

Abstract
Background and Purpose
Inflammation and thrombosis currently are recognized as critical contributors to the pathogenesis of ischemic stroke. CD147, also known as extracellular matrix metalloproteinase inducer, can function as a key mediator of inflammatory and immune responses. CD147 expression is increased in the brain after cerebral ischemia, but its role in the pathogenesis of ischemic stroke remains unknown. In this study, we show that CD147 acts as a key player in ischemic stroke by driving thrombotic and inflammatory responses.
Methods
Focal cerebral ischemia was induced in C57BL/6 mice by a 60-min transient middle cerebral artery occlusion (tMCAO). Animals were treated with anti-CD147 function blocking antibody (αCD147) or isotype control antibody. Blood-brain barrier permeability, thrombus formation, and microvascular patency were assessed 24h after ischemia. Infarct size, neurological deficits, and inflammatory cells invaded in the brain were assessed 72 hours after ischemia.
Results
CD147 expression was rapidly increased in ischemic brain endothelium after tMCAO. Inhibition of CD147 reduced infarct size and improved functional outcome on day 3 after tMCAO. The neuroprotective effects were associated with 1) prevented BBB damage, 2) decreased intravascular fibrin- and platelet- deposition, which in turn reduced thrombosis and increased cerebral perfusion, and 3) reduced brain inflammatory cell infiltration. The underlying mechanism may include reduced nuclear factor NF-κB activation, matrix metalloproteinase-9 (MMP-9) activity, and plasminogen activator inhibitor-1 (PAI-1) expression in brain microvascular endothelial cells.
Conclusions
Inhibition of CD147 ameliorates acute ischemic stroke by reducing thrombo-inflammation. CD147 might represent a novel and promising therapeutic target for ischemic stroke and possibly other thrombo-inflammatory disorders.
Keywords: CD147, inflammation, thrombosis, thrombo-inflammation, ischemic stroke
Introduction
Stroke is a leading cause of death and permanent disability worldwide. Reperfusion therapy with intravenous tissue plasminogen activator (tPA) initiated within 3–4.5 hours of stroke onset remains the only approved and validated therapy for acute ischemic stroke.1 However, a subset of patients still exhibit progressive neurological deterioration despite successful thrombolysis. Although the underlying mechanisms remain poorly understood, thrombotic events occurring in downstream cerebral microvessels may be of particular relevance for brain injury progression after stroke.2,3 Recent studies have suggested that thrombosis and inflammation are two closely intertwined processes that crucially contribute to ischemic brain injury and orchestrate stroke progression.4–7 These findings have given rise to the novel concept of “thrombo-inflammation” in which ischemic stroke is considered to be a thrombo-inflammatory disease.8,9 Accordingly, it has been recently proposed that simultaneous targeting of both thrombotic and inflammatory processes could represent a novel therapeutic strategy for acute ischemic stroke.9
CD147, a type I transmembrane glycoprotein of the immunoglobulin (Ig) superfamily, is broadly expressed on the surface of various cell types, including three major cell types (i.e. leukocytes, platelets, and endothelial cells) that are integrally involved in stroke-induced inflammation and thrombosis.10 Increased expression of CD147 has been implicated in many human diseases such as cancer, cardiovascular diseases, and neurological disorders. Therapeutic targeting of CD147 has yielded encouraging results in experimental models of human diseases, such as rheumatoid arthritis, asthmatic lung inflammation, myocardial ischemia/reperfusion injury, multiple sclerosis and experimental autoimmune encephalomyelitis.11–15 Although it has been reported that CD147 expression was increased in the brain following focal cerebral ischemia,16, 17 whether increased CD147 expression simply serves as an associative marker or substantially contributes to ischemic brain injury remains unknown. In this study, we tested the hypothesis that CD147 acts as a key player in ischemic stroke by driving thrombotic and inflammatory responses. We examined the therapeutic potential and mechanisms of neuroprotection by pharmacological inhibition of CD147 in mice following focal cerebral ischemia/reperfusion injury.
Materials and Methods
Details of materials and experimental procedures are available from the Online Supplements. This manuscript adheres to the AHA Journals’ implementation of the Transparency and Openness Promotion (TOP) Guidelines.
Stroke model and antibody treatment
Focal cerebral ischemia was induced in C57BL/6 mice by a 60-min transient middle cerebral artery occlusion (tMCAO) as described previously.18, 19. Two hours after tMCAO, the mice were randomly assigned to the following treatment groups: a rat anti-mouse CD147 monoclonal antibody (RL73.2, eBbioscience, named αCD147 mAb throughout this article) or isotype control antibody (rat IgG2a) administered via tail vein injection in 100 ul volume of PBS. This anti-CD147 antibody has been well characterized to block CD147 function in various mouse models.11–15 In the 24-hour experiments, a single dose of antibody was given at 4 h after onset of ischemia. In the 72-hour experiments, antibody treatment was initiated at 4 h and repeated at 24 h and 48 h after onset of ischemia.
Infarct volume and neurological deficits
Infarct volume was measured in TTC-stained coronal sections on day 3 after tMCAO.19 The modified Bederson score (global neurological function)20 and the grip strength test (motor function and coordination)21 were performed by a blinded investigator.
Blood Brain Barrier Permeability and Microvascular Perfusion
BBB permeability was determined by the extravasation of Evans Blue (EB, 961 Da) and sodium fluorescein (NaF, 376 Da) in the brain 24h after tMCAO.22 Cerebral microvascular perfusion was assessed by the FITC-dextran–labeled vessels.23
Western blot and gelatin zymography
Protein extracts were obtained from the cerebral cortices (bregma +2 to −3 mm) and the isolated cerebral microvessels. Two assays were performed as described previously.19, 24
Brain cell isolation and flow cytometry
Brain cell isolation and flow cytometric analysis was performed as described previously.25, 26 Flow cytometry was performed on a Becton Dickinson FACS Calibur and data was analyzed with CellQuest Pro software.
