

Abstract
Background
Studies of children with dilated cardiomyopathy (DCM) have suggested that improved survival has been primarily due to utilization of heart transplantation.
Objectives
We determined transplant-free survival for these children over 20 years and sought to identify the clinical characteristics at diagnosis that predicted death.
Methods
Children less than 18 years old with some type of DCM enrolled in the Pediatric Cardiomyopathy Registry were divided by year of diagnosis into an early cohort (1990–1999) and a late cohort (2000–2009). Competing risks and multivariable modeling were used to estimate the cumulative incidence of death, transplant, and echocardiographic normalization by cohort and to identify the factors associated with death.
Results
Of 1953 children, 1199 were in the early cohort and 754 were in the late cohort. Most children in both cohorts had idiopathic DCM (64% vs. 63%, respectively). Median age (1.6 vs. 1.7 years), left ventricular (LV) end diastolic z-scores (+4.2 vs. +4.2) and LV fractional shortening (16% vs. 17%) at diagnosis were similar between cohorts. Although the rates of echocardiographic normalization (30% and 27%) and heart transplantation (24% and 24%) were similar, the death rate was higher in the early cohort than in the late cohort (18% vs. 9%; p = 0.04). Being in the early cohort (hazard ratio [HR], 1.4; 95% CI, 1.04–1.9; p = 0.03) independently predicted death.
Conclusions
Children with DCM have improved survival in the more recent era. This appears to be associated with factors other than heart transplantation, which was equally prevalent in both eras.
Keywords: pediatrics, cardiomyopathy, heart failure, transplantation, echocardiography
INTRODUCTION
Dilated cardiomyopathy (DCM) is characterized by left ventricular (LV) dilation and systolic dysfunction (1,2). Nearly 40% of children with DCM undergo heart transplantation or die within 2 years of diagnosis (3). However, heart transplantation appears to have improved survival over the past 3 decades (4,5). The annual number of heart transplants has remained relatively stable over the past 20 years, likely due to limitations in donor heart availability (6). Although some children experience normalization of LV size and function, the percentage of such children has not markedly improved during this same period (7,8). Advanced medical therapies, in particular, ventricular assist devices, are now being used to treat children with heart failure (9).
We sought to determine whether rates of heart transplantation and death had changed over time in children diagnosed with DCM and enrolled in the large, multicenter Pediatric Cardiomyopathy Registry (PCMR), which has collected demographic and clinical data on pediatric cardiomyopathy patients since 1990 (10). We sought to identify baseline clinical characteristics that were associated with death. Specifically, we determined transplant-free survival in two separate decades to determine whether the management of these children has changed since 1990.
METHODS
Study Design
The PCMR is a National Heart, Lung, and Blood Institute-sponsored registry created to study the demographic features and clinical course of children with various cardiomyopathies (3,11). From 1990 to 2011, approximately 3,500 children less than 18 years old with different types of cardiomyopathies were enrolled by 98 pediatric cardiomyopathy centers in the United States and Canada. Baseline demographic, echocardiographic, and clinical data were recorded for all children at enrollment. Follow-up clinical and echocardiographic data were obtained by annual chart review.
All participating centers obtained Institutional Review Board approval for the study. The data in the PCMR database as of January 14, 2013 were analyzed in this study.
Patient Classification
For this study, we included only children with a diagnosis of pure DCM. Children with mixed forms of DCM (such as DCM with hypertrophic, restrictive, and/or non-compaction cardiomyopathy features) were excluded. The PCMR defines DCM by the following criteria: LV dilation with diminished LV systolic function; pathologic findings of DCM on autopsy or endomyocardial biopsy; or other clinical findings of DCM provided by the clinician at enrollment (11). We analyzed all etiologies of DCM and classified children into the following types at enrollment: idiopathic, myocarditis, neuromuscular disorder, familial, error of metabolism, or malformation syndrome (12). To determine changes over time, children with DCM were divided into two cohorts based on year of diagnosis: an early cohort (1990–1999) and a late cohort (2000–2009).
Left ventricular dilation was defined as a LV end-diastolic dimension (LVEDD) more than 2 standard deviations (SD) above normal (a z-score >+2) for body-surface area (BSA). Diminished LV systolic function was defined as LV fractional shortening (FS) or a LV ejection fraction (EF) more than 2 SD below normal for age (a z-score <−2). Left ventricular myocardial recovery was determined for each patient by comparing the echocardiographic values of LV dilation and LV systolic function obtained at least 30 days after the initial echocardiogram (7). “Echocardiographic normalization” was defined as a return to normal LV size (an LVEDD z-score ≤2) and normal LV systolic function (an LVFS or LVEF z-score ≥−2) on any subsequent echocardiogram. Children with persistently abnormal echocardiograms had continued LV dilation or diminished LV systolic function on follow-up echocardiograms.
Data Collection
Baseline demographic information (i.e., age, sex, race) was collected at enrollment. Clinical information on the type of DCM, evidence of heart failure, use of diuretic therapy, and echocardiographic measurements of LVEDD, LV end-systolic dimension (LVESD), LVFS, end-diastolic septal thickness (SWT) and posterior wall thickness (PWT), and LV mass were collected at enrollment and during annual follow-up visits. We calculated z-scores for LV measurements to adjust for age and BSA.
Statistical Methods
The descriptive statistics for each cohort include counts and percentages for categorical data and means and SD for normally distributed, continuous data. Skewed distributions are described with medians and interquartile ranges (IQR). Categorical variables were compared by Fisher exact tests. Normally distributed, continuous variables were compared with Student’s t tests, whereas skewed data were compared with Wilcoxon rank sum tests.
The time-related probability of death and transplant were estimated with the Kaplan-Meier method. Data were censored at the time of heart transplant in the freedom-from-death analysis. Nonparametric, competing-risks methodology was used to estimate the cumulative incidence of death versus heart transplant versus persistently abnormal echocardiogram versus echocardiographic normalization in each cohort (13,14). Risk factors for the outcome of death without heart transplant were identified with univariate Cox regression modeling. A multivariable Cox regression model was used to identify the factors independently associated with time-to-death without heart transplant. To adjust for variance among centers, the model accounted for subjects clustered within center by computing the robust sandwich covariance matrix estimate using “covs(aggregate)” and then adding center as a covariate.
Alpha was set at 0.05, and all tests were two tailed. All data were analyzed with the SAS statistical program, version 9.3 (SAS Institute Inc., Cary, North Carolina) by the Data Coordinating Center at the New England Research Institutes (Watertown, Massachusetts).
