

SUMMARY
Melanoma accounts for over 80% of skin cancer-related deaths and current therapies provide only short-term benefit to patients. Here, we show in melanoma cells that maternal embryonic leucine zipper kinase (MELK) is transcriptionally upregulated by the MAP kinase pathway via transcription factor E2F1. MELK knockdown or pharmacological inhibition blocked melanoma growth and enhanced the effectiveness of BRAFV600E inhibitor against melanoma cells. To identify mediators of MELK function, we performed stable isotope labeling with amino acids in cell culture (SILAC) and identified 469 proteins that had downregulated phosphorylation after MELK inhibition. Remarkably, 139 of these proteins were previously reported as substrates of BRAF or MEK, demonstrating that MELK is an important downstream mediator of the MAPK pathway. Furthermore, we show that MELK promotes melanoma growth by activating NF-κB pathway activity via Sequestosome 1 (SQSTM1/p62). Collectively, these results underpin an important role for MELK in melanoma growth, downstream of the MAPK pathway.
eTOC Blurb
Janostiak et al. find that MELK is overexpressed in melanoma and is necessary for melanoma growth. MELK regulates NF-κB pathway via SQSTM1, which in part is necessary for its ability to promote melanoma growth.
INTRODUCTION
Melanoma is the deadliest form of skin cancer, accounting for ~80% of skin cancer-related deaths (Miller and Mihm, 2006). Over 85% of melanomas are caused by mutations in BRAF or NRAS genes and mutation or deletion of the NF1 gene (Cancer Genome Atlas, 2015). These alterations can activate the MAP kinase pathway, which in turn promotes proliferation and facilitates melanoma initiation and progression (Downward, 2003; Karnoub and Weinberg, 2008; Wellbrock et al., 2004a; Wellbrock et al., 2004b).
After the initial discovery of BRAF mutations in a large percentage of melanomas (Davies et al., 2002), specific and highly-effective small-molecule inhibitors that target either BRAF or MEK mutants were developed and used to treat BRAF-mutant metastatic melanoma in clinic (Chapman et al., 2011; Flaherty et al., 2012). BRAF inhibitors alone or in combination with MEK inhibitors have shown some success; however, within months of treatment, drug resistance emerges and renders these drugs ineffective (Kim et al., 2013; Rizos et al., 2014; Shi et al., 2014). The alternative approach of targeting the MAP kinase (MAPK) pathway in NRAS-mutant and NF1-deficient melanoma has not proven effective (Ascierto et al., 2013; Whittaker et al., 2013). Similarly, new immunotherapeutic approaches, such as anti-CTLA-4 antibody (ipilimumab) and anti-PD1/PD-L1 antibodies (pembrolizumab or nivoluzumab), have benefited only a subset of patients (Hodi et al., 2010; Postow et al., 2015; Robert et al., 2015). Thus, new strategies for treating melanoma and improving patient survival are needed.
Maternal embryonic leucine zipper kinase (MELK) is a serine/threonine protein kinase that regulates cell cycle, stem cell renewal, and apoptosis (Badouel et al., 2006; Davezac et al., 2002; Jung et al., 2008; Nakano et al., 2005). Interestingly, MELK knockout mice are viable and display no adverse phenotypes (Wang et al., 2014). This information and the availability of small-molecule inhibitors of MELK with anti-cancer activity in breast and other cancers indicates that MELK might be a druggable target for cancer cell-selective therapy (Gray et al., 2005; Kohler et al., 2017; Nakano et al., 2005).
Here, we show that MELK is necessary for melanoma growth. We found that MELK regulated the phosphorylation of a large number of proteins, many of which were previously identified as substrates of BRAF and/or MEK. We also demonstrate that MELK regulation of the NF-κB pathway mediates, in part, the melanoma-promoting activity of MELK. Collectively, our studies identify MELK as an important regulator of melanoma growth downstream of the MAPK pathway.
RESULTS
MELK is overexpressed in melanoma by the MAPK pathway
MELK is highly overexpressed in several cancer types and its inhibition has been shown to block the tumor growth of some cancers (Inoue et al., 2016; Joshi et al., 2013; Kato et al., 2016; Wang et al., 2016; Wang et al., 2014). Interestingly, MELK knockout mice are viable and do not show any specific phenotypes (Wang et al., 2014). Therefore, MELK appears to be a potentially effective and cancer cell selective target. The role of MELK in melanoma has not been studied and very few MELK substrates have been identified thus far. Therefore, we asked if MELK plays a role in melanoma growth. We first analyzed the expression of MELK in previously published gene expression datasets of patient-derived melanoma samples. MELK was overexpressed in patient-derived melanoma samples compared to normal skin samples (Figure 1A and Figure S1A–C). Additionally, MELK expression significantly increased with melanoma spreading and metastatic melanoma had higher MELK expression than primary melanoma (Figure 1B and Figure S1B–C). Notably, a previous study identified increased expression of MELK and other genes as a genetic signature that predicts melanoma progression (Ryu et al., 2007). Collectively, these results suggest an important role for MELK in melanoma.
