akt and GSK-3β and decreased phosphorylation of GS (Fig. 3B,C). Therefore, these results suggest that FGF21 inhibits mTORC1 and acts as a hepatic insulin sensitizer, which may explain the beneficial effects of FGF21 on systemic glucose homeostasis.
FIG. 3.
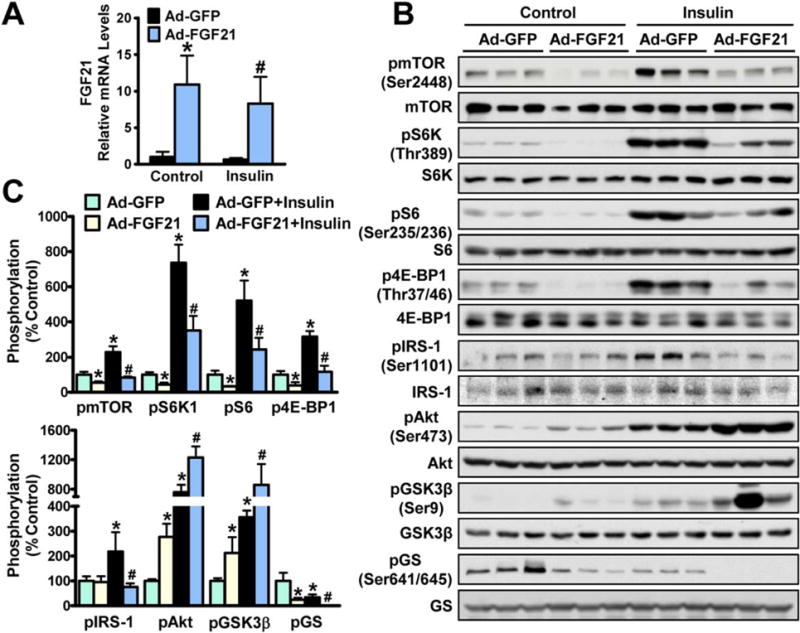
Activation of FGF21 is sufficient to repress mTORC1 activity and enhance insulin sensitivity in mice. Eight-week-old male C57BL/6 mice were treated with FGF21 or GFP adenoviruses by tail vein injection for 10 days, followed by intraperitoneal injection with insulin (1 U/Kg) or control (PBS) for 10 minutes; animals were killed and livers were collected. (A) Hepatic gene expression of FGF21. (B,C) Adenovirus-mediated overexpression of FGF21 potently attenuated insulin-induced activation of mTORC1 activity and stimulated insulin signaling in livers. Data are presented as the mean ± SEM; n = 6–7. *P < 0.05 versus Ad-GFP; #P < 0.05 versus Ad-GFP and insulin.
HEPATIC mTORC1 ACTIVITY AND INSULIN SIGNALING ARE ALTERED IN LIVERS OF FGF21 KNOCKOUT MICE
We next elucidated a causal relationship between FGF21 and mTORC1 complex and their effects on insulin signaling using FGF21 knockout mice. FGF21 deficiency was confirmed by hepatic FGF21 mRNA levels (Fig. 4A). No obvious changes in plasma lipid levels were evident (Supporting Fig. S3), which is consistent with early studies of FGF21-deficient mice fed on a chow diet.(14) In contrast to FGF21 overexpression, FGF21 deletion caused a profound activation of insulin-induced mTORC1 and phosphorylation of IRS-1, leading to decreased phosphorylation of Akt and GSK-3β and increased phosphorylation of GS (Fig. 4B,C). These studies indicate that FGF21 is necessary for suppression of mTORC1 and improvement in hepatic insulin sensitivity in vivo.
FIG. 4.