Immunohistochemistry
Double immunofluorescence staining was performed as described previously.19, 27 The number of vessels positively stained with fibrin/fibrinogen and thrombocytes and the number of occluded microvessels were counted in the ischemic boundary zone (both cortex and striatum). All immunostaining data was analyzed by a blinded investigator and data are presented as the density of immunoreactive vessels relative to the imaged area (mm2).27, 28
Statistical Analysis
All results were expressed as mean ± standard error of the mean (SEM). GraphPad Prism 5 software package was used for statistical analysis. Unless otherwise indicated, multiple comparisons were made using a 1-way analysis of variance (ANOVA) followed by the Bonferroni post hoc test. If only 2 groups were compared, unpaired, 2-tailed Student t test was applied. p<0.05 was considered statistically significant.
Results
CD147 expression is rapidly increased in brain microvessels after tMCAO
Immunofluoresence staining showed abundant expression of CD147 in the ischemic hemisphere at 24h after tMCAO, with the most prominent expression seen in the peri-infarct cortex, but only minimal expression seen in the contralateral hemisphere (Fig. 1A). Double staining further showed that CD147 colocalized extensively with CD31-positive microvascular endothelial cells in the peri-infarct cortex (Fig. 1B), but rarely colocalized with other brain cells (astrocytes, microglia, and neurons) (Fig. 1B; Suppl. Fig. IA and IB) at 24h after tMCAO. Interestingly, we observed that CD147 staining colocalized extensively with GFAP-positive astrocytes (Fig. 1B), partially with Iba1-positive microglia, but rarely with NeuN-positive neurons at 72h after tMCAO (Suppl. Fig. IA). Furthermore, western blotting was used to assess CD147 protein levels in the ischemic cerebral cortex and specifically in brain endothelial cells using isolated microvessels. The data showed that CD147 protein levels were significantly increased as early as 4 h and remained elevated 24 h after tMCAO in the ischemic cortex (Fig. 1C) and also in the microvascular endothelial cells (Fig. 1D). Collectively, these data indicate that brain microvascular endothelial cells are the major cellular source of increased CD147 expression in the ischemic brain during the first 24h after tMCAO.
Figure 1.
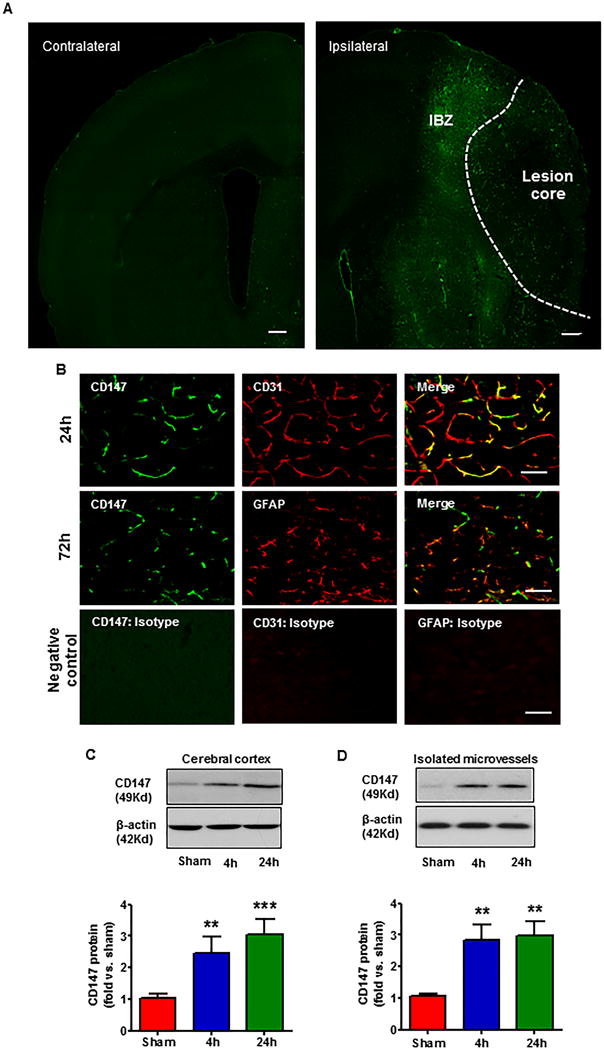
CD147 upregulation in the ischemic brain. A, Representative microphotograph of an intact coronal brain section (approximately +0.5 mm to bregma) showing the distribution of tMCAO-induced CD147 expression in the ipsilateral (ischemic) and contralateral hemisphere 24 h after onset of ischemia. CD147 was robustly induced in the ischemic hemisphere, prominently shown in microvessel-like structures. IBZ: ischemia boundary zone. Scale bar: 100 μm. B, Representative images of double immunostaining showing the co-localization of CD147 (green) with CD31 (endothelial marker, red) or GFAP (astrocytic marker, red) at 24h and 72h after tMCAO. n=5 per group. Images were acquired from the peri-infarct cortex. Scale bar: 50 μm. Negative control staining with isotype-matched control antibody did not show detectable labeling. (C, D), Representative images of western blots showing CD147 protein levels using protein extracts from ipsilateral cortices (C) or isolated brain microvessels (a pool of five mice per group) (D) at 4h and 24h after tMCAO. Semi-quantitation of immunoblots was analyzed by densitometry. Data are expressed as mean ± SEM from 3 independent experiments. **p<0.01, ***p<0.001 versus sham control.
Inhibition of CD147 improves acute stroke outcome
Acute stroke outcome was assessed on day 3 after tMCAO. Infarct volumes were significantly reduced in mice treated with αCD147 at a dose of 1–2 mg/kg (p<0.001), whereas a lower dose of 0.1 mg/kg αCD147 had no significant effect (p>0.05) compared to isotype-control treated mice (Fig. 2A, 2B). Importantly, the smaller infarct volumes translated into better neurological outcome. Mice treated with αCD147 (1 or 2 mg/kg) showed significant improvement in overall neurological function (Bederson score, Fig. 2C) as well as motor function and coordination (grip test, Fig. 2D) compared to isotype-controls. Based on these data, the dose 1.0 mg/kg of αCD147 or isotype antibody was used for further experiments. Physiological parameters remained within normal range in all experimental groups (Suppl. Table I).