Figure 1. MELK is upregulated in melanoma by the MAPK pathway through the transcription factor E2F1.

Indicated melanoma datasets were analyzed for MELK mRNA expression. Relative expression in patient-derived melanoma samples compared to normal skin (A) and in N1+ versus N0 or primary versus metastatic melanoma (B) is shown. C. MELK mRNA expression was measured after treatment with vemurafenib (2 μM) or trametinib (250 nM) for 24 h. Relative mRNA MELK expression is plotted in reference to DMSO treated melanoma cell lines. D. MELK protein expression was measured by immunoblotting in indicated melanoma cell lines after treatment with DMSO (−), vemurafenib (V; 2 μM), or trametinib (T; 250 nM) for 24 h. ACTINB was used as the loading control. E. mRNA expression for the indicated genes was measured in A375 cells 24 h after DMSO, vemurafenib (2 μM), or trametinib (250 nM) treatment. mRNA expression is shown relative to DMSO treated A375 cells. F. A375 cells expressing either E2F1 or non-silencing (NS) shRNA were analyzed for E2F1 (left) or MELK (right) mRNA expression using RT-qPCR. mRNA expression in E2F1 shRNA expressing cells is shown relative to NS shRNA expressing cells. G. Indicated protein levels were monitored in A375 cells expressing either E2F1 or NS shRNAs. ACTINB was used as a loading control. H. Relative MELK promoter-driven firefly luciferase (MELK-FLuc) activity is shown in A375 cells treated with DMSO or vemurafenib and transfected with or without a mutated E2F1 DNA binding site-containing MELK-FLuc construct. I. A375 cells treated with DMSO or vemurafenib (2 μM) for 24 h were analyzed for E2F1 recruitment on either the MELK or GAPDH promoter by chromatin immunoprecipitation assay. IgG antibody was used as a negative control. % enrichment relative to input under indicated conditions is shown. Data is presented as ±SD for three biological replicates. *, **, and *** represent p<0.05, p<0.01, and p<0.001, respectively. See also Figure S1.
We aimed to decipher the mechanism of MELK overexpression in melanoma. One of the most altered signaling pathways in melanoma is the MAPK pathway, which is constitutively active in over 85% of melanomas, because of mutations in BRAF/NRAS genes or inactivation of the NF1 gene (Cancer Genome Atlas, 2015). Therefore, we asked if the MAPK pathway is necessary for transcriptional upregulation of MELK in melanoma. We treated three BRAF-mutant melanoma cell lines (A375, M14 and SKMEL-28) with the BRAFV600E inhibitor vemurafenib or the MEK inhibitor trametinib. Treatment of these cells with either inhibitor reduced MELK mRNA (Figure 1C) and protein (Figure 1D) levels. Together, these results demonstrate that transcriptional upregulation of MELK in melanoma is primarily mediated by the MAPK pathway.
Transcription factor E2F1 is required for transcriptional upregulation of MELK in melanoma cells
To determine the mechanism of transcriptional upregulation of MELK, we analyzed the MELK promoter sequence using PROMO and rVista2.0. We identified conserved DNA binding sites for E2F and MYC transcription factors. We then asked if any of these transcription factors were upregulated, like MELK, by the action of the MAPK pathway. We treated A375 and M14 cells with vemurafenib or trametinib and analyzed the expression of E2F1-8 and MYC. Only MYC, E2F1, and E2F2 were significantly downregulated after treatment with vemurafenib or trametinib (Figure 1E and Figure S1D). Therefore, we individually knocked down MYC, E2F1, and E2F2 in melanoma cell lines using short hairpin RNAs (shRNAs) and analyzed the effect of these knockdowns on MELK expression. Knockdown of the transcription factor E2F1 significantly reduced MELK expression (Figure 1F–G), while E2F2 or MYC knockdown did not (Figure S1E–H).
