

Abstract
Chemical fixation is nearly indispensable in the biological sciences, especially in circumstances where cryo-fixation is not applicable. While universally employed for the preservation of cell organization, chemical fixatives often introduce artifacts that can confound identification of true structures. Since biological research is increasingly probing ever-finer details of the cellular architecture, it is critical to understand the nanoscale transformation of the cellular organization due to fixation both systematically and quantitatively. In this work, we employed Partial Wave Spectroscopic (PWS) Microscopy, a nanoscale sensitive and label-free live cell spectroscopic-imaging technique, to analyze the effects of the fixation process through three commonly used fixation protocols for cells in vitro. In each method investigated, we detected dramatic difference in both nuclear and cytoplasmic nanoarchitecture between live and fixed states. But significantly, despite the alterations in cellular nanoscale organizations after chemical fixation, the population differences in chromatin structure (e.g. induced by a specific chemotherapeutic agent) remains. In conclusion, we demonstrated that the nanoscale cellular arrangement observed in fixed cells was fundamentally divorced from that in live cells, thus the quantitative analysis is only meaningful on the population level. This finding highlights the importance of live cell imaging techniques with nanoscale sensitivity or cryo-fixation in the interrogation of cellular structure, to complement more traditional chemical fixation methods.
Keywords: Chromatin nanostructure, Fixation, Partial wave spectroscopy
1. Introduction
Direct cryo-fixation is regarded as the best method to preserve ultrastructure in biological samples [1–4]. Compared with other sample preparation protocols involving perfusions or immersions, cryo-fixation immobilizes molecules inside the sample within milliseconds [5] and does not alter the milieu in the specimen. In addition, structures prepared by cryo-fixation are free of artifacts caused by chemical fixation, dehydration, cryo-protectants, or resin embedding, as is often the case in other sample preparations [6]. In general, high-pressure freezing followed by direct imaging or freeze-substitution and ultramicrotomy is recommended for samples to be imaged with electron microscopy. However, there are circumstances where cryo-fixation is not possible or requires additional preparation. Large volume samples for instance, such as a whole mouse brain, can only be prepared by prolonged immersion for staining with osmium tetroxide before resin embedding, as has been done to completely map neuronal networks [7]. In biological studies, it is also frequently necessary to relate nanoscale structure with physical biochemical activity or functionality. To meet this end, tagging of specific molecules by gold [8] or fluorescence dyes [9,10], is almost inevitable. To arrest cell-dynamics and preserve the structure prior to tagging, in general, involves chemical fixation (e.g. using low percentage paraformaldehyde).
The realization that chemical fixation may cause unwanted structural or chemical changes inside biological specimens has seen much development in the past. On molecular level, extensive studies on the effects [11–17] of osmotic potentials, pH, and buffers of the fixatives on isolated proteins as well as tissue samples have been carried out. In clinical histology and cytology, the artifacts associated with fixation have been studied [18] and novel fixation-free methods have gained great interests through the years [19]. In terms of nanoscale cellular architecture, various research groups have reported morphological changes on specific organelles caused by fixations, such as the size and shape of vesicles and liposomes [16], using nano-resolution techniques like EM and AFM. Since most nano-resolution techniques are invasive to some extent (EM needs frozen or dehydrated samples, AFM requires mechanical perturbation), morphological changes are observed from limited numbers of unpaired samples and are at best indirect evidence of the amount of physical alteration due to fixation. To gain a better understanding of the true alterations to ultrastructure during fixation, a direct measurement of a physical quantity representing nanostructure before and after fixation is required.
To meet this end, we employed the state-of-the-art live cell Partial Wave Spectroscopic (PWS) microscopy to monitor cell changes during fixation [20]. As a novel optical cytometry, PWS couples spectroscopy with microscopy [21] and its ability to access the nanoscale is grounded in a previously overlooked difference between resolution and detectability. While sub-diffractional length scales are not resolvable, they are still detectable through spectral analysis, as in the case of live cell PWS with sensitivity from 20 nm to 200 nm [22]. At each pixel, an interference signal between the cell-substrate (glass or Petri dish) reflection and cellular refractive index (RI) variations is recorded.
The standard deviation of the interference spectra (Σ) is proportional to a measure of the heterogeneity of the RI distribution. For macromolecules, the refractive index is generally linearly related to the mass-density by n = n water + αρ, where α is the refractive increment ~ 0.17 cm3/mg for most biological specimens [23] (the special case includes pigmented molecules as hemoglobin and melanin). In other words, Σ represents the nanoscopic macromolecular arrangement: Σ increases with correlation length and standard deviation of the refractive index distribution, and refractive index distribution is proportional to the mass-density distribution of the sample. Alternatively, in a fractal medium such as chromatin, this mass density distribution can be understood as a proportional change in the fractal dimension, D. There are five major advantages of using live cell PWS microscopy to study the change of ultrastructure during fixation: 1. Non-invasive measurement enables a one-on-one comparison of ultrastructure of the same cells before and after treatment, 2. Native mass-density contrast imaging requires no additional treatment and introduces no structural alterations, 3. Nanoscale sensitivity reveals possible changes ranging from 20 nm to 200 nm, 4. High-throughput imaging of around 15 s per frame facilitates statistical analysis, 5. Cells measured by in-vitro PWS experiments in liquid are suitable for further treatment such as staining and dehydration, therefore it is possible to conduct correlative experiments on the same cells with nanoscale resolving techniques like electron microscopy. Due to these features, PWS microscopy is the ideal system for studying the effects of chemical fixation on the cellular nanostructure.