Figure 2.
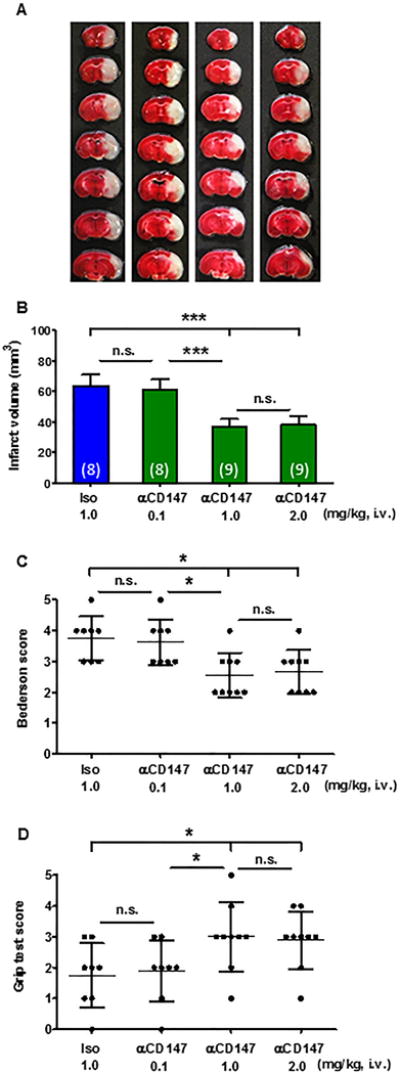
Inhibition of CD147 reduces infarct volumes and neurological deficits 72 h after tMCAO. A, Representative images of TTC-stained brain coronal sections of mice (left to right): receiving different doses (0.1, 1.0, 2.0 mg/kg) of the anti-CD147 function blocking antibody (αCD147) or 1.0 mg/kg of isotype control antibody (Iso). B, Quantitative analysis of infarct volumes. (C, D), Neurological deficits were assessed using the Bederson test (C) and the grip test (D). n=8-9/group. n.s.= not statistically significant; *P<0.05, ***p<0.001.
Inhibition of CD147 reduces BBB permeability through inhibition of microvascular MMP-9 activity
The BBB permeability was assessed by measuring extravasation of EB and NaF dyes. Isolated cerebral microvessels were analyzed by gelatin zymography for MMP-2/-9 MMP enzymatic activity. The amount of extravasated EB (Fig. 3A) and NaF (Fig. 3B) dyes in the ipsilateral hemispheres and MMP-9 activity (Fig. 3C, 3D) in isolated microvessels were significantly increased at 24h after tMCAO compared with sham controls. All of these measures were markedly reduced in the αCD147-treated mice compared with isotype-treated mice. Although tMCAO induced a mild but statistically significant increase in MMP-2 activity, there was no significant difference between the αCD147-treated and isotype-treated mice.
Figure 3.
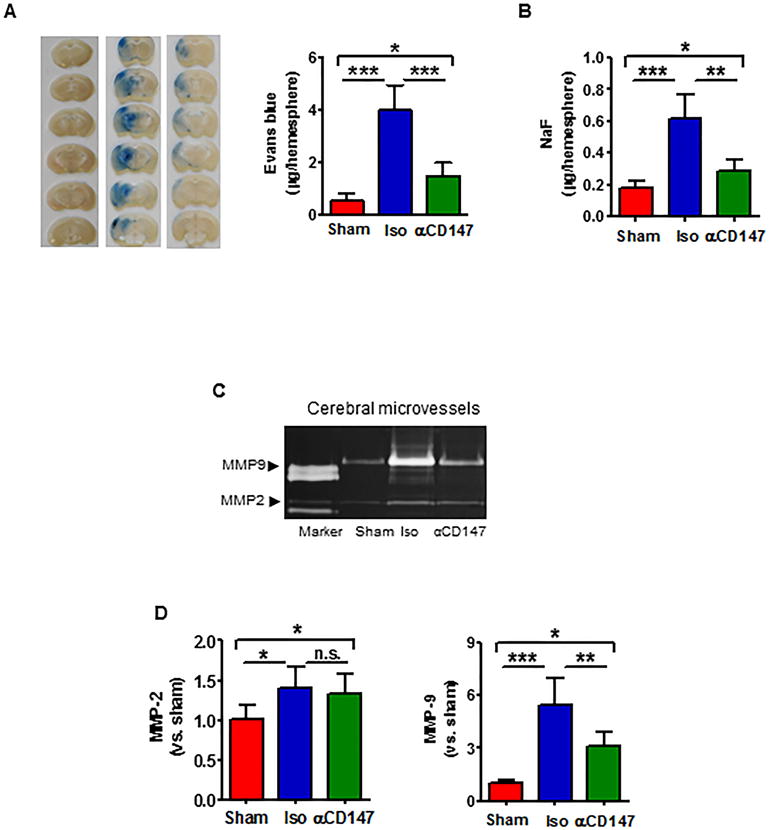
Inhibition of CD147 reduces BBB permeability through inhibition of MMP-9 in brain microvascular endothelial cells 24h after tMCAO. (A, B), (A, left panel) Representative pictures of Evans blue leakage in coronal sections from a sham control and tMCAO mice receiving 1.0 mg/kg of Iso or αCD147 treatment. Quantitative analysis of Evans blue (A, right) and sodium fluoride (NaF) (B) extravasation in the brain parenchyma. n=6 per group. C, Representative images of gelatin zymography for MMP-2 and -9 enzymatic activity using protein extracts from brain microvessels (a pool of five mice per group) isolated at 24 h after tMCAO. D, Semi-quantitation of MMP bands was analyzed by densitometry. Data are expressed as mean ±SEM from 3 independent experiments. n.s.= not statistically significant; *P<0.05, **p<0.01, ***P<0.001.