RESULTS
Patient Characteristics
Of the 1953 children with pure DCM diagnosed between 1990 and 2009, 1199 were in the early cohort (1990–1999) and 754 were in the late cohort (2000–2009; Table 1). Median ages were similar at the time of diagnosis. The cohorts had similar percentages of children with idiopathic DCM (64% vs. 63%), and the late cohort had a higher percentage of children with myocarditis (17% vs. 12%, respectively) and a lower percentage of children with neuromuscular disease (5% vs. 9%; p = 0.002). Both groups had a similar percentage of children with any heart failure symptoms at diagnosis (71%). The late cohort was more likely to present with New York Heart Association (NYHA) class IV symptoms (57% vs. 43%, p = 0.027) and to receive diuretic therapy (89% vs. 84%, p = 0.002) at time of diagnosis. An endomyocardial biopsy at time of diagnosis was more commonly performed in the early cohort (34% vs. 22%, p < 0.001). The cohorts had similar echocardiographic values at time of diagnosis, including mean LVFS, and LVEDD z-scores (Table 2). Variation by center was examined among the 12 largest sites with at least 50 patients. There were significant variations among centers in race, ethnicity, etiology, and echocardiographic parameters such as LVFS, LV posterior wall thickness, septal wall thickness, and LV mass z-scores.
Table 1.
Characteristics of 1953 Children with Dilated Cardiomyopathy, by Era of Diagnosis.
| Characteristic | Era of Diagnosis
|
p | |
|---|---|---|---|
| Early cohort, 1990–1999 n=1199 |
Late cohort, 2000–2009 n=754 |
||
| Age at diagnosis, median (IQR)1, years | 1.58 (0.34 to 11.3) | 1.68 (0.3 to 11.7) | 0.45 |
| Race, % | 0.006 | ||
| White | 58.1 | 50.2 | |
| Black | 20.1 | 22.8 | |
| Hispanic | 15.9 | 18.7 | |
| Other | 6.0 | 8.2 | |
| Male, % | 54.1 | 54.2 | 0.96 |
| Type, % | 0.002 | ||
| Idiopathic | 64.1 | 63.0 | |
| Myocarditis | 12.3 | 17.3 | |
| Neuromuscular disease | 9.2 | 4.9 | |
| Familial | 11.4 | 12.3 | |
| Error of metabolism | 2.4 | 1.9 | |
| Malformation syndrome | 0.6 | 0.5 | |
| Heart failure at diagnosis, % | 71.2 | 71.1 | 0.99 |
| NYHA2 class IV at diagnosis, % | 42.7 | 57.0 | 0.03 |
| Diuretic therapy, % | 83.8 | 88.9 | 0.002 |
| Endomyocardial biopsy, % | 33.5 | 22.3 | <0.001 |
| Follow up since diagnosis, median (IQR), years | 1.6 (0.5, 5.0) | 1.0 (0.5, 3.0) | |
IQR, interquartile range
NYHA, New York Heart Association functional classification
Table 2.
Left Ventricular Echocardiographic Profile of 1953 Children with Dilated Cardiomyopathy at Diagnosis, by Era of Diagnosis.
| Echocardiographic Characteristic | Era of Diagnosis
|
p | |
|---|---|---|---|
| Early cohort, n=1199 |
Late cohort, n=754 |
||
| LV end-diastolic dimension, mean (SD), z-score | 4.16 (2.7) | 4.19 (2.64) | 0.84 |
| LV end-systolic dimension, mean (SD), z-score | 5.93 (2.93) | 5.85 (3.05) | 0.63 |
| LV fractional shortening, mean (SD), % | 16.0 (8.3) | 16.5 (10.2) | 0.28 |
| LV fractional shortening, mean (SD), z-score | −8.42 (3.68) | −8.18 (4.41) | 0.25 |
| LV posterior wall thickness, mean (SD), z-score | −0.60 (2.62) | −0.38 (2.48) | 0.12 |
| Septal wall thickness, mean (SD), z-score | −0.86 (1.9) | −0.71 (1.96) | 0.18 |
| LV mass, mean (SD), z-score | 2.19 (2.82) | 2.37 (2.72) | 0.25 |
| LV PWT:EDD* ratio, mean (SD) | 0.13 (0.05) | 0.13 (0.05) | 0.38 |
PWT: posterior wall thickness, EDD: end-diastolic dimension