Next, we wanted to determine if the transcription factor E2F1 directly targets MELK. To this end, we cloned the MELK promoter with a E2F1 DNA binding site upstream of a firefly luciferase reporter gene. This MELK-FLuc construct was tested for responsiveness to the BRAF inhibitor vemurafenib. A375 cells transfected with the MELK-FLuc construct had reduced luciferase activity after vemurafenib treatment (Figure 1H). We also mutated the E2F1 DNA binding site on the MELK promoter and observed a substantial reduction of MELK promoter-driven reporter activity, making this construct non-responsive to vemurafenib treatment (Figure 1H). Finally, to determine if E2F1 directly associates with the MELK promoter in vivo, we performed chromatin immunoprecipitation (ChIP). We treated A375 cells with vemurafenib, or with dimethyl sulfoxide (DMSO) as a control, and performed ChIP for E2F1 for the MELK promoter or, as a control, the GAPDH promoter. E2F1 was significantly enriched at the MELK promoter compared to the negative control GAPDH promoter (Figure 1I). Additionally, E2F1 binding of the MELK promoter was inhibited by vemurafenib treatment (Figure 1I). Collectively, these results demonstrate that the MAPK pathway stimulates E2F1 expression, which in turn activates MELK transcription by directly binding to the MELK promoter in melanoma cells.
MELK inhibition blocks melanoma growth
Because MELK is a kinase that is highly expressed in melanoma cells, we asked if MELK is a potential target for melanoma therapy. To test whether MELK inhibition would block melanoma growth, we treated melanoma cell lines with the MELK inhibitor OTSSP167 (Chung et al., 2012; Kohler et al., 2017; Wang et al., 2014). OTSSP167 treatment significantly inhibited melanoma cell line proliferation (Figure 2A) and colony formation in a soft-agar assay (Figure 2B–C and Figure S2). To confirm that the growth inhibition was due to MELK kinase inhibition and not an off-target effect, we also treated cells with a second MELK inhibitor, MELK-8a (Toure et al., 2016). Consistent with our results with OTSSP167, MELK-8a inhibited melanoma cell growth in both the proliferation (Figure S3A) and soft-agar (Figure S3B–C) assays.
Figure 2. MELK inhibition blocks melanoma cell growth in culture.

Melanoma cell lines (A375, SKMEL-28, M14, YUGASP, and MeWo) were treated with indicated concentrations of OTSSP167 and analyzed for cell proliferation using the MTT assay. Relative proliferation (%) for each melanoma cell line relative to DMSO treated cells is shown. B. Melanoma cell lines (A375, SKMEL-28, M14, YUGASP, and MeWo) were treated with indicated concentrations of OTSSP167 and analyzed for anchorage-independent growth using the soft-agar assay. Representative images for indicated melanoma cell lines under indicated treatment conditions are shown. Scale bar: 200μm. C. Relative colony size (%) for indicated cell lines at indicated treatment conditions is shown. D, E. A375 cells expressing doxycycline-inducible MELK shRNA were infected with virus for expression of either MELK WT or MELK KD and grown without or with doxycycline and analyzed for soft-agar colony formation. Representative images are shown. Scale bar: 200μm. Data is presented as ±SD for three biological replicates. *, **, and *** represent p<0.05, p<0.01, and p<0.001, respectively. See also Figure S2 and Figure S3.
To determine if the ability of MELK to promote melanoma growth was dependent on its kinase activity, we performed rescue experiments with a wild-type MELK open reading frame or a kinase dead MELK mutant (MELK-D150A). Only wild-type MELK, and not the kinase dead mutant, was able to rescue growth in soft-agar assay, showing that the kinase activity of MELK is required for its ability to promote melanoma growth (Figure 2D–E and S3D–E).
MELK inhibition blocks the growth of vemurafenib-resistant cells and delays the emergence of vemurafenib resistance
Our results showed that MELK is a downstream target of the MAPK pathway and that MELK inhibition blocks melanoma growth. Therefore, we asked if vemurafenib-resistant melanoma cell lines could also be inhibited by MELK inhibitors. We analyzed A375 and SKMEL-239 parental cell lines and vemurafenib-resistant versions of these two cell lines. To test the effectiveness of MELK inhibitors for blocking vemurafenib-resistant cell lines, we treated parental and vemurafenib-resistant A375 and SKMEL-239 cell lines with vemurafenib alone or with MELK inhibitors (OTSSP167, MELK-8a). Treatment of parental cell lines (A375, SKMEL-239) with either vemurafenib or MELK inhibitors (OTSSP167, MELK-8a) inhibited proliferation and growth in soft agar (Figures 3A–F and Figure S4A–C). In vemurafenib-resistant cell lines, however, vemurafenib did not inhibit proliferation or growth in soft agar (Figure 3A–F). However, treatment with MELK inhibitors (OTSSP167, MELK-8a) did inhibit proliferation and growth in soft agar (Figure 3A–F and Figure S4A–C). Finally, we asked if MELK inhibition can forestall the emergence of vemurafenib resistance. We treated parental A375 melanoma cells with vemurafenib alone or in combination with OTSSP167 and performed a clonogenic assay to measure the emergence of vemurafenib resistance. After 6 weeks of treatment with these drugs, we visualized and quantified the number of drug resistant clones. Treating A375 cells with vemurafenib produced several vemurafenib-resistant colonies (Figure 3G). Strikingly, combined vemurafenib and OTSSP167 treatment did not yield any drug resistant colonies (Figure 3G). Collectively, these results demonstrate that MELK inhibition can inhibit the growth of vemurafenib-resistant melanoma and that the combination of vemurafenib and OTSSP167 can forestall the emergence of vemurafenib resistance.