Chemical fixation is conventionally employed for molecular biology applications like Hi-C, immunofluorescent, immunohistochemical experiments, clinical sample preservation, nanocytometry, and electron microscopy resin embedding. Consequently, the effects of the most commonly used protocols are of crucial importance in interpretation of the data. In this study, we employed PWS microscopy to analyze the changes in cellular ultrastructure induced by chemical fixation in the three most commonly used protocols: glutaraldehyde and formaldehyde fixed individual cells flat-embedded in Spurr resin for TEM [24], 4% PFA cell fixation [25] for immuno-labeling, and 95% ethanol (EtOH) cell fixation [26] for study of nucleic acids and clinical preparation. For the resin embedding study, we further prepared thin sections of the same cells imaged by PWS and obtained high-resolution TEM images of the cellular structure as a test of the quality of sample preparation. To quantitatively characterize the structural alterations, we compared the variously fixed cells directly to their live cells counterparts using the statistics of PWS Σ map in terms of mean value, coefficient of variance, skewness, kurtosis, and entropy. After aligning the PWS images at each fixation step, we calculated pixel-to-pixel cross correlation coefficient to quantify the similarity of the cellular structure before and after treatment. Finally, to analyze the ability of various fixatives (4% PFA, 2%GA + 2.5%FA, and 95% EtOH) to preserve clinically relevant differences between populations, we used as a model case-control system, HeLa cells treated with Daunorubicin, to compare with untreated partners before and after fixation. Daunorubicin (and doxorubicin) is commonly used chemotherapeutic agent known to induce a plurality of effects on cells, including intra-nuclear effects such as nucleosome expulsion, topoisomerase I inhibition, and DNA fragmentation [27]. Remarkably, even though chemically fixed cells are uncorrelated with their initial state, population level differences are maintained after cellular fixation. Consequently, this indicates that biologically relevant nanoscopic structural information can be extracted even after fixation. In summation, we found that qualitative analysis was accurate for some experiments involving chemical fixation, but quantitative analysis of nanoscale structure may not be possible for all fixatives and may depend on the underlying structural perturbation under investigation.
2. Results
2.1. PWS microscopy visualization of fixation process
In this work, we utilized live cell PWS microscopy to quantitatively study the effect of chemical fixation on the cellular nanoarchitecture. In brief, HeLa cells (ATCC) were grown on glass-bottom 5 ml petridishes and imaged as previously described at 37 C and 5% CO2 in cell appropriate media (for full description, see Materials and Methods). After acquisition of live cell structure, cells were processed with one of the tested protocols (4% PFA, 95% EtOH, and TEM resin-embedding). To control for possible changes in the cellular nano-architecture due to naturally occurring time-dependent reorganization, sequential live cell measurements were taken one minute apart for comparative analysis. While there are numerous criteria by which one could define whether the fixative employed preserved the underlying cellular features, we focused on understanding the effects on both spatially dependent and spatially independent (bulk) properties. As described above, statistical analysis of the underlying structural transformation was performed by measuring both changes in bulk properties (mean intensity, variance, skewness, kurtosis, and entropy) as well as by measuring spatial variations using a pixel-to-pixel cross correlation for various regions of interest (see Materials and Methods for complete characterization). An ideal fixative would preserve both the bulk properties and the spatial features of the live cell state.
The fixatives employed in this work had common minor effects on microscopic features, such as cell size, cell-cell contacts, or organelle location (Fig. 1). For instance, as is commonly seen, we observed that 95% EtOH caused significant cellular shrinkage due to dehydration (Fig. 1(D)), whereas 4% PFA had a negligible effect on cell size (Fig. 1(B)). Furthermore, while 4% PFA and 2% FA-2.5% GA have different utilizations, they produce similar microscopic cellular features (Fig. 1(F)), owing to their action as cross-linking agents.
Fig. 1.