Outcomes
Median follow-up was 1.6 years (IQR, 0.5 to 5.0 years) in the early cohort and 1.0 years (IQR 0.5 to 3.0 years) in the late cohort. Heart transplantation rates were similar between cohorts, but children in the early cohort were more likely to die without heart transplant (p <0.001; Figure 1). Heart transplantation and death rates varied by centers (p<0.01).
Figure 1. Estimated A) time to death (log rank p ≤ 0.001) and B) time to heart transplantation (log rank p = 0.12) in 1953 children with dilated cardiomyopathy, by cohort.
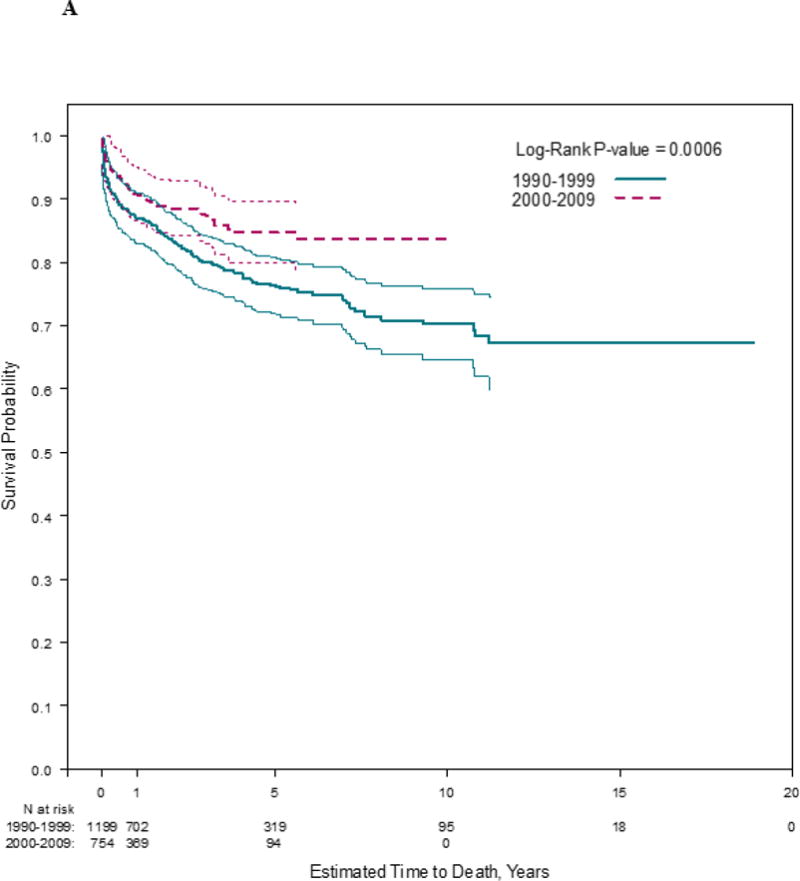
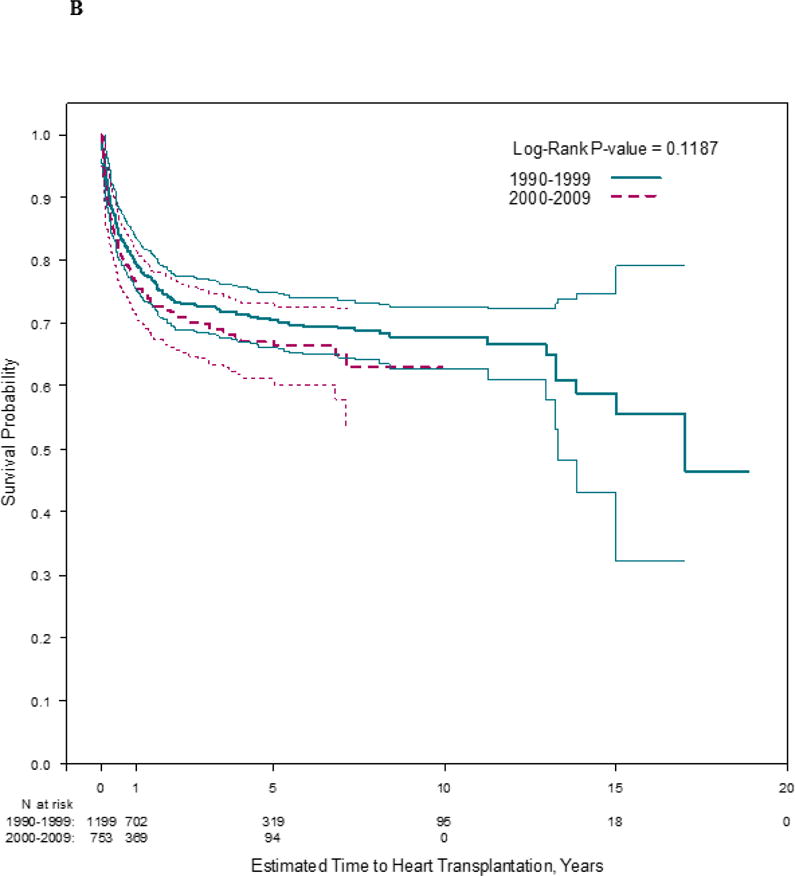
Follow-up times were censored at time of heart transplantation, where applicable.
Among the 1953 children, 73% had both LV dilation and depressed LV function at diagnosis as well as at least one follow-up echocardiogram acquired at least 30 days after the initial echocardiogram. Of the 882 children in the early cohort and the 549 children in the late cohort who met these criteria, 30% and 27%, respectively, recovered both normal LV function and size (median time from diagnosis to first echocardiographic normalization, 19.4 vs. 12.0 months), 24% in both cohorts underwent heart transplantation (median time from diagnosis to heart transplant, 5.0 vs. 3.8 months), and 18% and 9%, respectively, died (median time from diagnosis to death, 5.3 months vs. 3.2 months). Of the 317 children in the early cohort and the 205 children in the late cohort excluded for not meeting these echocardiographic criteria, 28% in both cohorts underwent heart transplantation (median time from diagnosis to heart transplant, 1.2 vs. 0.8 months), and 29% and 12%, respectively, died (median time from diagnosis to death, 0.5 vs, 0.4 months).
The cumulative incidence of the four competing outcomes (echocardiographic normalization, death, heart transplant, and persistently abnormal LV function or LV dilation) revealed that survival was better in the late cohort (Figure 2). At any given time point, the probabilities associated with the 4 states totals to 1.0. For example, at 3 years after diagnosis in the early cohort, 27% of children had normal echocardiographic values, 12% had died, 21% had undergone heart transplant, and 39% remained abnormal with respect to LV size or function. In comparison, at 3 years after diagnosis in the late cohort, 29% of children had normal echocardiographic values, 8% had died, 24% had undergone heart transplant, and 40% remained abnormal with respect to LV size or function. While the rates of echocardiographic normalization and heart transplantation were similar between cohorts (p = 0.22 and p = 0.35, respectively), the early cohort had a higher death rate in comparison to the late cohort (p = 0.04).
Figure 2. Estimated cumulative incidence rates for echocardiographic normalization in the presence of the competing risk for death or transplant among 1953 children with dilated cardiomyopathy. A) Early cohort (diagnosed between 1990 and 1999). B) Late cohort (diagnosed between 2000 and 2009).
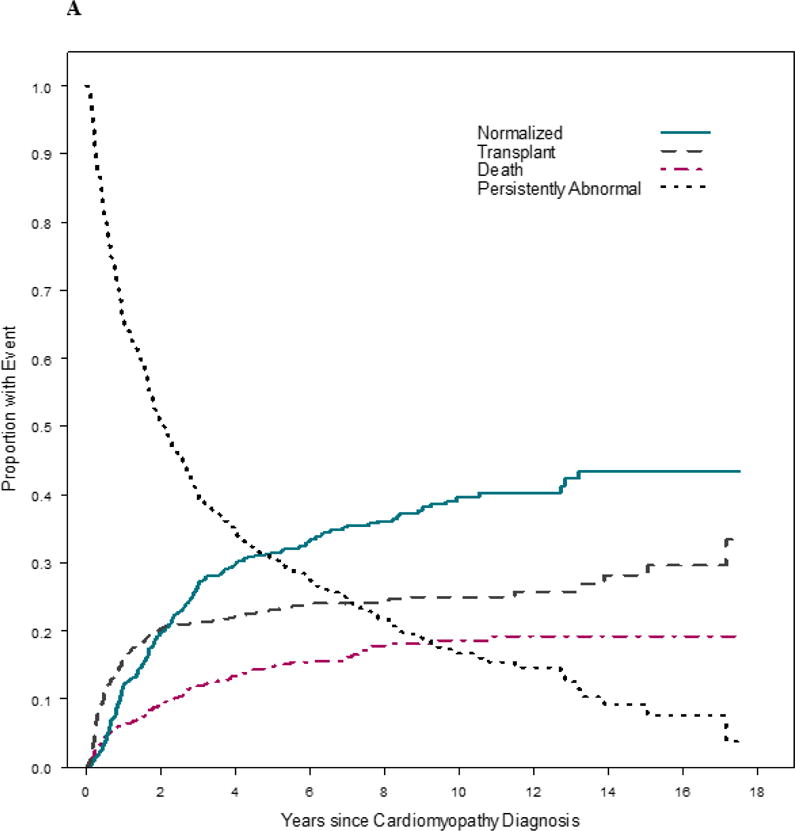
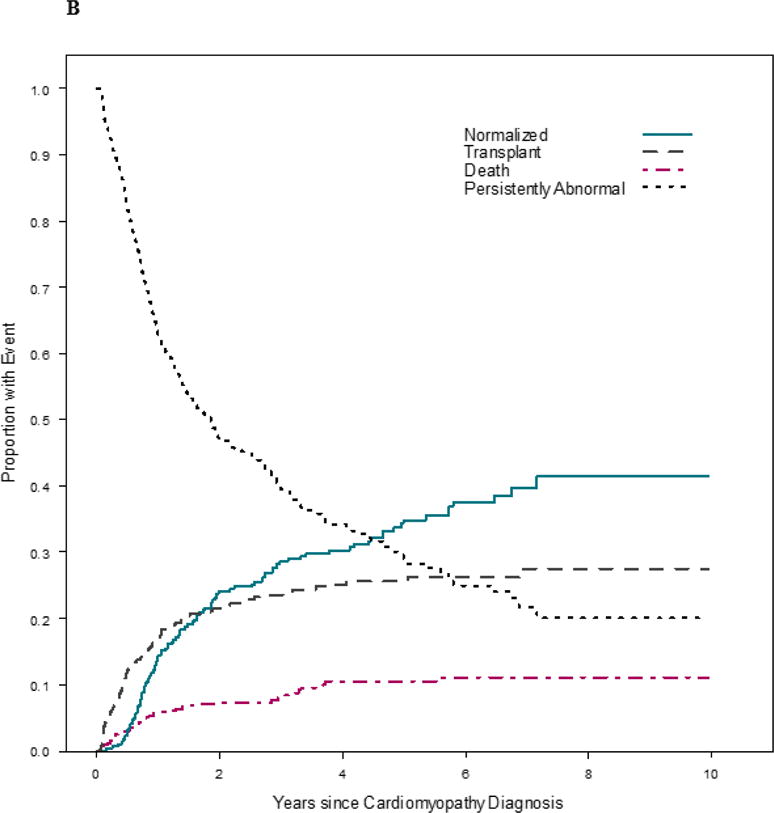
At any given time, the probabilities associated with the 4 states sum to 1.0. Although the rates of echocardiographic normalization and transplantation were similar between cohorts (p = 0.22 and p = 0.35, respectively), the early cohort had a higher death rate (p = 0.04).