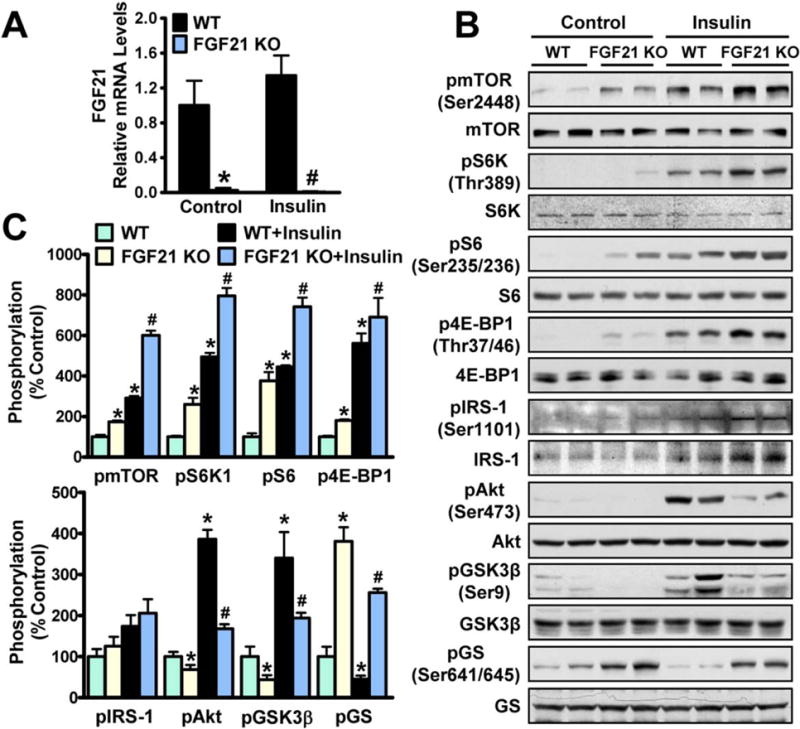
FGF21 deficiency results in activation of mTORC1 and hepatic IR in mice. FGF21 KO and WT mice at 5–6 months of age were injected with insulin (1 U/Kg) or control (PBS) intraperitoneally for 10 minutes, and then animals were killed. (A) Hepatic gene expression of FGF21. (B,C) Loss of FGF21 increased mTOR/S6K activity and suppressed hepatic insulin signaling. Data are presented as the mean ± SEM; n = 3. *P < 0.05 versus WT mice; #P < 0.05 versus WT mice treated with insulin. Abbreviations: KO, knockout; WT, wild type.
FGF21 INHIBITS mTORC1 ACTIVATION TO STIMULATE INSULIN ACTION AND PROMOTE GLYCOGEN SYNTHESIS IN VITRO
To investigate whether FGF21 is a driving force to inhibit hepatic mTORC1 and improve insulin sensitivity in vitro, effects of FGF21 overexpression were rigorously assessed in multiple hepatocytes. Compared with Ad-GFP, adenovirus-mediated overexpression of FGF21 potently inhibited insulin-stimulated mTORC1 activity (Fig. 5A and Supporting Fig. S4A). Notably, mTORC1 activity was blocked by rapamycin, an inhibitor of the mTORC1 protein kinase complex.(25) Consistent with in vivo results, FGF21 suppressed insulin-stimulated phosphorylation of IRS-1, resulting in an induction of phosphorylation of Akt and GSK-3β. Effects of FGF21 on mTORC1 activity and insulin signaling were confirmed in nutrient- and amino acid-stimulated HepG2 cells (Fig. 5B and Supporting Figs. S4B and S5A), in palmitate/BSA (bovine serum albumin)-induced insulin-resistant HepG2 cells (Fig. 5C) and further in human Huh7 hepatocytes (Supporting Fig. S5B). Notably, media levels of FGF21 were around 4,000 pg/mL in cells expressing Ad-FGF21 (Fig. 5D), suggesting that adenovirus-delivered expression of FGF21 may inhibit mTORC1 by hormonal stimulus.
FIG. 5.
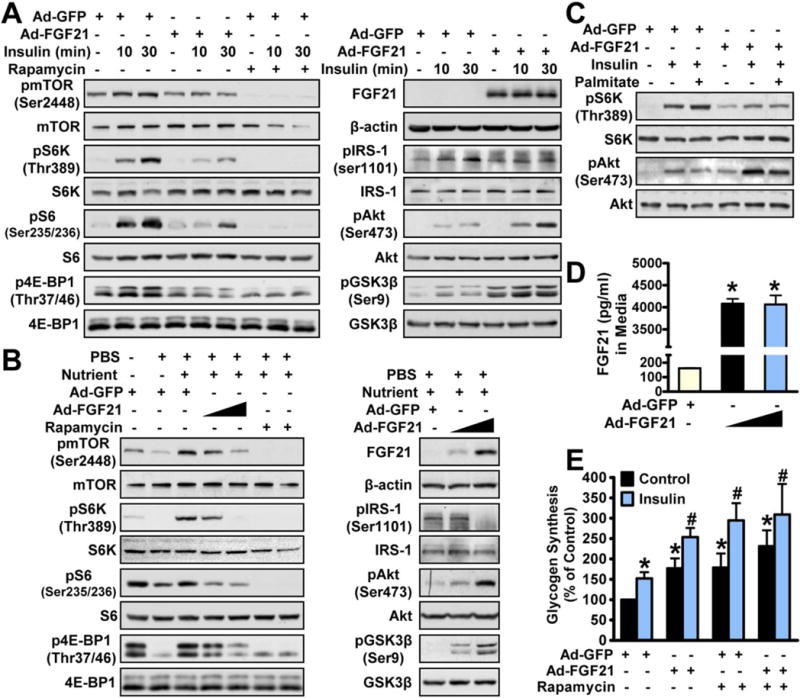
FGF21 stimulates hepatic insulin action and glycogen synthesis by repressing mTORC1 activation in hepatocytes. (A,B) Overexpression of FGF21 inhibited insulin-stimulated (A) and nutrient-stimulated (B) activation of mTORC1 to enhance phosphorylation of Akt in HepG2 cells. (C) FGF21 overexpression inhibited palmitate/BSA-induced mTORC1 activation and IR in HepG2 cells. (D) Media FGF21 levels were increased in HepG2 cells treated with Ad-FGF21. (E) Effects of FGF21 and rapamycin on insulin-induced glycogen synthesis in primary mouse hepatocytes. Cells were infected with adenoviruses for 48 hours and then incubated in serum-free medium overnight, followed by incubation with rapamycin (5 nM) or insulin (100 nM) for 3 hours as indicated. Data are represented as the mean ± SEM; n = 4–5. *P < 0.05 versus Ad-GFP; #P < 0.05 versus Ad-GFP and insulin.