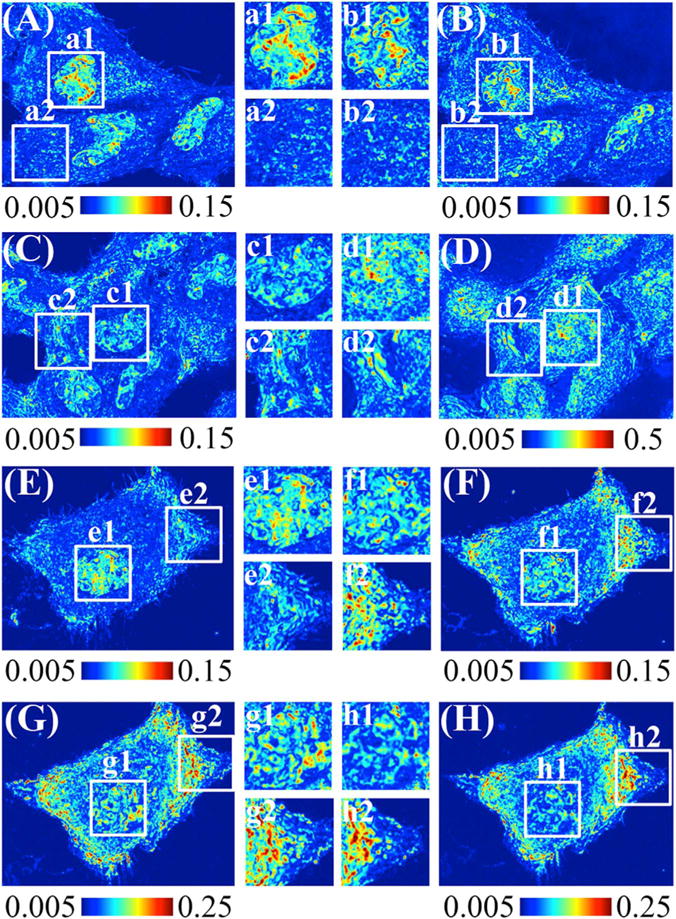
PWS measurement of Hela Cells at different stages of fixation. The higher value (redder) indicates higher level of mass-density heterogeneity. (A) (C) (E) show live Hela cells without any treatment. (B) After 4% PFA fixation. (D) After 95% EtOH fixation. (F)–(H), after 2.5% GA and 2% FA, after 1% OsO4, and after serial ethanol dehydration. Color scale: (A) (B) (E) (F) from 0.005 to 0.15, (D) from 0.005 to 0.5, (G) and (H) from 0.05 to 0.25. The inset squares are higher magnification images of nucleus and cytoplasm region.
Upon PFA fixation (Fig. 1(B)), micron-scale clustered regions formed, as indicated by the high value globules inside the nuclei, while the cytoplasm remained relatively intact. Comparatively, for the 95% ethanol fixed sample (Fig. 1(D)), we observed a significant increase of the overall Σ values throughout the cell. Finally, preparation of TEM resin-embedded cells required additional steps in addition to fixation: 1. 2.5% GA and 2% FA fixation (Fig. 1(F)), 1% OsO4 post-fixation (Fig. 1(G)), and serial ethanol dehydration (Fig. 1(H)). After GA-FA fixation, the nucleus showed similar changes as those observed in PFA fixation, with the formation of discrete micron-scale clusters. Σ intensity continued to increase after osmium staining, and increased furthermore after serial ethanol (Fig. 1(G) and Fig. 1(H)).
2.2. Correlation analysis of overall Σ distribution from cell to cell
To characterize the effects of these fixatives on the nanoscale organization, we analyzed properties of the cellular nanoarchitecture due to the live-to-fixed transition. To fully quantify changes in bulk statistical properties of the cellular mass-density distribution, we calculated the mean, coefficient of variance (CV), kurtosis, skewness, entropy of Σ for the nucleus and cytoplasm in at least 60 cells for each fixation group. We plotted the mean Σ in nuclei and cytoplasm respectively for all experimental conditions (Fig. 2) to compare the difference in the nanostructural alterations induced by different fixatives. The percentage change in mean Σ for nuclei and cytoplasm is listed in Table 1. For full characterization of bulk properties and number of cells measured, see SI Table 1–5.
Fig. 2.
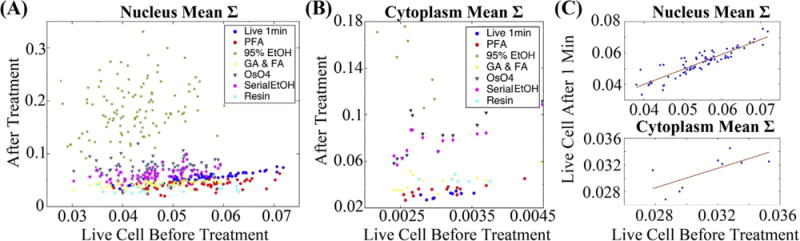
Correlation of the bulk properties between live cells and cells at different stage of treatments. We plotted the mean Σ in both nucleus (A) and cytoplasm (B) of live cell before treatment against the value after treatment. For (A), each data point represents one full nucleus. For (B), each data point represents one field of view in PWS measurement, may contain cytoplasm for multiple cells. (C) Enlarged nuclear and cytoplasmic mean Σ for live cells measured 1 min apart.
Table 1.