We analyzed the outcomes by cohort for each of the four largest types of DCM (idiopathic DCM, myocarditis, neuromuscular, and familial DCM), which included 97% of all children (Central Illustration). Heart transplantation rates were similar in children with idiopathic DCM in both cohorts, but children in the early cohort were more likely to die without heart transplant (p = 0.006). Children with myocarditis in the late cohort had higher transplantation rates, although both cohorts had similar death rates (p = 0.13). No other outcomes differed significantly between types of DCM.
Central Illustration. Survival in Pediatric Dilated Cardiomyopathy: Estimated time to death and time to heart transplantation among 1,953 children with dilated cardiomyopathy, by type.
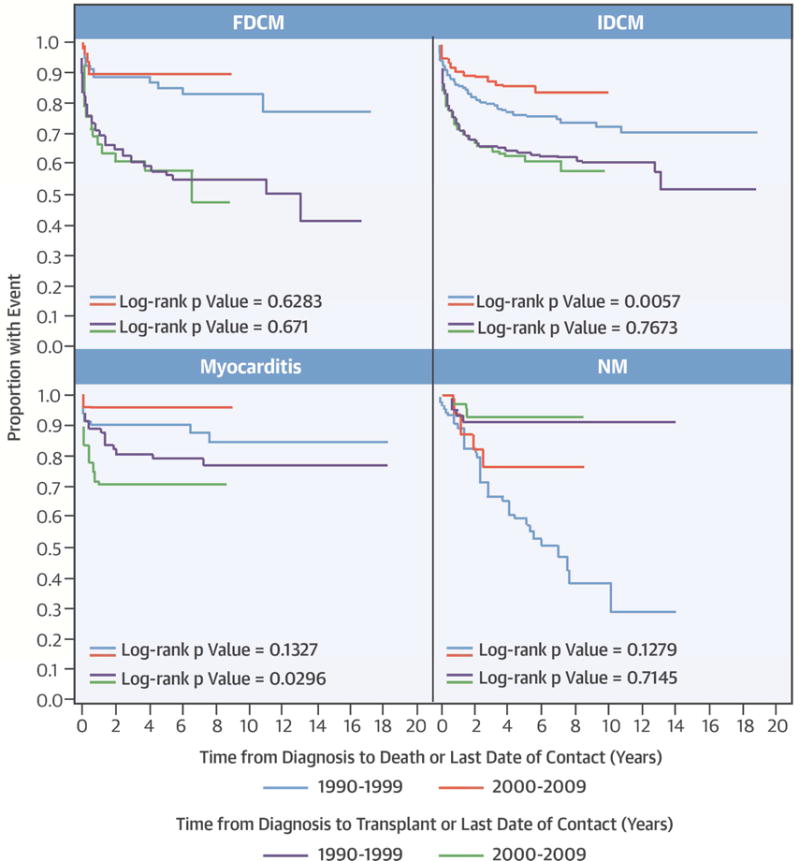
Follow-up times were censored at the time of heart transplantation, where applicable. FDCM: familial dilated cardiomyopathy; IDCM: idiopathic dilated cardiomyopathy, NM: neuromuscular disease
The cumulative incidences of the four competing outcomes were calculated for each of the four most common DCM types in each cohort. Rates of echocardiographic normalization, death, or transplant did not differ by cohort among children with idiopathic DCM, myocarditis, or familial DCM. The rates of death and heart transplantation were similar between cohorts in the neuromuscular group, but the late cohort had a higher rate of echocardiographic normalization (Figure 3).
Figure 3. Estimated cumulative incidence rates of echocardiographic normalization in the presence of the competing risk for death or transplant for children with dilated cardiomyopathy, by type. A) Early cohort (diagnosed between 1990 and 1999); B) Late cohort (diagnosed between 2000 and 2009).