Figure 3. MELK inhibition is sufficient to overcome vemurafenib resistance.

A–D. Parental and vemurafenib-resistant melanoma cell lines A375 and SKMEL-239 were treated with either DMSO or indicated concentrations of Vemurafenib or OTSSP167 and analyzed for proliferation using the MTT assay. Relative proliferation (%) for each cell line relative to DMSO treated cells is shown. E. Parental and vemurafenib-resistant melanoma cell lines A375 and SKMEL-239 were treated with 1 μM vemurafenib or 50 nM OTSSP167 and analyzed for anchorage-independent growth by the soft-agar assay. Representative images for indicated melanoma cell lines under indicated treatment conditions are shown. Scale bar: 200μm. F. Relative colony size (%) for indicated melanoma cell lines at indicated treatment conditions is shown. G. A375 melanoma cells were treated with 2 μM vemurafenib alone or in combination with 50 nM OTSSP167 over a period of four weeks. Images of representative plates and surviving colonies/cells are shown. Data is presented as ±SD for three biological replicates. *, **, and *** represent p<0.05, p<0.01, and p<0.001, respectively. See also Figure S4.
SILAC identifies cellular substrates of MELK
MELK is a serine/threonine kinase for which very few substrates are known. Therefore, to comprehensively identify MELK substrates, we performed a global phosphoproteomic analysis using stable isotope labeling with amino acids in cell culture (SILAC). There were two major goals for this experiment: 1) Characterize the diversity of proteins that are phosphorylated by MELK in melanoma cells; 2) Identify potential pathway(s) targeted by MELK to promote melanoma growth. To achieve these goals, we used two melanoma cell lines (A375 and M14) in which cell proliferation is inhibited by MELK inhibition. These cell lines were cultured in light medium, which contains light carbon (12C), light nitrogen (14N), lysine, and arginine, or in heavy medium, which contains heavy carbon (13C), heavy nitrogen (15N), lysine, and arginine. After five cell doublings incorporation of these amino acids exceeded 95% (Table S1), cells in light medium were treated with DMSO and cells in heavy medium were treated with MELK inhibitor OTSSP167 for 24 h. SILAC analysis was performed to identify potential MELK targets (Figure 4A). This analysis identified 469 proteins with reduced phosphorylation in both A375 and M14 cells on the same residues (Figure S5 and Table S2, S4 and S5). Strikingly, a comparative analysis showed that 139 substrates identified by our SILAC analysis were previously identified as MAPK pathway substrates (Figure 4B and Table S2, S4 and S5) (Galan et al., 2014; Stuart et al., 2015). This was not due to the reduced MAPK signaling because OTSSP167 treatment did not inhibit ERK1/2 phosphorylation (Figure S6A). We consider to be an important observation based on our findings that the MAPK pathway regulates MELK expression and might mediate a large part of the melanoma growth and progression promoting effect of MAPK pathway.
Figure 4. SILAC analysis identifies MELK targets.

Schematic representation of major steps of SILAC analysis to identify phosphopeptides that are altered after treatment with MELK inhibitor OTSSP167 in melanoma cell lines (A375 and M14). B. Venn diagram showing commonly identified proteins that overlap with previously identified BRAFV00E and MEK targets. C. Consensus site for MELK-mediated phosphorylation amino acid recognition motif is shown. D. Consensus site for MELK-mediated phosphorylation, recognition motif based on amino acid hydrophobicity is shown. E. Ingenuity pathway analysis of the MELK targets identified by SILAC analysis revealed eight NF-κB regulatory proteins that showed downregulated phosphorylation after treatment with MELK inhibitor OTSSP167. F. Site of phosphorylation on NF-κB regulatory proteins for which reduced phosphorylation was observed in SILAC for melanoma cell lines, A375 and M14, after treatment with MELK inhibitor OTSSP167. See also Figure S5, Table S1, Table S2, Table S4 and Table S5.