To further test the functional consequence of FGF21-mTORC1 on insulin sensitivity, glycogen synthesis activities were measured in primary hepatocytes. Hepatic glycogen synthesis was induced by insulin and synergistically induced by cotreatment with FGF21 (Fig. 5E). Likewise, inhibition of mTORC1 activity, using an acute and low dose of shows similar effects. Intriguingly, compared with FGF21 or rapamycin alone, cotreatment with FGF21 and rapamycin did not cause additional stimulation of glycogen synthesis, suggesting that FGF21 and rapamycin may promote glycogen synthesis through a similar mechanism of inhibiting mTORC1 in hepatocytes.
FGF21 IMPROVES HEPATIC INSULIN SENSITIVITY THROUGH INHIBITING mTORC1 ACTIVITY IN VIVO AND IN VITRO
Next, the downstream signaling in mediating FGF21’s inhibition of mTORC1 was vigorously explored. FGF21 attenuated insulin-stimulated phosphorylation of mTORC1 activity toward S6K and increased phosphorylation of Akt in TSC11/1 hepatocytes, whereas these effects were abrogated by TSC1 deficiency-induced hyperactivation of mTORC1 (Fig. 6A,B). Likewise, FGF21-induced glycogen synthesis was diminished in TSC1−/− cells (Fig. 6C), suggesting that FGF21 may act upstream of the TSC complex to inhibit mTORC1 and improve insulin action. Moreover, treatment with lentivirus encoding a constitutively active Rag mutant, RagAQ66L, potently stimulated phosphorylation of S6K in amino acid-free (-AA) media, consistent with a positive role of Rags in mediating amino acid—stimulated mTOR activation under nutrient-poor conditions (Fig. 6D,E).(26) Interestingly, treatment of FGF21 persistently reduced phosphorylation of S6K. Furthermore, FGF21’s inhibition of phosphorylation of S6K remained intact in the presence or absence of a selective ERK inhibitor (U0126)(27) (Fig. 6F). These results suggest that FGF21 inhibits mTORC1 in a Rag GTPase- or ERK-independent manner.
FIG. 6.
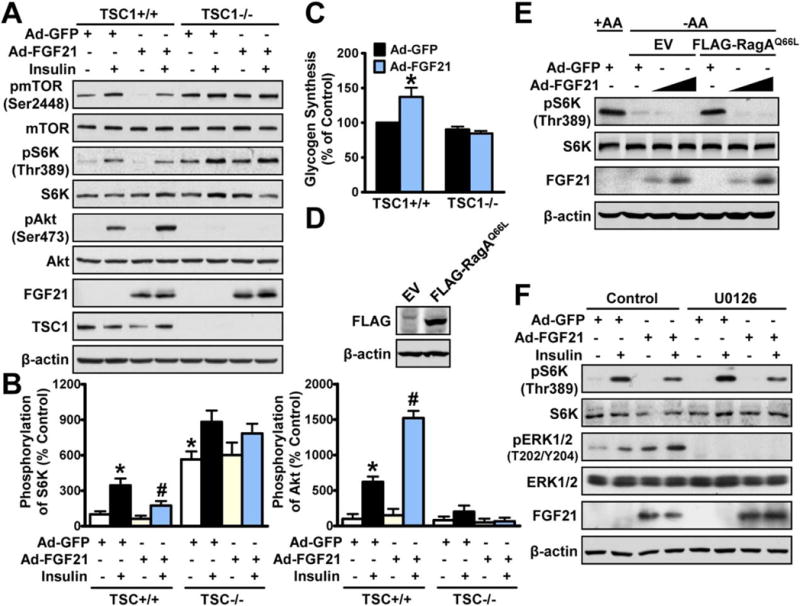
TSC mediates hepatic insulin-sensitizing effects of FGF21 in hepatocytes. (A,B) TSC1 deficiency abrogated FGF21-mediated inhibition of mTORC1 and augmentation of insulin sensitivity in hepatocytes (n = 3–4). (C) FGF21-stimulated glycogen synthesis in response to insulin was abrogated in TSC−/− hepatocytes (n = 4–5). Data are represented as the mean ± SEM. *P < 0.05 versus Ad-GFP; #P < 0.05 versus Ad-GFP and insulin. (D,E) FGF21-mediated inhibition of mTORC1 is independent of Rag GTPases in HepG2 cells. Cells expressing FLAG-tagged RagAQ66L or empty vector (EV) were infected with adenoviruses for 72 hours in DMEM (1AA), followed by incubation in amino acid-free DMEM (-AA) for 50 minutes. Overexpression of lentivirus encoding FLAG-RagAQ66L in HEK293T cells was confirmed by immunoblottings. (F) FGF21-mediated inhibition of mTORC1 remained intact in HepG2 cells treated with U0216 (10 μM). Cells were treated as indicated and representative immunoblottings are shown. Abbreviation: DMEM, Dulbecco’s modified Eagle’s medium.
To further demonstrate the causal link between FGF21’s inhibition of mTORC1 and augmentation of insulin sensitivity, in vitro and in vivo assays were performed. Constitutive activation of mTORC1’s downstream effector, S6K1 (Ad-S6K1), which has been shown to induce hepatic IR,(17) was confirmed by increased phosphorylation of S6 in HepG2 cells (Supporting Fig. S6A,B). Importantly, FGF21-stimulated phosphorylation of Akt and GSK-3β in the presence of insulin was abrogated by Ad-S6K1. Notably, phosphorylation of ERK1/2 was comparable between Ad-S6K1 and green fluorescent protein (GFP) treatment. Next, effects of mTOR/S6K activation on FGF21 actions were further investigated in diet-induced mice. Strikingly, Ad-S6K1 abrogated FGF21’s improvements in metabolic parameters in livers of HFHS diet-induced mice, such as fasting glucose, plasma insulin levels, HOMA-IR, hepatic glycogen levels, GTT, and ITT (Fig. 7A-C and Supporting Fig. S7). These studies suggest that FGF21’s effects on hepatic mTORC1 inhibition and insulin sensitization are causally linked.
FIG. 7.
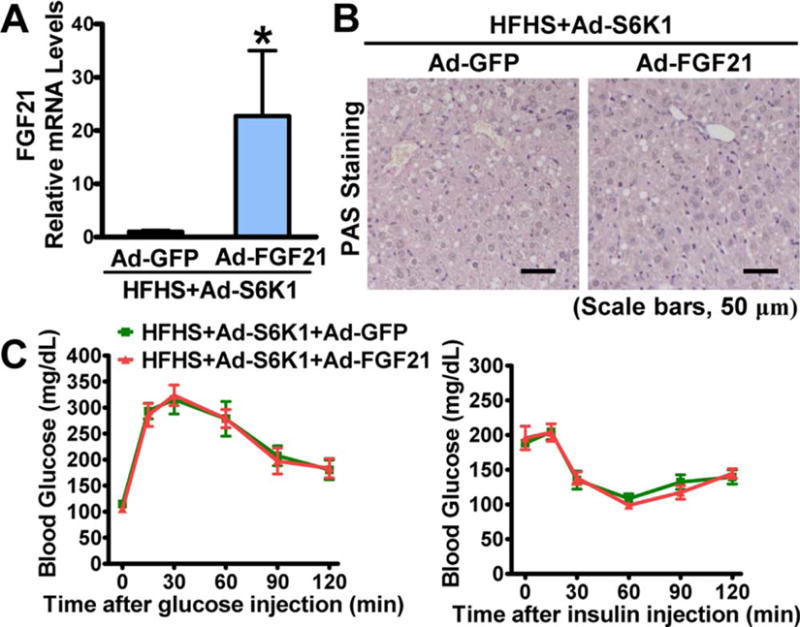
Hepatic overexpression of S6K1 abolishes hepatic insulin-sensitizing effects of FGF21 in HFHS diet-fed mice. Eight-week-old male C57BL/6 mice were fed on an HFHS diet for 11 weeks, followed by treatment with S6K1, FGF21, or GFP adenoviruses by tail vein injection for 10 days. (A) Hepatic gene expression of FGF21. (B) A representative PAS staining of liver sections is shown (scale bars, 50 μm). (C) GTTs (1 g/kg) or ITTs (1 U/ kg) were performed. Data are represented as the mean ± SEM; n = 4. *P < 0.05 versus Ad-GFP.