Changes and correlation coefficient of mean Σ in nucleus and cytoplasm compared to live cell.
| Live 1 min apart | PFA | 95% EtOH | GA and FA | OsO4 | Serial ETOH | Resin | |
|---|---|---|---|---|---|---|---|
| Nucleus | |||||||
| Mean Σ Change | −1.8% | −12.7% | 365.2% | −4.2% | 62.5% | 29.2% | −37.7% |
| Mean Σ Correlation | 0.9 | 0.42 | 0.31 | 0.43 | 0.22 | 0.44 | 0.33 |
| Cytoplasm | |||||||
| Mean Σ Change | 0% | 12.9% | 453.8% | 43.3% | 176.7% | 160% | 33.3% |
| Mean Σ Correlation | 0.71 | 0.83 | 0.79 | 0.65 | 0.68 | 0.78 | 0.33 |
In our control measurements of live cells imaged 1 min apart, we observed a conservation of mean Σ in both the nucleus (Fig. 2(A)) and the cytoplasm (Fig. 2(B)) with a Pearson correlation coefficient of 0.9 in the nucleus and 0.8 in the cytoplasm. Furthermore, other statistical properties of the nuclear and the cytoplasmic structure, such as coefficient of variation (CV), skewness, kurtosis, and entropy remain highly correlated and approach a Pearson correlation coefficient of 0.9 in live cell control measurements (Table 1, SI Fig. 4 and SI Table 6). Significantly, none of the fixatives employed in this study resulted in full preservation of all of the bulk statistical parameters in the nucleus and cytoplasm regions between live and fixed cells. For instance, 4% PFA fixation produced a relatively strong correlation (correlation coefficients were larger than or around 0.5) in the cytoplasm region except for entropy. The performance of PFA in nucleus was even less impressive than in the cytoplasm region (Table 1, SI Fig. 4). Likewise, 95% EtOH fixation produced a weak correlation (correlation coefficients were smaller than 0.4) for all bulk properties except for cytoplasm mean value and entropy. As shown in Fig. 2, mean nuclear and cytoplasmic Σ are weakly correlated after fixation with a Pearson correlation coefficient of 0.31 for the nucleus.
Finally, in the TEM resin-embedding protocol, the least significant changes occurred during GA-FA fixation, as the correlation coefficients of these stages are similar to PFA fixation, with correlation coefficient of 0.42 between live cell state for PFA fixation and 0.43 for GA/FA fixation. However, the correlation coefficient was 0.22 after Os staining and 0.44 after serial ethanol dehydration with the live cells. After resin infiltration, the correlation coefficient of mean value in the nuclear region dropped to 0.33.
2.3. Spatial cross correlation analysis of pixelated Σ distribution
To estimate the efficacy of fixation for quantitative studies, pixel-to-pixel cross correlation coefficient (CCC) was employed. We compared the absolute CCC between live cells and cells after fixation (Fig. 3(A)). Intuitively, the alignment of pre- and post-fixation PWS maps can significantly influence the spatial correlation. Therefore, before spatial CCC analysis, we performed a non-biased, optimal alignment of the pre- and post-fixation PWS maps by maximizing the cross-correlation of the bright field images (full details can be found in S.I. Fig. 1).
Fig. 3.
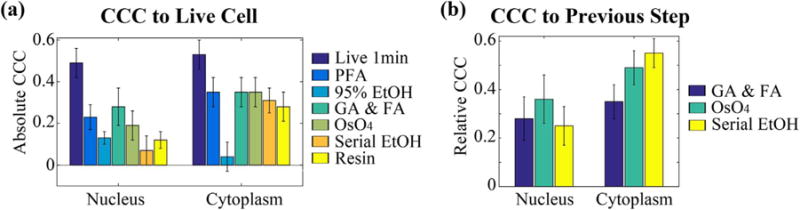
Pixel to pixel cross correlation coefficient (CCC) of PWS values in the nucleus and cytoplasm region. (A) Absolute CCC between live cells and cells at different fixation stage. (B) Relative CCC between cells at current and previous stage.
During the TEM-resin preparation, we further analyzed the influence of previous fixation step to the next fixation step, we also calculated the relative CCC between two subsequent steps (e.g. GA-FA to live cells, OsO4 to GA-FA, etc.) (Fig. 3(B)).
Under physiological growth conditions due to normal cellular dynamics, we observed the pixel-to-pixel CCC of around 0.5 for both nucleus and cytoplasm regions. For all fixations in this study, the absolute CCC after fixation was less than 0.4 in both the nucleus and the cytoplasm. 95% EtOH fixation, in particular, gave less than 0.1 with error bars extending into negative CCC for cytoplasm. Here, a negative correlation means there is a flip in the value distribution (relatively high values before fixation become relatively low values after fixation). Thus, the observance of a negative correlation indicates that fixation is potentially inverting the organization of underlying structures. However, as the magnitude of CCC is low, it is impossible to deduct the Σ (structure) distribution within population before fixation. Likewise, through analysis using the relative CCC, we observed CCC continued to drop as we went through GA-FA fixation, OsO4 post-fixation, and serial ethanol dehydration.