We next analyzed the SILAC data to predict the preferred amino acid motif for MELK-induced phosphorylation by a newly developed method. The MELK recognition site identified was very broad and most MELK-mediated phosphorylation of identified substrates occurred at serine (Figure 4C–D).
Finally, to identify the key pathways regulated by MELK-mediated phosphorylation, we performed Ingenuity pathway analysis. We identified the NF-κB pathway as an enriched pathway (Figure 4E). In total, eight proteins involved in NF-κB pathway regulation, which had decreased phosphorylation as a result of MELK inhibition, were identified by our SILAC experiments in both A375 and M14 cell lines (Figure 4F). We decided to further study MELK-mediated regulation of the NF-κB pathway because of the previously described role for this pathway in promoting melanoma tumor growth and progression (Dhawan and Richmond, 2002; Madonna et al., 2012; Ueda and Richmond, 2006).
MELK regulates NF-κB pathway via SQSTM1/p62
Based on our SILAC and Ingenuity pathway analysis results, we asked if MELK had a role in regulating the NF-κB pathway. Consistent with our SILAC results, treating A375 and M14 melanoma cell lines with the MELK inhibitor OTSSP167 resulted in attenuated NF-κB signaling, as assessed by decreased phosphorylation of IκBα (Figure 5A). A similar reduction in NF-κB signaling was observed in melanoma cells after MELK knockdown using doxycycline-inducible shRNAs (Figure 5B). Furthermore, melanoma cell lines that were treated with MELK inhibitor and cells that expressed MELK shRNAs both showed reduced luciferase activity when transfected with a NF-κB responsive reporter plasmid (pGL4.32[luc2P/NF-κB-RE/Hygro]) (Figure 5C). Similarly, known NF-κB transcriptional targets were downregulated after MELK knockdown (Figure 5D) and after OTSSP167 treatment (Figure 5E). We also found that treatment with another MELK inhibitor, MELK-8a, attenuated NF-κB pathway activity, as determined by decreased phosphorylation of IκBα and by decreased expression of NF-κB responsive genes (Figure S6B–C).
Figure 5. MELK regulates the NF-κB pathway via SQSTM1.

A. A375 and M14 cells were treated with OTSSP167 (50 nM) for 24 h and analyzed for indicated proteins by immunoblot analysis. ACTINB was used as the loading control. B. A375 cells expressing doxycycline-inducible MELK shRNAs that were either untreated or treated with doxycycline (2 μg/ml) and analyzed for indicated proteins by immunoblot analysis. ACTINB was used as loading control. C. (Left) A375 and M14 cells were transfected with NF-κB responsive F-Luc construct and treated with OTSSP167 (50 nM) for 24 h and analyzed for firefly luciferase activity. Relative firefly luciferase activity under indicated conditions is shown. (Right) A375 cells expressing MELK shRNAs were transfected with NF-κB responsive F-Luc construct and either remained untreated or treated with doxycycline and analyzed for firefly luciferase activity. Relative firefly luciferase activity under indicated conditions is shown. D. Indicated NF-κB target genes were analyzed by RT-qPCR in A375 cells expressing MELK shRNAs that were either left untreated or treated with doxycycline (2 μg/ml) for 72 h. Relative mRNA expression for indicated gene in relation to NS shRNA expressing cells is shown. E. A375 cells were either treated with DMSO or OTSSP167 (50 nM) for 24 h. Relative mRNA expression for indicated NF-κB target genes compared to DMSO treated cells is shown. F. Co-immunoprecipitation was performed using either MELK or, as a control, IgG antibodies. Immunoprecipitate and input were analyzed for indicated proteins. G. In vitro kinase assay was performed to determine the ability of recombinant MELK to phosphorylate SQSTM1. Autoradiograph for P32-labeled SQSTM1 is shown and the western blots for SQSTM1 and MELK are shown as controls. H. In vitro kinase assay using MELK inhibitor (left: OTSSP167 (50 nM), right: MELK-8a (500 nM)) was performed. Autoradiograph for P32-labeled SQSTM1 is shown and the western blot for SQSTM1 and MELK is shown. I. A375 cells expressing either a non-silencing (NS) or SQSTM1 shRNAs were analyzed for indicated protein using immunoblotting. ACTINB was used as a loading control. J. A375 cells expressing SQSTM1 shRNAs were transfected with NF-κB responsive F-Luc construct and analyzed for firefly luciferase activity 48 hours after transfection. Relative firefly luciferase activity under indicated conditions is shown. K. A375 cells expressing NS or SQSTM1 shRNAs were analyzed for the indicated NF-κB target genes by RT-qPCR analysis. Relative mRNA expression for the indicated genes in SQSTM1 shRNA expressing cells relative to NS shRNA expressing cells. Data is presented as ±SD for three biological replicates. *, **, and *** represent p<0.05, p<0.01, and p<0.001, respectively. See also Figure S6.