Inhibition of CD147 reduces microvascular thrombosis and improves microvascular patency
Fibrin and platelet deposition crucially contribute to secondary microvascular thrombosis after ischemic stroke.2,3 Double immunofluorescence staining showed that tMCAO induced extensive deposition of both fibrin/fibrinogen (Fig. 4A) and platelets (Fig. 4B) in brain microvessels, which was accompanied by increased microvascular occlusion (Fig. 4C) and reduced microvascular patency (Fig. 4D) at 24h after tMCAO. Importantly, inhibition of CD147 with αCD147 treatment (given at 4 h after onset of ischemia) profoundly reduced tMCAO-induced microvascular thrombosis thereby improving microvascular patency (Fig. 4 A–D).
Figure 4.
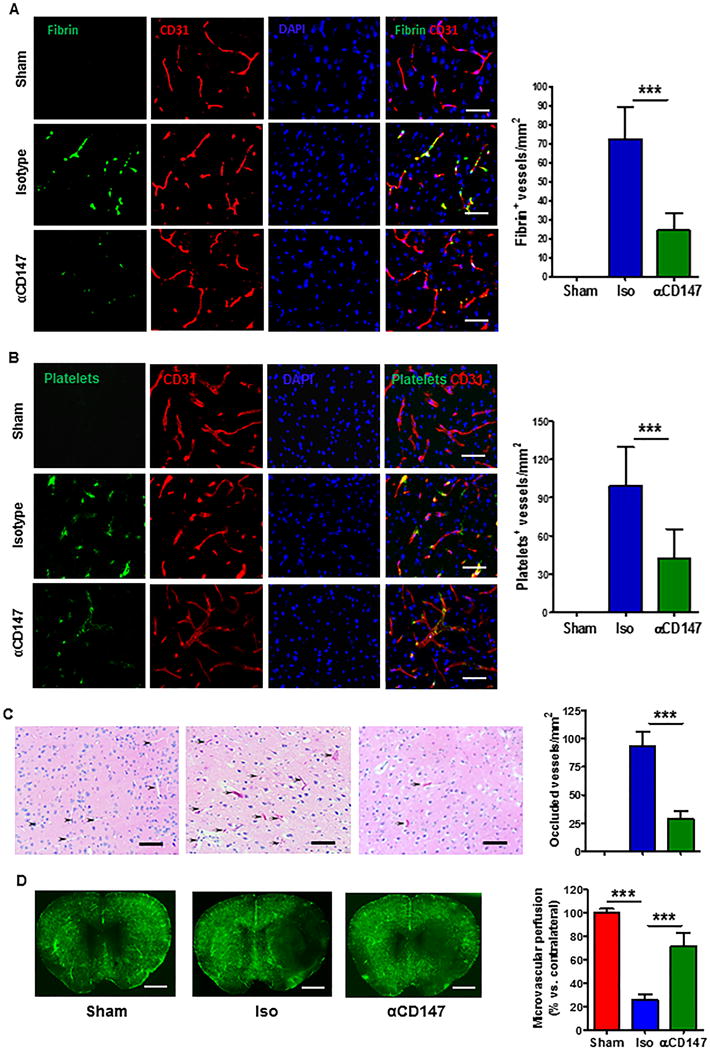
Inhibition of CD147 reduces intravascular fibrin and platelet deposition correlated with reduced microvascular thrombosis and improved microvascular perfusion 24h after tMCAO. (A, B), Representative images of double immunostaining showing the staining positive for fibrin/fibrinogen (green, in panel A) or thrombocytes (green, in panel B) in the cerebral microvessels (marked by CD31 staining, red) in the peri-infarct cortex. C, Hematoxylin and eosin staining showing occluded microvessels (indicated by arrows) and patent vessels (indicated by arrowheads) in the peri-infarct cortex. Data are presented as the number of fibrin/fibrinogen- (A, left) or thrombocyte/platelet-(B, left) positive vessels or occluded vessels (C, left) relative to the imaged area (mm2), as described in “Method”. Scale bars: 50 um, n=5/group, ***p<0.001. D, Representative images show microvascular FITC–dextran perfusion from the 3 groups of mice. Quantitative data are expressed as the percentage of the microvascular area perfused with FITC–dextran between the ipsilateral versus contralateral hemisphere. Scale bars: 2 mm, n=5/group, ***p<0.001.
Activation of NF-kB and upregulation of PAI-1 modulate inflammatory and thrombotic responses after ischemic stroke.29,30 Isolated cerebral microvessels were analyzed by western blot to evaluate changes in protein levels. Data showed that tMCAO robustly increased the phosphorylation of p65 (ser536) and its upstream signal IKKα (ser176) and IKKβ (ser177/181), as well as the expression of PAI-1 protein in brain microvascular endothelial cells. Importantly, all these effects were almost completely blocked with the αCD147 treatment (Fig. 5A, 5B).
Figure 5.
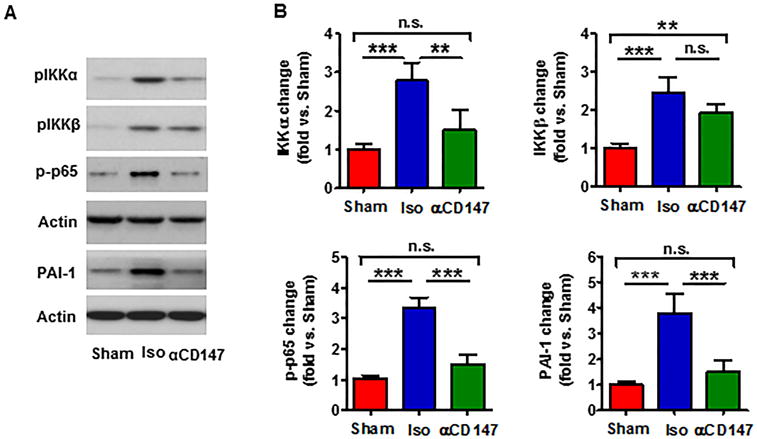
Inhibition of CD147 reduces NF-kB activation and PAI-1 expression in cerebral microvascular endothelial cells 24h after tMCAO. Protein extracts were prepared from isolated brain microvessels (a pool of five mice per group) at 24h after tMCAO. A, Representative images of western blots showing the phosphorylated proteins (IKKα, IKKβ and NF-қB p65) and PAI-1 protein levels. B, Semi-quantitation of immunoblots was analyzed by densitometry. Data are expressed as mean ± SEM from 3 independent experiments. n.s.= not statistically significant; **p<0.01, *** p<0.001.