2.4. Mean Σ difference for daunorubicin treatment and influence of ethanol fixation
In addition to alterations after fixation within individual cells, we also tested the ability of different fixatives to preserve the difference between populations. Daunorubicin, as an intercalating agent, would induce chromatin structural changes. We used Daunorubicin treated HeLa as a model case system and compared with the untreated HeLa cells. Interestingly, live cell PWS detects a dramatic change of Σ on the time-course of a few minutes (< 15 min) in live cells treated with Daunorubicin (Fig. 4). Using this as a model system to study fixation, we treated cells with 10 μM Daunorubicin for 15 min before fixation. Cells were then re-imaged after fixation and compared with untreated controls in both the live and fixed cell state. Surprisingly, EtOH fixation maintained population level differences in Σ (Fig. 4).
Fig. 4.
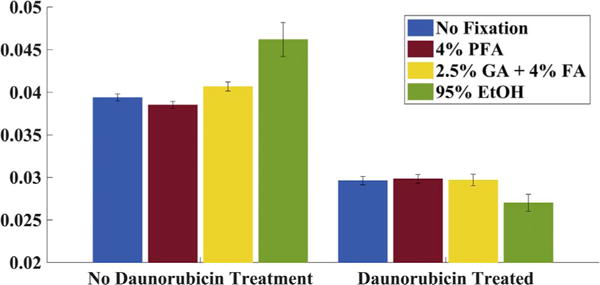
Mean Σ value of the nuclear region for HeLa cells. (A) Live HeLa cells before and after treated with Daunorubicin. (B) HeLa cells fixed by different fixatives without Daunorubicin and HeLa cells treated with Daunorubicin then fixed by EtOH.
3. Discussion
3.1. PWS microscopy visualization of fixation process
In live cells (Fig. 1(A)(C)(E)), nuclei were distinctly identifiable from the cytoplasm due to the inherent difference in their internal nanoscale structure. As described previously, higher Σ represents an increase in the nanoscale mass-density heterogeneity, which indicates more variations in nanoscale structure. In the nucleus, this heterogeneity arose in part due to differential chromatin compaction, with regions of dense chromatin clusters neighboring open surfaces [28]. Upon fixation, different levels of changes occurred throughout the cells. PFA and GA-FA fixation preserved on a macro scale the distinction between nucleus and cytoplasm, possibly due to their similar cross-linking interaction with cellular component. However, unlike with PFA fixation, GA-FA fixation caused asymmetric cytoplasmic changes. EtOH treatment blurred the spatial distinction greatly, especially within the peri-nuclear region, increased to comparable values of the nucleus (Fig. 1(D)). The values of Σ also increased significantly after EtOH fixation, which is possibly induced by volume-shrinkage (dehydration by EtOH). Qualitatively, 4% PFA and 2.5% GA and 2% FA fixations did not alter the microscopic cellular features, which was not the case in 95% ethanol fixation. However, while 4% PFA and 2.5% GA and 2% FA preserved the microscopic morphology, they produced distinct influences on the spatial distribution of the underlying nanoscale topology.
3.2. Correlation analysis of overall Σ distribution from cell to cell
As described previously, an ideal fixative solution would at least maintain overall properties of the cellular nanoarchitecture. In particular, bulk features such as the mean and CV of the nuclear or cytoplasmic Σ should remain correlated from cell-to-cell after fixation and spatial features. For example, in live cells measured 1 min apart, both the correlation coefficient and linear coefficient of the mean Σ value should approach 1. In this context, a high correlation coefficient indicates the cellular topology (nanoscale mass-density distribution) remains intact after fixation. However, the fact that none of the fixatives tested in this work preserved all of the bulk statistical parameters indicates that considerable structural changes occurred during fixation process. Interestingly, in comparison of 95% EtOH fixation alone with TEM resin-embedding serial EtOH dehydration, the serially dehydrated samples are significantly more correlated with live cells (correlation coefficient after 95% EtOH fixation was 0.31), potentially caused by a partial preservation of those cellular structures by GA-FA and OsO4 fixation.
3.3. Spatial cross correlation analysis of pixelated Σ distribution
In quantitative structure-function relation studies, the usage of fixation is not only to preserve the structure, but also to near-instantaneously arrest cell-dynamics. In this context, studies of the nanoscopic structure-function relationship in eukaryotic cells depend on accurately preserving the spatial distribution of nanoscale features (ideally, the nanoscale mass-density distribution should remain exactly the same before and after fixation). In ideal circumstances, the pixel-to-pixel CCC after fixation should be higher than the CCC between two live cell measurements taken 1 min apart, as the structure in live cells is constantly evolving even at these short time-scales. As shown in Fig. 3(A), the CCC of live cells imaged 1 min apart is around 0.5 for both nucleus and cytoplasm region. So an ideal fixation would yield a higher CCC value, as it should arrest cellular dynamics. However, for all fixations in this study, the less than 0.4 CCC and even less than 0.1 CCC in the case of EtOH for cytoplasm, suggests extremely weak or no correlation at all before and after fixation. This means that even though the macroscale features are likely to be preserved, the nanoscale spatial structural distribution has gone through dramatic changes during fixation, and it is challenging to relate the cellular nanostructures before and after fixation.