ADMINISTRATION OF FGF21 ATTENUATES THE DEVELOPMENT OF NONALCOHOLIC STEATOHEPATITIS
To investigate the role of FGF21 on nonalcoholic steatohepatitis (NASH), an MCD diet-induced mouse model(11) was used. Administration of FGF21 showed decreased pathological features of NASH in mice fed with MCD diet, including decreased HS and hepatocellular ballooning, and reduced perisinusoidal fibrosis as evidenced by Sirius Red staining (Fig. 8A–C). FGF21 caused a significant reduction of the NASH Clinical Research Network (NASH CRN) scores, a semiquantification analysis of NASH.(29) Notably, plasma alanine aminotransferase (ALT) levels were decreased by FGF21, suggesting decreased hepatocyte damage. These results are consistent with a previous observation showing that FGF21 protects against NASH in mice.(11) Intriguingly, FGF21 treatment decreased mTORC1 activity toward S6K (Fig. 8D). Further investigation is needed to examine whether mTORC1 inhibition mediates FGF21’s improvements in NASH.
FIG. 8.
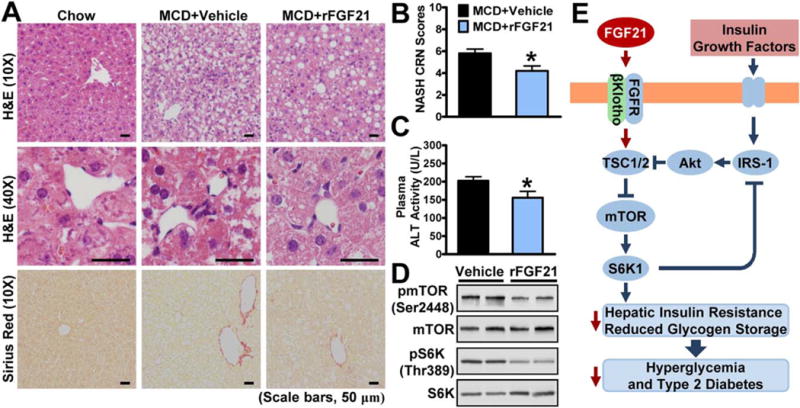
Administration of FGF21 improves MCD diet-induced steatohepatitis in mice. Eleven-week-old male C57BL/6 mice were fed MCD diet for 3 weeks and then treated with recombinant FGF21 (0.6 mg/kg/day) or vehicle (PBS) by subcutaneous injection once-daily for 10 days. (A) Representative H&E and Sirius Red stainings of liver sections (scale bars, 50 μm). (B,C) Reduction of histological NASH CRN scores (B) and ALT levels (C) in mice treated with FGF21. Data are represented as the mean ± SEM; n = 5–7. *P < 0.05 versus MCD and vehicle. (D) FGF21 inhibited mTORC1 activity in livers, and representative immunoblottings are shown. (E) The proposed model for regulation of hepatic insulin sensitivity by FGF21-βKlotho-mediated inhibition of mTORC1 activation. Excessive nutrient- and hormonal-dependent activation of mTORC1 causes insulin resistance through a negative feedback loop by mediating IRS-1 serine phosphorylation, whereas administration of FGF21 represses mTORC1 and serine phosphorylation of IRS-1, leading to improved hepatic insulin sensitivity and glucose homeostasis. The FGF21-mTORC1 axis may represent a novel drug target for treating hepatic IR and T2D.
Discussion
This study demonstrates that the hepatocyte-derived hormone, FGF21, acts as a novel inhibitor of the nutrient sensor mTORC1 complex and functions as an autocrine/paracrine modulator of hepatic insulin sensitivity and glucose homeostasis. Although pharmacological and physiological studies have demonstrated beneficial functions of FGF21 in the liver, the downstream signaling pathways mediating these activities remain largely unknown. We demonstrate, for the first time, that the FGF21-βKlotho pathway is sufficient and necessary to repress mTOR/S6K activity. Moreover, FGF21’s inhibition of mTORC1 and augmentation of hepatic insulin sensitivity are causally linked. FGF21-mediated inhibition of mTORC1 may represent a molecular mechanism by which pharmacological and genetic manipulation of FGF21 ameliorate hepatic insulin resistance, hyperglycemia, and T2D.