Inhibition of CD147 reduces inflammatory infiltrate in the postaischemic brain
Flow cytometry was employed to assess inflammatory infiltrate in the mouse brain 72h after tMCAO. Fig. 6A showed the flow cytometry strategy for gating and identifying immune cells. The number of infiltrating immune cells including neutrophils (Gr1+), total T cells (CD3+), T helper (CD3+CD4+), T cytotoxic (CD3+CD8+) cells, macrophages (CD11b+CD45high), as well as brain resident microglia (CD11b+CD45low) were markedly increased in the ischemic hemispheres compared to their respective sham controls (Fig. 6B). These data are generally consistent with the results of previous studies.31,32 Importantly, we found that inhibition of CD147 with αCD147 treatment (initiated at 4h and repeated at 24 and 48h after onset of ischemia) profoundly reduced the accumulation of these inflammatory cells in the ischemic brain, compared with isotype-control treatment (Fig. 6B).
Figure 6.
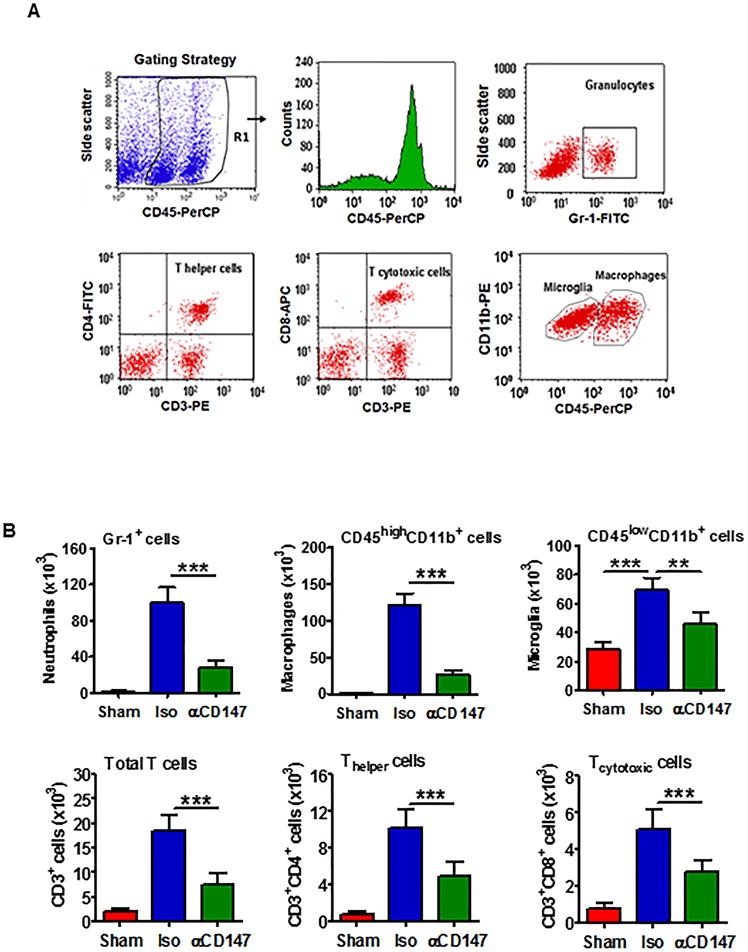
Inhibition of CD147 reduces inflammatory cell infiltration in the ischemic brain 72h after tMCAO. Immune cells were isolated from the ipsilateral hemispheres (a pool of three mice per group) at 72h after tMCAO. Single-cell suspensions were analyzed by flow cytometry. A, Gating strategy for identifying immune cell populations: granulocytes/neutrophils (Gr-1+), total T cells (CD3+), T helper- (CD3+CD4+) and T cytotoxic- (CD3+CD8+) cells, and microglia (CD11b+CD45low) and macrophages (CD11b+CD45high). CD45+ cells were gated for analysis of viable immune populations. B, Flow cytometric quantitation of cell subpopulations in the 3 groups studied. Data are expressed mean ± SEM from 3 independent experiments. **P<0.01, ***P<0.001.
Discussion
The present study, for the first time, demonstrates that CD147 acts detrimentally in ischemic stroke by driving brain inflammation and microvascular thrombosis. Inhibition of CD147 with αCD147 treatment initiated 4 hours after stroke onset substantially reduced infarct volume and neurological deficits. The observed beneficial effects are likely attributed to reduced BBB damage, inflammation, thrombosis, and improved microvascular patency.
Endothelial cells represent important targets for therapeutic intervention in many cardiovascular and neurological diseases. After ischemic stroke, brain microvascular endothelial cells are rapidly converted into a pro-inflammatory and pro-thrombotic state, which potentiate microvascular damage and infarct progression.33 Therefore, targeting brain microvascular endothelial cells represents a promising therapeutic approach for ischemic stroke. In the present study, we provided the first evidence that targeting CD147 may have therapeutic potential for endothelial dysfunction in ischemic stroke. This is supported by the following findings: (1). CD147 expression was rapidly increased in the ischemic brain microvascular endothelial cells after tMCAO; (2). Inhibition of CD147 with αCD147 treatment (given at 4h after ischemia onset) reduces MMP-9 activity and PAI-1 expression in brain microvascular endothelial cells by analyzing brain microvessels isolated at 24h after tMCAO, which is likely associated with profound inhibition of tMCAO-induced activation of NF-κB signaling (p65 and its upstream IKKα). Activation of nuclear factor kappa B (NF-kB) signaling is required for the transcriptional induction of many pro-inflammatory and pro-thrombotic mediators involved in ischemic stroke.29 Considering that significant increase in expression of CD147 in other brain cells (reactive astrocytes and activated microglia) occurred relatively late (72h) after tMCAO and antibodies are large molecules that cannot easily cross the BBB, we speculate that the observed anti-inflammatory and anti-thrombotic actions by αCD147 treatment are mediated mainly through direct targeting of endothelial CD147 in the brain microvessels at least in the early phase (within 24h) after tMCAO.