3.4. Mean Σ difference for daunorubicin treatment and influence of chemical fixation
Surprisingly, all three fixations (4% PFA, 2%GA + 2.5%FA, 95% Ethanol) maintained population level differences in the nanoscopic structural information, as it preserved the differences in nuclear mean Σ (Fig. 4). This suggests that while nanoscopic organization is transformed during fixation, it may be possible to still extract clinically meaningful nanoscopic information after fixation. As a result, PWS measurement presents a robust method for systematically analyzing preparation protocols aimed at studying nanoscopic structural features when fixation cannot be avoided.
4. Conclusion
Using live cell PWS, a nanoscale live-cell microscope utilizing spectral analysis, we presented the first direct comparison between cells at different stages of fixation as well as a systematic analysis of the difference in the nanoscale topology of live and fixed cells. In summary, PFA and GA & FA fixations are more capable of preserving the overall cellular structures and are sufficient in enabling qualitative structural analysis than 95% EtOH fixation. For quantitative analysis, we found that all fixation protocols tested showed low spatial correlation (pixel-to-pixel cross correlation) in their mass-density distribution, and it is not intuitive to build a straightforward quantitative relation between the structures measured before and after fixation. While this may not be possible in all cases, these results indicate that live cell imaging or cyro-fixation should be used whenever possible in place of chemical fixation. Additionally, while direct fixation with commonly used fixatives (PFA, GA, EtOH) causes dramatic changes in cellular topology, it can still be used for comparative population analysis of the chromatin nanoarchitecture.
5. Materials and methods
5.1. Cell culture
Hela Cells (ATCC) were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) between passages 5–20. Unless otherwise noted, PWS microscopy measurements were obtained from live cells grown on uncoated size-1 glass coverslips attached to 50 mm petri dishes (MatTek, Ashland Massachusetts). Petri dishes were seeded with between 10,000 and 50,000 cells in 2 ml of RPMI-1640 at the time of passage. For correlative, step-by-step analysis with transmission electron microscopy, cells were seeded at the time of platting onto gridded size 2 german-glass bottom Petri dishes (MatTek Corp.), with cells and the grid on different focal planes. Cells were allowed at least 24 h to adhere to the glass bottom and grow. All cells were imaged when confluence was between 40–60% of the surface area using a stage-top incubator (In Vivo Inc.) at 37 °C in a 5% CO2.
5.2. PWS microscopy measurement
The PWS system was built onto a commercial Leica DMIRB microscope as previously described. We used broadband light (500–700 nm) from a xenon lamp, and focused this light onto the same plane as Kohler illumination (NA of illumination = 0.15, NA of collection = 1.25) by an objective lens. The backscattered image from the cells passed through a liquid crystal runnable filter (Cri) tuned 500–700 nm with spectral resolution of 7 nm. We then projected the resulting monochromatic image with a 40× magnification CMOS camera (Hamamatsu ORCA-Flash 2.8). The pixel size of the object plane is 250 nm by 250 nm.
5.3. Live cell time lapse PWS imaging as control
As the structure of cells can evolve in time, Hela cells in PWS built-in incubator 1 min apart as control to characterize influence of the live cell activity on cellular mass-density distribution change (SI. Fig. 2).
5.4. Paraformaldehyde (PFA) and ethanol fixation study
For PFA study, we fixed Hela cells with 4% PFA (EMS) for immunofluorescence study for 20 min at room temperature. For EtOH study, we fixed the Hela cells with 95% ethanol for 20 min at room temperature. We performed PWS measurement before and after treatment for both experiments.
5.5. Flat embedding in Spurr’s Resin for TEM fixation study
The experiments conducted to test the structural changes during TEM resin section sample preparation are shown in Fig. 1.
5.5.1. Fixation of Glutaraldehyde (GA) and Formaldehyde (FA)
Fixative was consisted of 2.5% GA (EMS) and 2% FA (EMS) in 1× PBS (Dulbecco’s Phosphate-Buffered Saline) with pH adjusted to 7.4 by NaOH (Sigma Aldrich). Cells were immersed for 1 h at room temperature for sufficient fixation.
5.5.2. Fixation of Osmium Tetroxide (OsO4)
We drained the GA and FA fixative and rinsed the cells with DI water for 3 times, 5 min each time. We post-fixed the cells with 1% OsO4 (EMS) prepared by DI water for 45 min at room temperature. After post-fixation, we removed OsO4 solution and rinsed the cells with DI water for three changes of 10 min.
5.5.3. Serial ethanol dehydration
To not disturb the cells’ attachment to the glass bottom, we performed serial ethanol dehydration followed by flat embedding directly in the original Petri dish. Cells are gently dehydrated by immersion in ethanol prepared by DI water of different gradient (30%, 50%, 70 &, 85%, 95%) for 10 min each before fully dehydrated in 100% ethanol for four changes of 10 min. Step 1, 2, 3 and PWS measurements were conducted in sequence as shown in Fig. 5.
Fig. 5.

Workflow of monitoring TEM Resin section sample preparation by PWS.