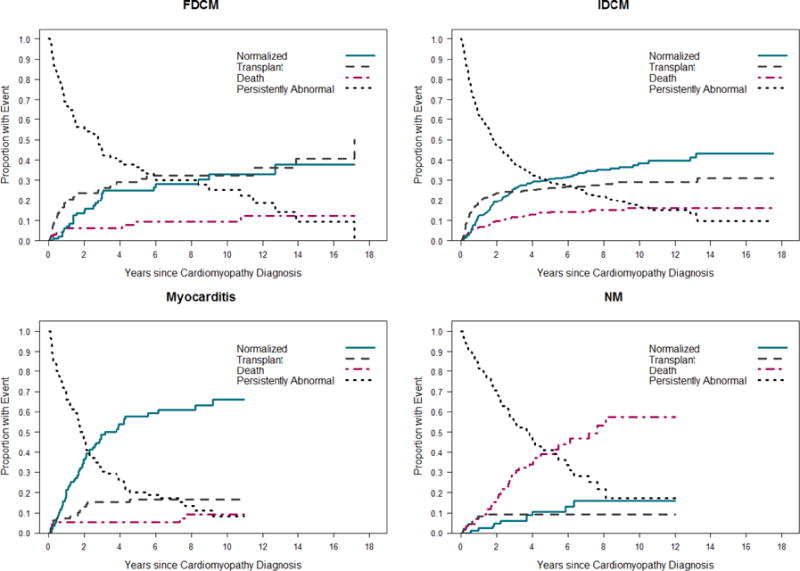
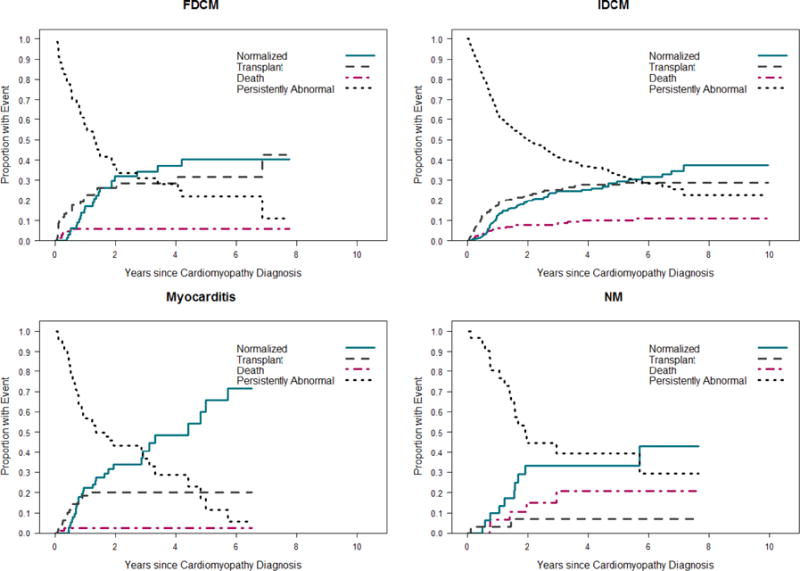
At any given time, the probabilities associated with the 4 states sum to 1.0. Rates of echocardiographic normalization, death and transplantation did not differ by cohort among children with familial DCM, idiopathic DCM, or myocarditis. Although the rates of death and transplantation were similar between cohorts among children with neuromuscular disease, the late cohort had a higher rate of echocardiographic normalization (p<0.001). FDCM, familial dilated cardiomyopathy; IDCM, idiopathic dilated cardiomyopathy, NM, neuromuscular disease.
Predictors at Diagnosis for Death without Heart Transplant
We also analyzed the baseline characteristics of children who died without heart transplant in each cohort to identify potential risk factors for early death. More children died in the early cohort (220 [18%] vs. 71 [9%]), but baseline characteristics did not differ between cohorts, including median age, heart failure symptoms, NYHA classification, type of DCM, and echocardiographic values, such as LVEDD z-score and LVFS.
We performed univariate modeling to identify predictors of death in the entire sample. Cohort, age, heart failure symptoms, certain DCM types (neuromuscular disease, myocarditis, and familial), and echocardiographic values such as LVESD z-score, LV end-diastolic SWT z-score, and LVFS z-score were associated with death (Table 3). Multivariable analysis identified early cohort, presence of heart failure symptoms at diagnosis, neuromuscular disease, and lower LVFS z-score as independent risk factors for death. Myocarditis was associated with better survival (Table 4). After controlling for these variables and variation by center, children in the early cohort were 40% more likely to die than were children in the late cohort (hazard ratio [HR] = 1.4; 95% CI, 1.04 to 1.89; p = 0.028).
Table 3.
Unadjusted Cox Proportional Hazards Regression Analysis of Predictors of Death among 1953 Children with Dilated Cardiomyopathy not Undergoing Heart Transplantation.
| N | Hazard ratio (95% CI) | p | |
|---|---|---|---|
| Age, year | 1953 | 1.02 (1.003, 1.04) | 0.02 |
| Male | 1953 | 1.09 (0.87, 1.38) | 0.45 |
| Race | 1917 | 0.13 | |
| White | Reference | ||
| Black | 1.35 (1.02, 1.78) | ||
| Hispanic | 0.94 (0.67, 1.31) | ||
| Other | 0.93 (0.55, 1.55) | ||
| Era of diagnosis | 1953 | <0.001 | |
| Early cohort (1990–1999) | 1.59 (1.22, 2.09) | ||
| Late cohort (2000–2010) | Reference | ||
| Type | 1952 | <0.001 | |
| Idiopathic | Reference | ||
| Myocarditis | 0.48 (0.30, 0.76) | ||
| Neuromuscular disease | 1.86 (1.36, 2.54) | ||
| Familial | 0.42 (0.19, 0.94) | ||
| Error of metabolism | 1.39 (0.76, 2.55) | ||
| Malformation syndrome | 2.09 (0.78, 5.61) | ||
| Heart failure at diagnosis | 1943 | 1.80 (1.37, 2.37) | <0.001 |
| Diuretic therapy | 1890 | 1.00 (0.73, 1.38) | 0.67 |
| Family history of sudden death | 1121 | 1.14 (0.71, 1.81) | 0.51 |
| LVEDD‡ z-score | 1527 | 1.04 (0.99, 1.09) | 0.12 |
| LVESD§ z-score | 1338 | 1.07 (1.01, 1.12) | 0.01 |
| LV** fractional shortening z-score | 1580 | 0.93 (0.89, 0.96) | <0.001 |
| LV posterior wall thickness z-score | 1235 | 0.94 (0.89, 1.01) | 0.08 |
| Septal thickness z-score | 1127 | 0.91 (0.84, 0.99) | 0.04 |
| LV mass z-score | 1217 | 1.01 (0.95, 1.06) | 0.86 |
LVEDD, left ventricular end-diastolic dimension
LVESD, left ventricular end-systolic dimension
LV, left ventricular
Table 4.
Final Multivariable Cox Regression Model of Predictors of Death among 1953 Children with Dilated Cardiomyopathy not Undergoing Heart Transplantation.
| Characteristic at diagnosis | N | Hazard ratio (95% CI) | p |
|---|---|---|---|
| Era of diagnosis | 1953 | 0.028 | |
| 1990–1999 | 1.40 (1.04, 1.89) | ||
| 2000–2010 | Reference | ||
| Type | 1952 | <0.001 | |
| Idiopathic | Reference | ||
| Myocarditis | 0.44 (0.28, 0.69) | ||
| Neuromuscular disease | 3.37 (2.37, 4.80) | ||
| Familial | 0.56 (0.27, 1.13) | ||
| Error of metabolism | 1.71 (1.04, 2.83) | ||
| Malformation syndrome | 2.76 (0.96, 7.94) | ||
| Heart failure at diagnosis | 1943 | 2.34 (1.72, 3.18) | <0.001 |
| LV fractional shortening z-score | 1580 | 0.93 (0.88, 0.98) | 0.007 |
DISCUSSION
Children with DCM have a poor prognosis, with about half dying or requiring heart transplantation within 5 years of diagnosis (3). Improved survival in these children has been primarily attributed to heart transplantation (15,16). Although transplantation has remained the preferred treatment for end-stage heart failure, children listed for heart transplantation have some of the highest waitlist mortality rates in solid-organ transplantation (17). Thus, the survival advantage associated with transplantation is limited by donor availability.