We demonstrated that αCD147 treatment protects against BBB disruption following acute ischemic stroke through inhibition of MMP-9 in ischemic brain microvascular endothelial cells. MMP-9 is known to promote BBB disruption in acute ischemic stroke.34,35 Notably, although well known as an inducer of extracellular MMPs, CD147 may not always necessarily be associated with induction of MMPs under certain pathological conditions. It has been reported that antibody blockade of CD147 does not influence MMP activity in several mouse models.11–13 For instance, Seizer et al have reported that inhibition of CD147 with αCD147 treatment had no impact on MMP-2 and MMP-9 activity in the myocardium after ischemia/reperfusion injury.13 In both mouse and rat tMCAO stroke models, we and others have previously demonstrated that MMP-9 expression was markedly increased in ischemic brain microvessels during the first 24 h after stroke, whereas infiltrating leukocytes (especially neutrophils) appear to be the major cellular source of brain MMP-9 at later time points (24–72h) after stroke.19,36 In the present study, we further demonstrate that CD147 is highly expressed in ischemic brain microvessels and inhibition of CD147 with αCD147 treatment (given at 4h after onset of ischemia) profoundly suppressed MMP-9 activity detected in isolated brain microvessels that was markedly increased 24h after tMCAO. These data provide strong, albeit indirect, evidence that CD147 acts as an inducer of MMP-9 in brain microvascular endothelial cells after ischemic stroke. Nevertheless, further research is needed to investigate if and how CD147 blockade directly inhibits MMP-9 enzymatic activity in brain endothelial cells using in vitro brain endothelial cell culture models. In addition, αCD147 treatment also reduced brain MMP-9 levels at 72h after tMCAO (Suppl. Fig. II), which could be attributed reduced leukocyte (mainly neutrophils) infiltration.
Despite successful thrombolysis approximately 25% of patients with acute ischemic stroke do not show any clinical improvement due to incomplete reperfusion.37,38 Downstream microvascular thrombosis is recognized as a key contributing factor to incomplete reperfusion after ischemic stroke.37,38 Corroborating these clinical findings, experimental studies have shown that even if the site of major arterial occlusion is reopened, microvascular thrombosis continues to occur at distal sites, which in turn reduces microvascular potency and contributes to neurological progression after ischemic stroke.39–41 Considering that microvascular thrombosis occurs immediately after onset of ischemia39,40 and that a salvageable penumbra may exist for at least 4.5 hours in some stroke patients,41 preventing microvascular thrombosis is considered to be essential for successful neuroprotection in treatment of ischemic stroke.42 It has been reported that intravascular fibrin- and platelet- deposition critically contribute to downstream microvascular obstruction following acute ischemic stroke.43,44 Upregulation of PAI-1 expression in brain endothelial cells after ischemic stroke foster intravascular fibrin and platelet deposition, contributing to microvascular obstruction.28,30 In the present study, we demonstrate that inhibition of CD147 with αCD147 treatment exerts potent anti-thrombotic effects by reducing intravascular fibrin- and platelet- deposition, which in turn reduces downstream microvascular thrombosis and improves microvascular patency 24h after tMCAO. This finding suggests that CD147 targeted therapy represents a promising approach to prevent microvascular thrombosis thus conferring microvascular protection in acute ischemic stroke.
Brain inflammation has been implicated as a secondary injury mechanism following ischemic stroke, where the main cellular players are immune cells including activated resident microglial cells and infiltrating leukocytes, mainly neutrophils, monocytes/macrophages, and T cells.45 These inflammatory cells are involved in stroke progression by producing reactive oxygen species (ROS), pro-inflammatory cytokines and chemokines, MMPs, and other neurotoxic factors.45 Moreover, neutrophil infiltration can impair microvascular perfusion by occluding cerebral capillaries, a phenomenon commonly referred to as “no reflow”.46,47 It has been reported that inhibition of CD147 with αCD147 treatment via interrupting CD147-cyclophilin interaction reduces leukocyte infiltration into inflamed tissues in several mouse models.11–13 In the present study, we demonstrate that inhibition of CD147 with αCD147 treatment (initiated at 4h and repeated at 24h and 48h after onset of ischemia) profoundly reduced inflammatory cell infiltration including neutrophils, T cells, and macrophages/activated microglia invaded in the ischemic brain 72 h after tMCAO. Interestingly, intravital microscopy showed that αCD147 treatment almost completely abrogated leukocyte adhesion in the brain microvasculature at 24h tMCAO (Suppl. Fig. III). Taken together, our findings suggest that targeting CD147 may represent an effective approach to reduce inflammatory cell infiltration in acute ischemic stroke.
It should be noted, however, that the mechanisms whereby anti-CD147 treatment protects against brain injury after ischemic stroke should be much more complex than those we examined in this study. Although the findings of this study suggest that brain endothelial cells may be a primary target of anti-CD147 treatment, we cannot exclude the possibility that CD147 blockade also works importantly by targeting other cells, such as leukocytes or even platelets. Leukocytes and platelets are central to both inflammation and thrombosis, two important pathological pathways of ischemic stroke. Further research is needed to answer the following questions: (1) the time-dependent expression profiles of CD147 in blood leukocyte subtypes and platelets after ischemic stroke; (2) the effects of CD147 blockade on circulating platelet and leukocyte activation and their interaction, as well as their interaction with brain endothelial cells and subsequent leukocyte trafficking into the ischemic brain; (3) the relative importance of CD147 expressed in the brain cells versus blood cells by selective targeting of blood leukocytes (or platelets) versus brain endothelial cells.
In summary, pharmacological inhibition of CD147 improves stroke outcome by a combined anti-thrombotic and anti-inflammatory mechanism and thereby represents a novel and promising therapeutic strategy for ischemic stroke and possibly other thrombo-inflammatory disorders.
Supplementary Material
Acknowledgments
Sources of Funding
This work was supported by National Institutes of Health grants NS088719 and NS089991 (Dr. Li).