THE LIVER IS A DIRECT TARGET OF FGF21 ACTIONS TO IMPROVE HEPATIC INSULIN SENSITIVITY AND GLUCOSE HOMEOSTASIS
The present study utilizes in vivo and in vitro approaches to demonstrate previously unrecognized mechanisms of FGF21 directly in the liver. First, hepatic knockdown of βKlotho exacerbates hepatic IR and abolishes glucose-lowering effects of FGF21 in diet-induced IR mice. These results are consistent with recent findings that βKlotho is essential for FGF21 activity(2,30) and that FGF21 deficiency in the liver, instead of adipose tissue, impairs insulin sensitivity and glucose homeostasis in DIO mice.(15) Second, the present study utilizes cell-based assays, such as mouse primary hepatocytes and human HepG2 and Huh7 hepatocytes, to rigorously demonstrate the downstream signaling of FGF21, which circumvents FGF21’s endocrine actions in other tissues. These data demonstrate that FGF21 inhibits mTORC1 activity to improve insulin signaling in a hepatocyte-autonomous manner. Third, this study identifies, for the first time, that administration of FGF21 induces hepatic glycogen synthesis both in vivo and in vitro, which is consistent with increased glycogen storage in FGF21 transgenic mice(31) and ob/ob mice treated with recombinant FGF21.(13) This study defines FGF21 as a potent activator of hepatic glycogen synthesis. Although these data appear to contradict the previous study showing that FGF21 improves hyperglycemia in hepatic IR liver-specific insulin receptor-deficient mice,(32) the intact IRS in the mice that mediates FGF21-mTORC1 action on normalization of hyperglycemia may be a contributing factor to this difference. Taken together, in addition to regulation of fatty acid oxidation, FGF21 may promote glucose utilization and control peripheral glucose homeostasis by stimulating hepatic glycogen storage.
FGF21 SERVES AS A NEGATIVE REGULATOR OF mTORC1 KINASE COMPLEX
The present study establishes the nutrient sensor mTORC1 complex as a bona-fide downstream target of FGF21. FGF21 gain- and loss-of-function approaches have demonstrated the inhibitory action of FGF21 on mTORC1 in mouse livers, primary hepatocytes, and multiple liver cell lines in response to insulin-resistant challenges. These findings are consistent with our previous observation that SIRT1, a potent upstream inducer of FGF21,(7) inhibits mTORC1 in liver of insulin-resistant mice.(23) Further investigation is needed to examine whether FGF21 inhibition of mTORC1 mediates the insulin-sensitizing effects of SIRT1. In obese animals as well as T2D and nonalcoholic fatty liver diseasee patients, hepatic and circulating levels of FGF21 are elevated,(33) whereas mTOR/ S6K activity is overactivated in obesity and insulin-resistant rodents.(18) There are several reasons that may account for the seeming discrepancy between FGF21 levels and mTORC1 activity in obesity and T2D. First, activation of the mTOR/S6K pathway by nutrient excess may induce production and secretion of FGF21 in the liver, which is consistent with the recent findings that hepatic mTORC1 induces FGF21.(34) Second, FGF21 resistance in obesity(35) may account for compensatory overproduction of FGF21 in the circulation. Therefore, it is likely that the ability of FGF21 to repress hepatic mTORC1 is diminished because of FGF21 resistance in obesity.
One of the most important findings is that FGF21 regulates mTORC1, at least in part, through TSC1/2. The fact that FGF21-mediated inhibition of mTORC1 is abolished in TSC1-deficient hepatocytes suggests the presence of an inhibitory site upstream of TSC1/2. Moreover, FGF21 is sufficient to inhibit mTORC1 in HepG2 cells in the presence of the constitutive active Rag GTPase, RagAQ66L, and the selective ERK inhibitor, U0126, suggesting a Rag- or ERK-independent mechanism in mediating FGF21’s inhibition of mTORC1. These results are consistent with previous findings that FGF21’s beneficial effects are independent of ERK.(16) Taken together, this study demonstrates an essential role of TSC1/2 in mediating FGF21’s inhibition of mTORC1, although the molecular link between FGF21-βKlotho and TSC1/2 needs further investigation.
mTORC1 INHIBITION MEDIATES EFFECTS OF FGF21 ON IMPROVING HEPATIC INSULIN SENSITIVITY
There are several reasons that FGF21’s inhibition of mTORC1 and augmentation of insulin sensitivity are causally linked. First, hepatic overexpression of S6K1 abolishes metabolic effects of FGF21 in diet-induced insulin-resistant mice and in hepatocytes. Second, mTORC1 activation by TSC1 deficiency abrogates FGF21-stimulated glycogen synthesis in hepatocytes. Moreover, compared with treatment of FGF21 or rapamycin alone, cotreatment with FGF21 and rapamycin does not cause additional stimulation of glycogen synthesis in hepatocytes, suggesting that FGF21 may repress mTORC1 to induce glycogen synthesis through a mechanism similar to rapamycin.