We found that children with DCM in the late cohort survived longer, despite stable rates of heart transplantation in both cohorts. Our competing risks analysis revealed a 33% relative risk reduction (8% vs. 12%) in the cumulative incidence of death at 3 years after diagnosis in the more recent cohort. No differences in baseline characteristics between cohorts could explain the difference in survival. In multivariable modeling, the diagnosis of DCM in the early cohort remained an independent predictor of death.
A few baseline characteristics did differ between cohorts (Table 1). Although the late cohort had fewer white and more Hispanic children, univariate analysis (Table 3) indicated that Hispanic race was not associated with death. The late cohort also had more children with myocarditis and fewer children with neuromuscular disease. We hypothesize that this difference might be explained by improved recognition and diagnosis of myocarditis (e.g., increased use of cardiac magnetic resonance imaging, MRI) in the late cohort. In addition, clinicians in the later period may have been early adopters and proponents of using heart failure medications in children with neuromuscular DCM, possibly leading to less LV dysfunction and dilation in this cohort (18–21).
Multivariable modeling of the entire sample (Table 4) revealed that being in the early cohort remained an independent predictor of death, even after controlling for the improved survival of children with myocarditis and poorer survival in children with neuromuscular DCM. Finally, children in the early cohort were more likely to have a cardiac biopsy performed at diagnosis. This change in practice is likely due to more recent efforts to incorporate non-invasive tests, such as cardiac MRI, into clinical diagnostic algorithms in cases of suspected myocarditis (22). The higher use of cardiac biopsy in the early cohort is unlikely to affect differences in survival between cohorts because the incidence of procedure-related mortality is estimated to be about 0.1% (23).
Competing risk analysis in each of the four most common types of DCM (idiopathic, myocarditis, neuromuscular, and familial) revealed no statistically significant differences in the death rate by cohort, despite each type in the late cohort having consistently lower cumulative incidence rates of death. Because combining all DCM types reduced the death rate in the late cohort (p = 0.04), we hypothesize that statistical power was insufficient to detect a difference in death rate in each type by cohort. We acknowledge the possibility that this cohort effect on survival might be the result of chance, given the borderline significant p value. Our competing risk analysis also identified an improved rate of echocardiographic normalization in the neuromuscular group in the late cohort. This improvement may be explained by improvements in medical management of these children, such as increased use of steroids and angiotensin-converting enzyme inhibitors (24).
Prior studies have shown that heart transplant-free survival in children with DCM has not improved over time. In a retrospective, single institutional study of children with DCM diagnosed between 1976 and 2005, survival did not improve over time (25). In contrast, other studies have found that improved survival in these children was primarily related to increased heart transplantation rates. In the Pediatric Heart Transplant Study (PHTS) database, waitlist mortality declined from 16% to 7% as the heart transplantation rate increased from 68% to 77% between 1995 and 2005 (16).
In our study, the incidence of echocardiographic normalization at 3 years was similar in both cohorts (27% vs. 29%). This is similar to the results of other studies evaluating the incidence of cardiac improvement (26). An early study of the PCMR evaluating children with idiopathic DCM reported that younger age, specifically less than 10 years of age, and less LV dilation at diagnosis independently predicted echocardiographic normalization within 2 years of presentation (7). A population-based study from Australia reported echocardiographic normalization in 33% of children 15 years after diagnosis, with higher LVFS z-scores at diagnosis predicting an increased likelihood of normalization (27).
Heart failure symptoms at diagnosis, seen in 71% of our children, independently predicted death. Similarly, in a retrospective, single-institutional study of 142 consecutive children with idiopathic DCM, NYHA class IV symptoms at presentation independently predicted death (28). Asymptomatic children likely have a better prognosis because they are usually diagnosed through screening mechanisms, such as a positive family history, and thus tend to have less severe disease at diagnosis.
In our study, children with neuromuscular DCM were 3.4 times as likely to die than were children with idiopathic DCM, a finding also reported in other studies of children with neuromuscular DCM (29). Progressive neuromuscular diseases tend to affect other organ systems (such as respiratory failure), and children with these diseases are less likely to be eligible for heart transplantation. In contrast, in both our and other studies, children with myocarditis were much less likely to die (30–32, 22). A retrospective, multicenter, database study of 216 children with myocarditis reported a survival-to-hospital-discharge rate of 92% (33).
Worse LV systolic function at diagnosis (a lower LVFS z-score) independently predicted death in our study and in others (27, 34–37). A prospective, multicenter study of 127 children with DCM determined that an LVEF of less than 38% was associated with disease progression and worse prognosis (38).
The improved heart transplant-free survival in our late cohort may be attributed to multiple factors. The increased use of ventricular assist devices has greatly improved waitlist mortality in children with DCM (39). Although this increase might lead to increased heart transplantation rates, some children recover enough LV function with ventricular assist devices that they no longer need heart transplantation (40, 41). Another possibility is that changing medical management of these children has improved transplant-free survival. In the past decade, more children have been treated early with beta-blockers and angiotensin-converting enzyme inhibitors for extended periods of time (25, 42, and 43). In addition, the improved recognition and management of acute heart failure at initial diagnosis or at exacerbation with enhanced intensive care unit level care may have led to better transplant-free survival in the more recent decade. Finally, more pediatric centers are now providing multidisciplinary heart failure care for these children which may lead to improved outcomes in these patients (44).
Limitations of the Study
Although we were able to demonstrate an era effect in terms of transplant-free survival in the final multivariable model, this effect was not as robust as other predictors such as etiology of DCM and presence of heart failure symptoms when comparing the HR and p values. In addition, the observational design of the PCMR data set does not allow associations between survival and various therapeutic modalities to be assessed. In addition, information on the clinical care of these children after diagnosis is limited. Knowing the use and duration of inotropic support, mechanical ventilation, and mechanical circulatory support might have clarified the improved survival in the late cohort. In addition, data on potentially important echocardiographic variables, such as mitral or tricuspid regurgitation, were not available for most children. Similarly, although beta-blockers and angiotensin-converting enzyme inhibitors have improved morbidity and mortality in large studies of adults in heart failure (42), data on the use of these drugs were not collected in more than half the children and thus were not included as possible predictors of death in the multivariable model.
CONCLUSIONS
In this large, multicenter registry study, 754 children with DCM diagnosed between 2000 and 2009 survived longer than did the 1199 children diagnosed between 1990 and 1999. This improvement appears to be related to treatment modalities other than heart transplantation, the rates of which were the same in both cohorts. Even when controlling for the degree of LV systolic dysfunction, the cause of DCM, and the presence of heart failure symptoms at diagnosis, children with DCM in the early cohort were 40% more likely to die than were children in the late cohort. Further investigations into the changing management of DCM children, including the use newer ventricular assist devices and medication therapy, are warranted to better elucidate this improvement in transplant-free survival.