In our SILAC analysis, we identified SQSTM1 as a protein with decreased phosphorylation after MELK inhibition. SQSTM1 has been shown to be involved in the regulation of NF-κB signaling (Long et al., 2010; Wooten et al., 2005; Zotti et al., 2014). SQSTM1 is also shown to be important for NF-κB mediated tumorigenesis (Duran et al., 2008). We hypothesized that MELK phosphorylates SQSTM1 to stimulate the NF-κB pathway. To test this, we performed co-immunoprecipitation (co-IP) to detect whether MELK interacts with and directly phosphorylates SQSTM1. Our co-IP experiments showed that SQSTM1 interacts with MELK (Figure 5F). Next, we performed an in vitro kinase assay using recombinant MELK and SQSTM1 proteins, as described previously (Canman et al., 1998), to test if MELK directly phosphorylates SQSTM1. Consistent with our SILAC experiments, MELK directly phosphorylated SQSTM1 (Figure 5G). To confirm that MELK inhibition reduced SQSTM1 phosphorylation, we performed the in vitro kinase assay using the MELK inhibitors, OTSSP167 and MELK-8a. As anticipated, inhibition of MELK kinase activity led to decreased SQSTM1 phosphorylation (Figure 5H). Based on these results, we examined the activity of NF-κB pathway upon SQSTM1 knockdown. Similar to the effect of MELK inhibition, shRNA-induced knockdown of SQSTM1 inhibited the NF-κB signaling pathway and expression of NF-κB target genes (Figure 5I–K).
Constitutively active IKKβ partially rescues NF-κB signaling and melanoma growth after MELK inhibition
Because IKKβ acts downstream of SQSTM1, we asked if overexpression of constitutively active IKKβ could rescue the inhibition of NF-κB signaling caused by MELK and SQSTM1 inhibition. Ectopic expression of constitutively active IKKβ partially rescued impaired NF-κB signaling caused by MELK inhibition and by downregulation of MELK expression, as assessed by phosphorylation of IκBα and NF-κB responsive reporter activity (Figure 6A–C). Expression of constitutively active IKKβ also rescued the effects of SQSTM1 knockdown in melanoma cells, which indicates that the NF-κB pathway is a downstream effector of SQSTM1 function (Figure 6D).
Figure 6. Overexpression of constitutively active IKKβ (IKKβ CA) restores NF-κB signaling in MELK-inhibited melanoma cells.

A, B. (Left) A375 cells expressing doxycycline-inducible MELK shRNA were transfected with IKKβ CA or empty vector. Cells were either untreated or treated with doxycycline (2 μg/ml) for 72 h and analyzed for indicated proteins by immunoblot analysis. ACTINB was used as a loading control. (Right) A375 cells expressing MELK shRNAs were transfected with IKKβ CA or empty vector and NF-κB responsive F-Luc construct. Cells were either remained untreated or treated with doxycycline and analyzed for firefly luciferase activity. Relative firefly luciferase activity under indicated conditions is shown. C. (Left) A375 cells were transfected with constitutively active IKKβ (IKKβ CA) or empty vector and subsequently treated either with DMSO or OTSSP167 (50 nM) for 24 h and analyzed for indicated proteins by immunoblot analysis. ACTINB was used as a loading control. (Right) A375 cells were transfected with IKKβ CA or empty vector and NF-κB responsive F-Luc construct and treated with OTSSP167 (50 nM) for 24 h and analyzed for firefly luciferase activity. Relative firefly luciferase activity for indicated conditions is shown. D. A375 cells expressing SQSTM1 shRNA were transfected with constitutively active IKKβ (IKKβ CA) or empty vector and analyzed for indicated proteins by immunoblot analysis 48 h after transfection. ACTINB was used as a loading control. E. A375 melanoma cells were transfected with constitutively active IKKβ (IKKβ CA) or empty vector and treated with OTSSP167 (25 nM) and analyzed for anchorage-independent growth using soft-agar assay. Representative images for soft-agar colonies for indicated melanoma cell lines for the indicated treatment conditions are shown. Scale bar: 200μm. F. Relative colony size (%) for A375 cells expressing empty vector or constitutively active IKKβ and were DMSO or OTSSP167 (25 nM) treated. Data is presented as ±SD for three biological replicates. *, **, and *** represent p<0.05, p<0.01, and p<0.001, respectively.