Footnotes
Disclosures
None
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2015 update: a report from the American Heart Association. Circulation. 2015;131:e29–322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Stoll G, Kleinschnitz C, Nieswandt B. Molecular mechanisms of thrombus formation in ischemic stroke: novel insights and targets for treatment. Blood. 2008;112:3555–3562. doi: 10.1182/blood-2008-04-144758. [DOI] [PubMed] [Google Scholar]
- 3.van der Spuy WJ, Pretorius E. Interrelation between inflammation, thrombosis, and neuroprotection in cerebral ischemia. Rev Neurosci. 2012;23:269–278. doi: 10.1515/revneuro-2012-0028. [DOI] [PubMed] [Google Scholar]
- 4.Kraft P, Göb E, Schuhmann MK, Göbel K, Deppermann C, Thielmann I, et al. FTY720 ameliorates acute ischemic stroke in mice by reducing thrombo-inflammation but not by direct neuroprotection. Stroke. 2013;44:3202–3210. doi: 10.1161/STROKEAHA.113.002880. [DOI] [PubMed] [Google Scholar]
- 5.Göb E, Reymann S, Langhauser F, Schuhmann MK, Kraft P, Thielmann I, et al. Blocking of plasma kallikrein ameliorates stroke by reducing thromboinflammation. Ann Neurol. 2015;77:784–803. doi: 10.1002/ana.24380. [DOI] [PubMed] [Google Scholar]
- 6.Dhanesha N, Prakash P, Doddapattar P, Khanna I, Pollpeter MJ, Nayak MK, et al. Endothelial Cell-Derived von Willebrand factor is the major determinant that mediates von Willebrand factor-dependent acute ischemic stroke by promoting postischemic thrombo-inflammation. Arterioscler Thromb Vasc Biol. 2016;36:1829–1837. doi: 10.1161/ATVBAHA.116.307660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuhmann MK, Guthmann J, Stoll G, Nieswandt B, Kraft P, Kleinschnitz C. Blocking of platelet glycoprotein receptor Ib reduces “thrombo-inflammation” in mice with acute ischemic stroke. J Neuroinflammation. 2017;14:18. doi: 10.1186/s12974-017-0792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nieswandt B, Kleinschnitz C, Stoll G. Ischaemic stroke: a thrombo-inflammatory disease? J Physiol. 2011;589:4115–4123. doi: 10.1113/jphysiol.2011.212886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke. 2016;47:1165–1172. doi: 10.1161/STROKEAHA.115.011238. [DOI] [PubMed] [Google Scholar]
- 10.Zhu X, Song Z, Zhang S, Nanda A, Li G. CD147: a novel modulator of inflammatory and immune disorders. Curr Med Chem. 2014;21:2138–2145. doi: 10.2174/0929867321666131227163352. [DOI] [PubMed] [Google Scholar]
- 11.Damsker JM, Okwumabua I, Pushkarsky T, Arora K, Bukrinsky MI, Constant SL. Targeting the chemotactic function of CD147 reduces collagen-induced arthritis. Immunology. 2009;126:55–62. doi: 10.1111/j.1365-2567.2008.02877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gwinn WM, Damsker JM, Falahati R, Okwumabua I, Kelly-Welch A, Keegan AD, et al. Novel approach to inhibit asthma-mediated lung inflammation using anti-CD147 intervention. J Immunol. 2006;177:4870–4879. doi: 10.4049/jimmunol.177.7.4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seizer P, Ochmann C, Schönberger T, Zach S, Rose M, Borst O, et al. Disrupting the EMMPRIN (CD147)-cyclophilin A interaction reduces infarct size and preserves systolic function after myocardial ischemia and reperfusion. Arterioscler Thromb Vasc Biol. 2011;31:1377–1386. doi: 10.1161/ATVBAHA.111.225771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agrawal SM, Silva C, Wang J, Tong JP, Yong VW. A novel anti-EMMPRIN function-blocking antibody reduces T cell proliferation and neurotoxicity: relevance to multiple sclerosis. J Neuroinflammation. 2012;9:64. doi: 10.1186/1742-2094-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Agrawal SM, Silva C, Tourtellotte WW, Yong VW. EMMPRIN: a novel regulator of leukocyte transmigration into the CNS in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci. 2011;31:669–677. doi: 10.1523/JNEUROSCI.3659-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burggraf D, Liebetrau M, Martens HK, Wunderlich N, Jäger G, Dichgans M, et al. Matrix metalloproteinase induction by EMMPRIN in experimental focal cerebral ischemia. Eur J Neurosci. 2005;22:273–277. doi: 10.1111/j.1460-9568.2005.04187.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhu W, Khachi S, Hao Q, Shen F, Young WL, Yang GY, et al. Upregulation of EMMPRIN after permanent focal cerebral ischemia. Neurochem Int. 2008;52:1086–1091. doi: 10.1016/j.neuint.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin R, Yu S, Song Z, Quillin JW, Deasis DP, Penninger JM, et al. Phosphoinositide 3-kinase-gamma expression is upregulated in brain microglia and contributes to ischemia-induced microglial activation in acute experimental stroke. Biochem Biophys Res Commun. 2010;399:458–464. doi: 10.1016/j.bbrc.2010.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin R, Song Z, Yu S, Piazza A, Nanda A, Penninger JM, et al. Phosphatidylinositol-3-kinase gamma plays a central role in blood-brain barrier dysfunction in acute experimental stroke. Stroke. 2011;42:2033–2044. doi: 10.1161/STROKEAHA.110.601369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 21.Moran PM, Higgins LS, Cordell B, Moser PC. Age-related learning deficits in transgenic mice expressing the 751-amino acid isoform of human amyloid precursor protein. Proc Natl Acad Sci U S A. 1995;92:5341–5345. doi: 10.1073/pnas.92.12.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, et al. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. 2016;47:1068–1077. doi: 10.1161/STROKEAHA.115.010835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desilles JP, Loyau S, Syvannarath V, Gonzalez-Valcarcel J, Cantier M, Louedec L, et al. Alteplase Reduces Downstream Microvascular Thrombosis and Improves the Benefit of Large Artery Recanalization in Stroke. Stroke. 2015;46:3241–3248. doi: 10.1161/STROKEAHA.115.010721. [DOI] [PubMed] [Google Scholar]