CLINICAL PERSPECTIVES.
Competency in Medical Knowledge: Children with dilated cardiomyopathy have improved survival in the more recent era. This appears to be associated with factors other than heart transplantation, which was equally prevalent in both eras.
CONDENSED ABSTRACT.
Studies of children with dilated cardiomyopathy have suggested that improved survival has been primarily due to utilization of heart transplantation. A NHLBI-funded registry was analyzed to compare 1,199 children diagnosed with dilated cardiomyopathy in the years 1990–1999 to 754 children diagnosed with dilated cardiomyopathy in the years 2000–2009. Children diagnosed with dilated cardiomyopathy in the early cohort were 40% more likely to die as compared with the late cohort. This appears to be associated with factors other than the availability of heart transplantation, which was equally prevalent in both eras.
Acknowledgments
We thank the participating centers for patient recruitment and follow-up data collection. We also thank the CCF for their ongoing support of the Pediatric Cardiomyopathy Registry.
Funding: Supported by grants from the National Heart, Lung, and Blood Institute (NHLBI) HL53392 and the Children’s Cardiomyopathy Foundation (CCF) located in Tenafly, New Jersey. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NHLBI or CCF.
Abbreviations
- DCM
Dilated Cardiomyopathy
- LV
Left Ventricular
- BSA
Body Surface Area
- PCMR
Pediatric Cardiomyopathy Registry
- LVEDD
Left Ventricular End-Diastolic Dimension
- FS
Fractional Shortening
- EF
Ejection Fraction
- LVESD
Left Ventricular End-Systolic Dimension
- NYHA
New York Heart Association
- MRI
Magnetic Resonance Imaging
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors report no apparent competing interests.
Clinical Trial Registration: NCT00005391; https://clinicaltrials.gov/ct2/show/NCT00005391?term=NCT00005391&rank=1
References
- 1.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies. Circulation. 2006;113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 2.Richardson P, McKenna W, Bristow M, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–2. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 3.Towbin JA, Lowe AM, Colan SD, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–76. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 4.Hsu DT, Canter CE. Dilated cardiomyopathy and heart failure in children. Heart Fail Clin. 2010;6:415–32. vii. doi: 10.1016/j.hfc.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 5.Tsirka AE, Trinkaus K, Chen S-C, et al. Improved outcomes of pediatric dilated cardiomyopathy with utilization of heart transplantation. J Am Coll Cardiol. 2004;44:391–7. doi: 10.1016/j.jacc.2004.04.035. [DOI] [PubMed] [Google Scholar]
- 6.Dipchand AI, Edwards LB, Kucheryavaya AY, et al. The Registry of the International Society for Heart and Lung Transplantation: Seventeenth official pediatric heart transplantation report—2014; focus theme: Retransplantation. J Heart Lung Transplant. 2014;33:985–95. doi: 10.1016/j.healun.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Everitt MD, Sleeper LA, Lu M, et al. Recovery of echocardiographic function in children with idiopathic dilated cardiomyopathy: results from the Pediatric Cardiomyopathy Registry. J Am Coll Cardiol. 2014;63:1405–13. doi: 10.1016/j.jacc.2013.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.den Boer SL, Lennie van Osch-Gevers M, van Ingen G, et al. Management of children with dilated cardiomyopathy in The Netherlands: implications of a low early transplantation rate. J Heart Lung Transplant. 2015;34:963–9. doi: 10.1016/j.healun.2015.01.980. [DOI] [PubMed] [Google Scholar]
- 9.Blume ED, Naftel DC, Bastardi HJ, Duncan BW, Kirklin JK, Webber SA, Pediatric Heart Transplant Study Investigators Outcomes of children bridged to heart transplantation with ventricular assist devices: a multi-institutional study. Circulation. 2006;113:2313–9. doi: 10.1161/CIRCULATIONAHA.105.577601. [DOI] [PubMed] [Google Scholar]
- 10.Wilkinson JD, Landy DC, Colan SD, et al. The Pediatric Cardiomyopathy Registry and heart failure: key results from the first 15 years. Heart Fail Clin. 2010;6:401–3. doi: 10.1016/j.hfc.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grenier MA, Osganian SK, Cox GF, et al. Design and implementation of the North American Pediatric Cardiomyopathy Registry. Am Heart J. 2000;139:S86–S95. doi: 10.1067/mhj.2000.103933. [DOI] [PubMed] [Google Scholar]
- 12.Lipshultz SE, Sleeper LA, Towbin JA, et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N Engl J Med. 2003;348:1647–55. doi: 10.1056/NEJMoa021715. [DOI] [PubMed] [Google Scholar]
- 13.Lunn M, McNeil D. Applying Cox regression to competing risks. Biometrics. 1995;51:524–32. [PubMed] [Google Scholar]
- 14.Haller B, Schmidt G, Ulm K. Applying competing risks regression models: an overview. Lifetime Data Anal. 2013;19:33–58. doi: 10.1007/s10985-012-9230-8. [DOI] [PubMed] [Google Scholar]
- 15.McMahon AM, van Doorn C, Burch M, et al. Improved early outcome for end-stage dilated cardiomyopathy in children. J Thorac Cardiovasc Surg. 2003;126:1781–7. doi: 10.1016/j.jtcvs.2003.07.029. [DOI] [PubMed] [Google Scholar]
- 16.Kirk R, Naftel D, Hoffman TM, et al. Outcome of pediatric patients with dilated cardiomyopathy listed for transplant: a multi-institutional study. J Heart Lung Transplant. 2009;28:1322–8. doi: 10.1016/j.healun.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Almond CSD, Thiagarajan RR, Piercey GE, et al. Waiting list mortality among children listed for heart transplantation in the United States. Circulation. 2009;119:717–27. doi: 10.1161/CIRCULATIONAHA.108.815712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogata H, Ishikawa Y, Ishikawa Y, Minami R. Beneficial effects of beta blockers and angiotensin-converting enzyme inhibitors in Duchenne muscular dystrophy. J Cardiol. 2009;53:72–8. doi: 10.1016/j.jjcc.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Matsumura T, Tamura T, Kuru S, Kikuchi Y, Kawai M. Carvedilol can prevent cardiac events in Duchenne muscular dystrophy. Intern Med. 2010;49:1357–63. doi: 10.2169/internalmedicine.49.3259. [DOI] [PubMed] [Google Scholar]
- 20.Kajimoto H, Ishigaki K, Okumura K, et al. Beta-blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ J. 2006;70:991–4. doi: 10.1253/circj.70.991. [DOI] [PubMed] [Google Scholar]