To determine whether forced NF-κB pathway activation in melanoma cells could also rescue the melanoma growth inhibition caused by MELK inhibition, we expressed constitutively active IKKβ in the A375 melanoma cell line and analyzed the growth of melanoma cells in soft-agar assay. Overexpression of constitutively active IKKβ stimulated the growth of A375 cells in soft agar, even in the presence of the MELK inhibitor OTSSP167 (Figure 6E–F). Similarly, overexpression of constitutively active IKKβ restored the growth of A375 melanoma cells in the presence of the second MELK inhibitor MELK-8a (Figure S6D). In contrast, expression of an empty vector in the presence of OTSSP167 or MELK-8a did not rescue the growth of A375 cells (Figure 6E–F and Figure S6D–E). Collectively, these results demonstrate that attenuation of NF-κB signaling is partly responsible for blocking melanoma growth inhibition after MELK inhibition (Figure 7).
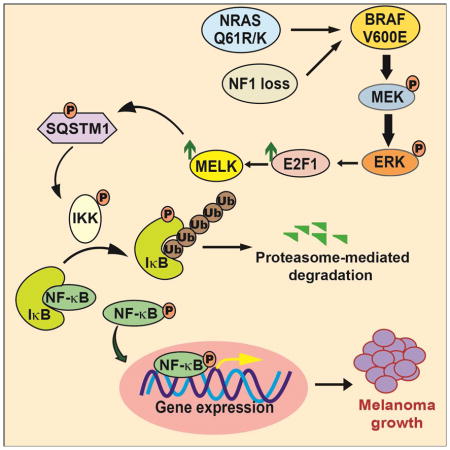
Figure 7. Model elucidating the role of MELK in melanoma.

Activated MAP kinase signaling pathway leads to the upregulation of MELK expression via the transcription factor E2F1. MELK in turn phosphorylates the adapter protein SQSTM1, which stimulates NF-κB pathway activity, which is necessary for stimulating melanoma growth.
DISCUSSION
In this study, we show that MELK is important for melanoma growth that functions, in part, by facilitating NF-κB pathway activity. Our study allows us to draw several important conclusions. First, MELK expression was activated by the MAPK pathway and it was necessary for melanoma growth. Second, we unexpectedly found that MELK phosphorylated many proteins that were previously reported to be BRAF or MEK substrates. Third, MELK inhibition blocked the growth of melanoma that was resistant to the BRAF inhibitor vemurafenib. Finally, MELK regulation of the NF-κB pathway occurred via SQSTM1, partly accounting for its role in promoting melanoma growth. These results are important because they describe a role for MELK in melanoma as a survival kinase. This work also demonstrates that pharmacological inhibition of MELK with a highly potent MELK inhibitor can exert strong inhibitory effects on tumor growth in a wide variety of melanoma types, including NRAS-mutant, NF1-deficient, and vemurafenib-resistant melanoma.
MELK inhibition blocks melanoma growth
Melanoma is an aggressive form of skin cancer, as illustrated by a 5–year survival rate of only 15–20% for stage IV melanoma (Sandru et al., 2014). Only a small fraction of patients experience long-term benefits from current targeted therapies and immunotherapies (Johnson and Sosman, 2015). Therefore, alternative methods to effectively treat melanoma need to be developed. We found that MELK is a survival kinase for melanoma and that MELK inhibition, by either genetic or pharmacological methods, blocked growth of melanoma cells. Furthermore, MELK inhibition in melanoma cells inhibited tumor growth in a broad variety of genotypes, including BRAF-mutant, NRAS-mutant, and NF1-deficient melanoma. Additionally, vemurafenib-resistant melanoma cells were sensitive to MELK inhibitors and we observed that MELK inhibitors forestalled the emergence of vemurafenib resistance in melanoma cells. Notably, MELK knockout mice are viable and do not show any obvious defects. Collectively, these observations suggest that MELK is an important and broadly applicable therapeutic target in melanoma.