- 24.Liu W, Sood R, Chen Q, Sakoglu U, Hendren J, Cetin O, et al. Normobaric hyperoxia inhibits NADPH oxidase-mediated matrix metalloproteinase-9 induction in cerebral microvessels in experimental stroke. J Neurochem. 2008;107:1196–1205. doi: 10.1111/j.1471-4159.2008.05664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campanella M, Sciorati C, Tarozzo G, Beltramo M. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;22:586–592. doi: 10.1161/hs0202.103399. [DOI] [PubMed] [Google Scholar]
- 26.Arac A, Brownell SE, Rothbard JB, Chen C, Ko RM, Pereira MP, et al. Systemic augmentation of alphaB-crystallin provides therapeutic benefit twelve hours post-stroke onset via immune modulation. Proc Natl Acad Sci U S A. 2011;108:13287–13292. doi: 10.1073/pnas.1107368108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Chopp M, Teng H, Ding G, Jiang Q, Yang XP, et al. Combination treatment with AcSDKP and tissue plasminogen activator provides potent neuroprotection in rats after stroke. Stroke. 2014;45:1108–1114. doi: 10.1161/STROKEAHA.113.004399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Cui X, Zacharek A, Roberts C, Chopp M. eNOS mediates TO90317 treatment-induced angiogenesis and functional outcome after stroke in mice. Stroke. 2009;40:2532–2538. doi: 10.1161/STROKEAHA.108.545095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harari OA, Liao JK. Nf-kappab and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang ZG, Chopp M, Goussev A, Lu D, Morris D, Tsang W, et al. Cerebral microvascular obstruction by fibrin is associated with upregulation of PAI-1 acutely after onset of focal embolic ischemia in rats. J Neurosci. 1999;19:10898–10907. doi: 10.1523/JNEUROSCI.19-24-10898.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liesz A, Zhou W, Mracskó É, Karcher S, Bauer H, Schwarting S, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134:704–720. doi: 10.1093/brain/awr008. [DOI] [PubMed] [Google Scholar]
- 32.Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849–1857. doi: 10.1161/STROKEAHA.108.534503. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Tsuji K, Lee SR, Ning M, Furie KL, Buchan AM, et al. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke. 2004;35:2726–2730. doi: 10.1161/01.STR.0000143219.16695.af. [DOI] [PubMed] [Google Scholar]
- 34.Chaturvedi M, Kaczmarek L. MMP-9 inhibition: a therapeutic strategy in ischemic stroke. Mol Neurobiol. 2014;49:563–573. doi: 10.1007/s12035-013-8538-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jin R, Yang G, Li G. Molecular insights and therapeutic targets for blood-brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis. 2010;38:376–385. doi: 10.1016/j.nbd.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao BQ, Wang S, Kim HY, Storrie H, Rosen BR, Mooney DJ, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat Med. 2006;12:441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- 37.De Silva DA, Fink JN, Christensen S, Ebinger M, Bladin C, Levi CR, et al. Assessing reperfusion and recanalization as markers of clinical outcomes after intravenous thrombolysis in the echoplanar imaging thrombolytic evaluation trial (EPITHET) Stroke. 2009;40:2872–2874. doi: 10.1161/STROKEAHA.108.543595. [DOI] [PubMed] [Google Scholar]
- 38.Alexandrov AV, Hall CE, Labiche LA, Wojner AW, Grotta JC. Ischemic stunning of the brain: early recanalization without immediate clinical improvement in acute ischemic stroke. Stroke. 2004;35:449–452. doi: 10.1161/01.STR.0000113737.58014.B4. [DOI] [PubMed] [Google Scholar]
- 39.Choudhri TF, Hoh BL, Zerwes HG, Prestigiacomo CJ, Kim SC, Connolly ES, Jr, et al. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor-mediated platelet aggregation. J Clin Invest. 1998;102:1301–1310. doi: 10.1172/JCI3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desilles JP, Loyau S, Syvannarath V, Gonzalez-Valcarcel J, Cantier M, Louedec L, et al. Alteplase reduces downstream microvascular thrombosis and improves the benefit of large artery recanalization in stroke. Stroke. 2015;46:3241–348. doi: 10.1161/STROKEAHA.115.010721. [DOI] [PubMed] [Google Scholar]
- 41.Dalkara T, Arsava EM. Can restoring incomplete microcirculatory reperfusion improve stroke outcome after thrombolysis? J Cereb Blood Flow Metab. 2012;32:2091–2099. doi: 10.1038/jcbfm.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gursoy-Ozdemir Y, Yemisci M, Dalkara T. Microvascular protection is essential for successful neuroprotection in stroke. J Neurochem. 2012;123(Suppl 2):2–11. doi: 10.1111/j.1471-4159.2012.07938.x. [DOI] [PubMed] [Google Scholar]
- 43.Okada Y, Copeland BR, Fitridge R, Koziol JA, del Zoppo GJ. Fibrin contributes to microvascular obstructions and parenchymal changes during early focal cerebral ischemia and reperfusion. Stroke. 1994;25:18471–1853. doi: 10.1161/01.str.25.9.1847. [DOI] [PubMed] [Google Scholar]
- 44.Zhang ZG, Zhang L, Tsang W, Goussev A, Powers C, Ho KL, et al. Dynamic platelet accumulation at the site of the occluded middle cerebral artery and in downstream microvessels is associated with loss of microvascular integrity after embolic middle cerebral artery occlusion. Brain Res. 2001;912:181–194. doi: 10.1016/s0006-8993(01)02735-4. [DOI] [PubMed] [Google Scholar]
- 45.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang CM. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion in baboons. Stroke. 1991;22:1276–1283. doi: 10.1161/01.str.22.10.1276. [DOI] [PubMed] [Google Scholar]
- 47.Arumugam TV, Salter JW, Chidlow JH, Ballantyne CM, Kevil CG, Granger DN. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am J Physiol Heart Circ Physiol. 2004;287:H2555–2560. doi: 10.1152/ajpheart.00588.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.