- 21.Viollet L, Thrush PT, Flanigan KM, Mendell JR, Allen HD. Effects of angiotensin-converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am J Cardiol. 2012;110:98–102. doi: 10.1016/j.amjcard.2012.02.064. [DOI] [PubMed] [Google Scholar]
- 22.Foerster SR, Canter CE, Cinar A, et al. Ventricular remodeling and survival are more favorable for myocarditis than for idiopathic dilated cardiomyopathy in childhood: an outcomes study from the Pediatric Cardiomyopathy Registry. Circ Heart Fail. 2010;3:689–97. doi: 10.1161/CIRCHEARTFAILURE.109.902833. [DOI] [PubMed] [Google Scholar]
- 23.Pophal SG, Sigfusson G, Booth KL, et al. Complications of endomyocardial biopsy in children. J Am Coll Cardiol. 1999;34:2105–10. doi: 10.1016/s0735-1097(99)00452-0. [DOI] [PubMed] [Google Scholar]
- 24.Judge DP, Kass DA, Thompson WR, Wagner KR. Pathophysiology and therapy of cardiac dysfunction in Duchenne muscular dystrophy. Am J Cardiovasc Drugs. 2011;11:287–94. doi: 10.2165/11594070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.Kantor PF, Abraham JR, Dipchand AI, Benson LN, Redington AN. The impact of changing medical therapy on transplantation-free survival in pediatric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:1377–84. doi: 10.1016/j.jacc.2009.11.059. [DOI] [PubMed] [Google Scholar]
- 26.Lewis AB. Late recovery of ventricular function in children with idiopathic dilated cardiomyopathy. Am Heart J. 1999;138:334–8. doi: 10.1016/s0002-8703(99)70121-3. [DOI] [PubMed] [Google Scholar]
- 27.Alexander PM, Daubeney PE, Nugent AW, et al. Long-term outcomes of dilated cardiomyopathy diagnosed during childhood: results from a national population-based study of childhood cardiomyopathy. Circulation. 2013;128:2039–46. doi: 10.1161/CIRCULATIONAHA.113.002767. [DOI] [PubMed] [Google Scholar]
- 28.Azevedo VM, Santos MA, Albanesi Filho FM, Castier MB, Tura BR, Amino JG. Outcome factors of idiopathic dilated cardiomyopathy in children—a long-term followup review. Cardiol Young. 2007;17:175–84. doi: 10.1017/S1047951107000170. [DOI] [PubMed] [Google Scholar]
- 29.Connuck DM, Sleeper LA, Colan SD, et al. Characteristics and outcomes of cardiomyopathy in children with Duchenne or Becker muscular dystrophy: a comparative study from the Pediatric Cardiomyopathy Registry. Am Heart J. 2008;155:998–1005. doi: 10.1016/j.ahj.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.English RF, Janosky JE, Ettedgui JA, Webber SA. Outcomes for children with acute myocarditis. Cardiol Young. 2004;14:488–93. doi: 10.1017/S1047951104005049. [DOI] [PubMed] [Google Scholar]
- 31.Lee KJ, McCrindle BW, Bohn DJ, et al. Clinical outcomes of acute myocarditis in childhood. Heart. 1999;82:226–233. doi: 10.1136/hrt.82.2.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gagliardi MG, Bevilacqua M, Bassano C, et al. Long term follow up of children with myocarditis treated by immunosuppression and of children with dilated cardiomyopathy. Heart. 2004;90:1167–71. doi: 10.1136/hrt.2003.026641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klugman D, Berger JT, Sable CA, He J, Khandelwal SG, Slonim AD. Pediatric patients hospitalized with myocarditis: a multi-institutional analysis. Pediatr Cardiol. 2010;31:222–8. doi: 10.1007/s00246-009-9589-9. [DOI] [PubMed] [Google Scholar]
- 34.Alvarez JA, Wilkinson JD, Lipshultz SE. Outcome predictors for pediatric dilated cardiomyopathy: a systematic review. Prog Pediatr Cardiol. 2007;23:25–32. doi: 10.1016/j.ppedcard.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Andrews RE, Fenton MJ, Ridout DA, Burch M, British Congenital Cardiac Association New-onset heart failure due to heart muscle disease in childhood: a prospective study in the United Kingdom and Ireland. Circulation. 2008;117:79–84. doi: 10.1161/CIRCULATIONAHA.106.671735. [DOI] [PubMed] [Google Scholar]
- 36.Daubeney PE, Nugent AW, Chondros P, et al. National Australian Childhood Cardiomyopathy Study. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. 2006;114:2671–8. doi: 10.1161/CIRCULATIONAHA.106.635128. [DOI] [PubMed] [Google Scholar]
- 37.Alvarez JA, Orav EJ, Wilkinson JD, et al. for the Pediatric Cardiomyopathy Registry Investigators Competing risks for death and cardiac transplantation in children with dilated cardiomyopathy: results from the Pediatric Cardiomyopathy Registry. Circulation. 2011;124:814–23. doi: 10.1161/CIRCULATIONAHA.110.973826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molina KM, Shrader P, Colan SD, et al. Predictors of disease progression in pediatric dilated cardiomyopathy. Circ Heart Fail. 2013;6:1214–22. doi: 10.1161/CIRCHEARTFAILURE.113.000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zafar F, Castleberry C, Khan MS, et al. Pediatric heart transplant waiting list mortality in the era of ventricular assist devices. J Heart Lung Transplant. 2015;34:82–8. doi: 10.1016/j.healun.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 40.Cavigelli-Brunner A, Schweiger M, Knirsch W, et al. VAD as bridge to recovery in anthracycline-induced cardiomyopathy and HHV6 myocarditis. Pediatrics. 2014;134:e894–9. doi: 10.1542/peds.2013-2272. [DOI] [PubMed] [Google Scholar]
- 41.George CL, Ameduri RK, Reed RC, Dummer KB, Overman DM, St Louis JD. Long-term use of ventricular assist device as a bridge to recovery in acute fulminant myocarditis. Ann Thorac Surg. 2013;95:e59–e60. doi: 10.1016/j.athoracsur.2012.09.036. [DOI] [PubMed] [Google Scholar]
- 42.Harmon WG, Sleeper LA, Cuniberti L, et al. Treating children with idiopathic dilated cardiomyopathy (from the Pediatric Cardiomyopathy Registry) Am J Cardiol. 2009;104:281–6. doi: 10.1016/j.amjcard.2009.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shaddy RE, Boucek MM, Hsu DT, et al. Carvedilol for children and adolescents with heart failure. JAMA. 2007;298:1171–9. doi: 10.1001/jama.298.10.1171. [DOI] [PubMed] [Google Scholar]
- 44.Rossano JW, Shaddy RE. Heart failure in children: etiology and treatment. J Pediatr. 2014;165:228–33. doi: 10.1016/j.jpeds.2014.04.055. [DOI] [PubMed] [Google Scholar]
- 45.Kantor PF, Mertens LL. Clinical practice: heart failure in children. Part II: current maintenance therapy and new therapeutic approaches. Eur J Pediatr. 2010;169:403–10. doi: 10.1007/s00431-009-1133-7. [DOI] [PubMed] [Google Scholar]