5.5.4. Flat embedding
Dehydrated cells were infiltrated with Spurr’s resin (EMS) in ethanol (1:1) for 1 h at room temperature and then switched to Spurr’s resin in ethanol with higher concentration (2:1) overnight at room temperature. At this step, we conducted PWS measurement to visualize the cell structure with resin infiltration. Further infiltration by 100% Spurr’s resin for 2 h was conducted on the second day. Fresh Spurr’s resin was used for flat embedding and polymerized for 2 days in a 65 °C oven.
5.5.5. TEM sample preparation and imaging
After polymerization, 200 nm sections were obtained by ultramicrotome (Leica UC7) with a diamond knife (Diatome) and post stained with 1% uranium acetate aqueous solution (EMS) for nanostructure imaging by TEM to examine the quality of fixation in this protocol. HT7700 (Hitachi) TEM with a bottom-mounted CCD camera (Orius) was operated at 120 kV to capture high-contrast micrographs for cellular nanostructure analysis. For each cell on the sections, we stitched high-magnification images (usually 10–20 pictures) with a pixel size of 5 nm with MosaicJ [29] in FIJI [30] to form high-resolution images of the whole cell (SI. Fig. 3).
5.6. Chemical fixation influence on daunorubicin treatment study
Three fixatives were tested separately (4% PFA, 2.5%GA+2%FA, 95% EtOH). Live Hela cells were treated with 10 μM Daunorubicin for 15 min before fixation. PWS measurements were conducted at three time-points: before any treatment, after Daunorubicin before fixation, and after fixation. Another batch of live Hela cells were chemically fixed only and imaged by PWS.
5.7. Quantification of structural changes
First of all, we aligned the PWS images of the same cells by MATLAB (The MathWorks Inc.) by picking the combination of rotation and translation with the highest cross correlation coefficient of the two bright field images (SI Fig. 1). After alignment, we carefully cropped the nuclei from cytoplasm using Photoshop CS6 (Adobe). To quantify the structural changes during the transition from live to fixed cells, we compared the spatial distribution of mass-density before and after fixation in terms of mean value, coefficient of variance, skewness, kurtosis, and entropy in nuclei and cytoplasm separately. Correlation of the bulk properties between live cells and treated cells was estimated by correlation coefficient and linear coefficient. We also calculated the pixel-to-pixel cross correlation coefficient of nuclei to quantify the microscopic changes caused by sample preparation.
Supplementary Material
Acknowledgments
This work was funded by NIH Grants nos. R01CA200064, R01CA165309, and R013B016983, NSF Grant no. CBET-1240416, and LUNgevity Foundation (2015–2016 Early Detection Award, no. 2015-04). The TEM word made use of the EPIC facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN.
Appendix A. Supplementary material
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.yexcr.2017.06.022.
References
- 1.Dubochet J, McDowall A, Menge B, Schmid E, Lickfeld KG. Electron microscopy of frozen-hydrated bacteria. J Bacteriol. 1983;155(1):381–390. doi: 10.1128/jb.155.1.381-390.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santana BP, Nedel F, Perelló Ferrúa C, da Silva AF, Demarco FF, Lenin Villarreal Carreño N. Comparing different methods to fix and to dehydrate cells on alginate hydrogel scaffolds using scanning electron microscopy. Microsc Res Tech. 2015 doi: 10.1002/jemt.22508. [DOI] [PubMed] [Google Scholar]
- 3.Studer D, Graber W, Al‐Amoudi A, Eggli P. A new approach for cryofixation by high‐pressure freezing. J Microsc. 2001;203(3):285–294. doi: 10.1046/j.1365-2818.2001.00919.x. [DOI] [PubMed] [Google Scholar]
- 4.Plattner H, Artalejo AR, Neher E. Ultrastructural organization of bovine chromaffin cell cortex—analysis by cryofixation and morphometry of aspects pertinent to exocytosis. J Cell Biol. 1997;139(7):1709–1717. doi: 10.1083/jcb.139.7.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Müller M, Moor H. Cryofixation of Thick Specimens by High Pressure Freezing, The Science of Biological Specimen Preparation. SEM AFM OÕHare, Chicago, IL. 1984, pp:131–138. [Google Scholar]
- 6.Ebersold HR, Cordier JL, Lüthy P. Bacterial mesosomes: method dependent artifacts. Arch Microbiol. 1981;130(1):19–22. doi: 10.1007/BF00527066. [DOI] [PubMed] [Google Scholar]
- 7.Mikula S, Binding J, Denk W. Staining and embedding the whole mouse brain for electron microscopy. Nat Methods. 2012;9(12):1198–1201. doi: 10.1038/nmeth.2213. [DOI] [PubMed] [Google Scholar]
- 8.Amiry-Moghaddam M, Ottersen OP. Immunogold cytochemistry in neuroscience. Nat Neurosci. 2013;16(7):798–804. doi: 10.1038/nn.3418. [DOI] [PubMed] [Google Scholar]
- 9.de Souza N. Super-resolution CLEM. Nat Methods. 2015;12(1):37. [Google Scholar]