Interestingly, the recent study showed that activation of mTORC1 by TSC1 deficiency induces FGF21 under starvation.(34) Our studies may provide a mechanistic insight into a role of FGF21 in counteracting mTORC1-induced desensitization of IRS and IR in obesity and T2D (Fig. 8E). The interplay between FGF21 and mTORC1 may provide a finely tuned mechanism for insulin signaling in response to nutrient availability. It is conceivable that hepatic insulin sensitivity is likely controlled by the precise physiological context, which may be determined by net effects of the negative feedback loop of mTORC1 activity toward S6K-IRS and the positive feedback loop through inducing FGF21. The implication that FGF21 inhibits mTORC1 also provides a possible explanation for the opposite metabolic effects of FGF21 and mTORC1 on energy expenditure and growth.(22,25,36,37) Collectively, the present study indicates that FGF21 enhances insulin sensitivity through inhibition of mTORC1 in livers. It may also represent a mechanism in the regulation of metabolism in other tissues to control whole-body physiology, which would be of interest and is to be further determined.
In summary, the present study has identified a central role of mTORC1 in mediating therapeutic actions of FGF21 on hepatic and systemic IR. Moreover, it is intriguing that FGF21 is sufficient to ameliorate MCD diet-induced NASH. These findings provide a novel mechanism by which FGF21 regulates metabolic effects by its actions in hepatocytes and livers. The FGF21-mTORC1 axis may represent a novel drug target to combat hyperglycemia, T2D, and potentially the development of steatohepatitis.
Supplementary Material
Acknowledgments
We appreciate Qiwei Zhai (Institute for Nutritional Sciences, Shanghai, INS) for helpful discussion. We are grateful to Leping Cheng (Institute of Neuroscience, Shanghai, ION), Jiawei Zhou (ION), Shu Zhuo (INS), Yifan Bu (INS), Huating Li (The University of Hong Kong, China, HKU), Pengchen Zhou (HKU), and Hao Zhong (Jinhai, China) for technical assistance and Zhonghui Weng (INS) for animal studies.
This work is supported by grants from the National Natural Science Foundation of China (nos. 81270930 and 31471129) and the Hundred Talents Program of the Chinese Academy of Sciences (2013OHTP04) to Y.L. This work was also supported by the Research Grants Council of Hong Kong (C7055-14G and HKU783413) to A.X.
Abbreviations
- 4E-BP1
eIF4E-binding protein 1
- Ad
adenovirus
- Akt
protein kinase B
- ALT
alanine aminotransferase
- BSA
bovine serum albumin
- DIO
diet-induced obese
- ERK
extracellular signal-regulated kinase
- FGF21
fibroblast growth factor 21
- FGFR
FGF receptor
- GFP
green fluorescent protein
- GS
glycogen synthase
- GSK-3
glycogen synthase kinase 3
- GTT
glucose tolerance test
- H&E
hematoxylin and eosin
- HFHS
high fat, high sucrose
- HOMA-IR
homeostasis model assessment of IR
- HS
hepatic steatosis
- IR
insulin resistance
- IRS-1
insulin receptor substrate 1
- ITT
insulin tolerance test
- KLB
βKlotho
- MCD
methionine and choline deficient
- mRNA
messenger RNA
- mTORC1
mammalian target of rapamycin complex 1
- NASH
nonalcoholic steatohepatitis
- NASH CRN
NASH Clinical Research Network
- NC
negative control
- PAS
Periodic Acid-Schiff
- PBS
phosphate-buffered saline
- PFU
plaque-forming units
- S6
ribosomal protein S6
- S6K1
ribosomal protein S6 kinase 1
- shRNAs
short-hairpin RNAs
- SIRT1
sirtuin 1
- TSC
tuberous sclerosis complex
- Rag GTPase
Ras-related GTP-binding protein GTPase
- T2D
type 2 diabetes
Footnotes
Additional Supporting Information may be found in the online version of this article at http://onlinelibrary.wiley.com/doi/10.1002/hep.28523/suppinfo.
Potential conflict of interest: Nothing to report.
Author names in bold designate shared co-first authorship.
Supporting Information
Additional Supporting Information may be found in the online version of this article at http://onlinelibrary.wiley.com/doi/10.1002/hep.28523/suppinfo.
References
- 1.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPAR[alpha] and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Ding X, Boney-Montoya J, Owen Bryn M, Bookout Angie L, Coate Katie C, Mangelsdorf David J, et al. βKlotho Is required for fibroblast growth factor 21 effects on growth and metabolism. Cell Metab. 2012;16:387–393. doi: 10.1016/j.cmet.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams AC, Coskun T, Irizarry Rovira AR, Schneider MA, Raches DW, Micanovic R, et al. Fundamentals of FGF19 & FGF21 action in vitro and in vivo. PLoS One. 2012;7:e38438. doi: 10.1371/journal.pone.0038438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, et al. Molecular cloning and expression analyses of mouse βklotho, which encodes a novel Klotho family protein. Mech Dev. 2000;98:115–119. doi: 10.1016/s0925-4773(00)00439-1. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146:539–549 e7. doi: 10.1053/j.gastro.2013.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPAR[alpha]-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Wong K, Walsh K, Gao B, Zang M. Retinoic acid receptor β stimulates hepatic induction of fibroblast growth factor 21 to promote fatty acid oxidation and control whole-body energy homeostasis in mice. J Biol Chem. 2013;288:10490–10504. doi: 10.1074/jbc.M112.429852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher FM, Chui PC, Nasser IA, Popov Y, Cunniff JC, Lundasen T, et al. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology. 2014;147:1073–1083 e6. doi: 10.1053/j.gastro.2014.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernardo B, Lu M, Bandyopadhyay G, Li P, Zhou Y, Huang J, et al. FGF21 does not require interscapular brown adipose tissue and improves liver metabolic profile in animal models of obesity and insulin-resistance. Sci Rep. 2015:5. doi: 10.1038/srep11382. Article number: 11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berglund ED, Li CY, Bina HA, Lynes SE, Michael MD, Shanafelt AB, et al. Fibroblast growth factor 21 controls glycemia via regulation of hepatic glucose flux and insulin sensitivity. Endocrinology. 2009;150:4084–4093. doi: 10.1210/en.2009-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150:4931–4940. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markan KR, Naber MC, Ameka MK, Anderegg MD, Mangelsdorf DJ, Kliewer SA, et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. 2014;63:4057–4063. doi: 10.2337/db14-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Z, Tian H, Lam Karen SL, Lin S, Hoo Ruby CL, Konishi M, et al. Adiponectin mediates the metabolic effects of fgf21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17:779–789. doi: 10.1016/j.cmet.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Um SH, D’Alessio D, Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab. 2006;3:393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, Roden M, et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2007;104:14056–14061. doi: 10.1073/pnas.0706517104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1–2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwiatkowski DJ, Zhang H, Bandura JL, Heiberger KM, Glogauer M, el-Hashemite N, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11:525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, et al. FGF21 regulates PGC-1a and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 2012;26:271–281. doi: 10.1101/gad.177857.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011;25:1664–1679. doi: 10.1096/fj.10-173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 25.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 26.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nojima H, Adachi M, Matsui T, Okawa K, Tsukita S, Tsukita S. IQGAP3 regulates cell proliferation through the Ras/ERK signalling cascade. Nat Cell Biol. 2008;10:971–978. doi: 10.1038/ncb1757. [DOI] [PubMed] [Google Scholar]
- 28.Ye D, Li FY, Lam KSL, Li H, Jia W, Wang Y, et al. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut. 2012;61:1058–1067. doi: 10.1136/gutjnl-2011-300269. [DOI] [PubMed] [Google Scholar]
- 29.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. HEPATOLOGY. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 30.Adams AC, Cheng CC, Coskun T, Kharitonenkov A. FGF21 requires βklotho to act in vivo. PLoS One. 2012;7:e49977. doi: 10.1371/journal.pone.0049977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, et al. FGF21 induces PGC-1a and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emanuelli B, Vienberg SG, Smyth G, Cheng C, Stanford KI, Arumugam M, et al. Interplay between FGF21 and insulin action in the liver regulates metabolism. J Clin Invest. 2014;124:515–527. doi: 10.1172/JCI67353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. 2010;139:456–463. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cornu M, Oppliger W, Albert V, Robitaille AM, Trapani F, Quagliata L, et al. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc Natl Acad Sci. 2014;111:11592–11599. doi: 10.1073/pnas.1412047111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisher FM, Chui PC, Antonellis PJ, Bina HA, Kharitonenkov A, Flier JS, et al. Obesity Is a fibroblast growth factor 21 (fgf21)-resistant state. Diabetes. 2010;59:2781–2789. doi: 10.2337/db10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inagaki T, Lin VY, Goetz R, Mohammadi M, Mangelsdorf DJ, Kliewer SA. Inhibition of growth hormone signaling by the fasting-induced hormone FGF21. Cell Metab. 2008;8:77–83. doi: 10.1016/j.cmet.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polak P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, Hall MN. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008;8:399–410. doi: 10.1016/j.cmet.2008.09.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.