- 10.Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529(7586):418–422. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caulfield JB. Effects of varying the vehicle for OsO4 in tissue fixation. J Biophys Biochem Cytol. 1957;3(5):827. doi: 10.1083/jcb.3.5.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finean J. The effects of osmium tetroxide fixation on the structure of myelin in sciatic nerve. Exp Cell Res. 1954;6(2):283–292. doi: 10.1016/0014-4827(54)90175-5. [DOI] [PubMed] [Google Scholar]
- 13.Iqbal S, Weakley B. The effects of different preparative procedures on the ultrastructure of the hamster ovary. Histochemistry. 1974;38(2):95–122. doi: 10.1007/BF00499659. [DOI] [PubMed] [Google Scholar]
- 14.Johnson TJ. Aldehyde fixatives: quantification of acid‐producing reactions. J Electron Microsc Tech. 1985;2(2):129–138. [Google Scholar]
- 15.Mason JT, O’leary TJ. Effects of formaldehyde fixation on protein secondary structure: a calorimetric and infrared spectroscopic investigation. J Histochem Cytochem. 1991;39(2):225–229. doi: 10.1177/39.2.1987266. [DOI] [PubMed] [Google Scholar]
- 16.Murk J, Posthuma G, Koster A, Geuze H, Verkleij A, Kleijmeer M, et al. Influence of aldehyde fixation on the morphology of endosomes and lysosomes: quantitative analysis and electron tomography. J Microsc. 2003;212(1):81–90. doi: 10.1046/j.1365-2818.2003.01238.x. [DOI] [PubMed] [Google Scholar]
- 17.Schnell U, Dijk F, Sjollema KA, Giepmans BN. Immunolabeling artifacts and the need for live-cell imaging. Nat Methods. 2012;9(2):152–158. doi: 10.1038/nmeth.1855. [DOI] [PubMed] [Google Scholar]
- 18.Rastogi V, Puri N, Arora S, Kaur G, Yadav L, Sharma R. Artefacts: a diagnostic dilemma–a review. J Clin Diagn Res: JCDR. 2013;7(10):2408. doi: 10.7860/JCDR/2013/6170.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fereidouni F, Datta-Mitra A, Demos S, Levenson R, editors. SPIE BiOS: International Society for Optics and Photonics. 2015. Microscopy with UV Surface Excitation (MUSE) for slide-free histology and pathology imaging. [Google Scholar]
- 20.Almassalha LM, Bauer GM, Chandler J, Gladstein S, Cherkezyan L, Stypula-Cyrus Y, Weiberg S, Zhang D, Thusgaard Ruhoff P, Roy H, Subramanian H, Chandel N, Szleifer I, Backman V. Label-free imaging of the native, living cellular nano-architecture using partial-wave spectroscopic microscopy. Proc Natl Acad Sci USA. 2016;113(42):E6372–E6381. doi: 10.1073/pnas.1608198113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Subramanian H, Pradhan P, Liu Y, Capoglu IR, Li X, Rogers JD, et al. Optical methodology for detecting histologically unapparent nanoscale consequences of genetic alterations in biological cells. Proc Natl Acad Sci. 2008;105(51):20118–20123. doi: 10.1073/pnas.0804723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cherkezyan L, Capoglu I, Subramanian H, Rogers J, Damania D, Taflove A, et al. Interferometric spectroscopy of scattered light can quantify the statistics of subdiffractional refractive-index fluctuations. Phys Rev Lett. 2013;111(3):033903. doi: 10.1103/PhysRevLett.111.033903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies H, Wilkins M, Chayen J, La Cour L. The use of the interference microscope to determine dry mass in living cells and as a quantitative cytochemical method. Q J Microsc Sci. 1954;3(31):271–304. [Google Scholar]
- 24.Reymond OL, Pickett‐Heaps JD. A routine flat embedding method for electron microscopy of microorganisms allowing selection and precisely orientated sectioning of single cells by light microscopy. J Microsc. 1983;130(1):79–84. doi: 10.1111/j.1365-2818.1983.tb04200.x. [DOI] [PubMed] [Google Scholar]
- 25.Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, et al. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods. 2013;10(4):329–331. doi: 10.1038/nmeth.2374. [DOI] [PubMed] [Google Scholar]
- 26.Ludyga N, Grünwald B, Azimzadeh O, Englert S, Höfler H, Tapio S, et al. Nucleic acids from long-term preserved FFPE tissues are suitable for downstream analyses. Virchows Arch. 2012;460(2):131–140. doi: 10.1007/s00428-011-1184-9. [DOI] [PubMed] [Google Scholar]
- 27.Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim Biophys Acta. 2014;1845(1):84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JS, Pradhan P, Backman V, Szleifer I. The influence of chromosome density variations on the increase in nuclear disorder strength in carcinogenesis. Phys Biol. 2011;8(1):015004. doi: 10.1088/1478-3975/8/1/015004. [DOI] [PubMed] [Google Scholar]
- 29.Thévenaz P, Unser M. User‐friendly semiautomated assembly of accurate image mosaics in microscopy. Microsc Res Tech. 2007;70(2):135–146. doi: 10.1002/jemt.20393. [DOI] [PubMed] [Google Scholar]
- 30.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.