MELK regulates a large number of previously reported BRAF-MEK-ERK substrates
MELK is a serine/threonine protein kinase that regulates the cell cycle, stem cell renewal, and apoptosis (Badouel et al., 2006; Davezac et al., 2002; Jung et al., 2008; Nakano et al., 2005). Previous studies have identified some MELK substrates, including ASK1, ZNF622, BCL2L14, and CDC25B (Davezac et al., 2002; Jung et al., 2008; Lin et al., 2007; Seong et al., 2002). The apoptotic functions of MELK are mediated by ASK1 and BCL2L14 regulation (Jung et al., 2008; Lin et al., 2007), while its cell cycle regulatory effects are proposed to be mediated by its phosphorylation of CDC25B (Davezac et al., 2002; Mirey et al., 2005). In addition to regulating apoptosis and cell cycle, MELK also regulates other aspects of cell biology. For example, MELK has been shown to inhibit spliceosome assembly during mitosis by phosphorylating ZNF622, thereby contributing to its redirection to the nucleus. Using SILAC, we identified 469 proteins with downregulated phosphorylation after MELK inhibition. Remarkably, we also noted that a large number of proteins (139 proteins) were previously identified as potential MEK and BRAF substrates (Galan et al., 2014; Stuart et al., 2015). Because MELK expression is regulated by the MAPK pathway, these findings suggest MELK is a major mediator of MAPK pathway function that promotes melanoma growth.
MELK is a regulator of NF-κB pathway
The NF-κB pathway is a major tumor promotion pathway in melanoma and several other cancer types (Dhawan and Richmond, 2002; Erstad and Cusack, 2013; Liu et al., 2015; Madonna et al., 2012; Pikarsky et al., 2004). We found that MELK regulates the NF-κB pathway by phosphorylating SQSTM1/p62, which is consistent with a previous study that showed that SQSTM1 is important for NF-κB-mediated tumorigenesis (Duran et al., 2008). Additionally, we demonstrated that MELK inhibition decreased the expression of NF-κB transcriptional targets, and we partially rescued diminished melanoma growth after MELK inhibition by expressing constitutively active IKKβ. Collectively, these results identify MELK as a regulator of the NF-κB pathway and show that MELK at least partly promotes melanoma growth by activating the NF-κB pathway.
EXPERIMENTAL METHODOLOGY
Cell culture
A375, M14, SKMEL28, and MeWo cell lines were obtained from American Type Culture Collection (ATCC). YUGASP cells were obtained from Yale SPORE in Skin Cancer. SKMEL239 cell lines (parental, vemurafenib-resistant) are described previously (Poulikakos et al., 2011) and were a kind gift from Drs. David Solit and Neal Rosen. A375, MeWo and YUGASP were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and 1% Penicillin/Streptomycin antibiotics. M14, SKMEL28, SKMEL239 were grown in RPMI supplemented with 10% FBS and 1% Penicillin/Streptomycin antibiotics.
SILAC
Cell labeling
Cells were seeded at 15% confluency in the respective complete medium (For A375: DMEM+10% dialyzed FBS, 1% PenStrep; for M14: RPMI+10% dialyzed FBS, 1% PenStrep). All media was deficient in lysine and arginine, and supplemented with light- or heavy-labeled lysine (13C6 15N2) and light- or heavy-labeled arginine (13C6 15N4). Cells were subsequently cultured for at least five doublings in light or heavy medium, which achieved over 95% labeling efficiency for us in pilot experiments. After labeling, cells were treated for 24 h with 25 nM (A375) or 50 nM (M14) of OTSSP167. After treatment, cells were trypsinized and counted to obtain a cell pellet of 2 × 107 cells/condition and subjected to SILAC analysis using mass spectrometry.
Sample preparation
The heavy and light cells pellets were lysed in RIPA buffer spiked with protease and phosphatase inhibitors, using short 15 sec sonication bursts. Lysates were centrifuged at 14,000 rpm for 20 min. After centrifugation, the supernatants were collected and protein concentration was measured using a Hitachi L-8900 Amino Acid Analyzer. From each sample, 200 μg of proteins were aliquoted, combined, and precipitated using a methanol-chloroform precipitation method. The protein pellets were resuspended in 8 M urea/0.4 M ammonium bicarbonate buffer, reduced with 45 mM DTT for 30 min at 37°C, alkylated with 100 mM iodoacetamide for 30 min in the dark at room temperature, and digested with Lys-C protease (1:20 w/w) by incubating overnight (~16 h) at 37°C. The Lys-C digest was further diluted and digested with trypsin (1:20 w/w) by incubating for 8 h at 37°C. The digest was desalted with MacroSpin column (The Nest Group, Inc., Southboro, MA) and dried down in a SpeedVac concentrator. Desalted peptides were then phosphopeptide enriched using titanium dioxide resin imbedded in 10-μl tips (Glygen Corp., Columbia, MD). Flow-throughs were reserved and enriched peptides were eluted using 1:33 ammonium hydroxide:water. The SpeedVac dried flow-through and elution fractions were resuspended in buffer A (0.1% formic acid in water) and subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
Mass spectrometry data acquisition and analysis
The samples were analyzed by LC-MS/MS on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Scientific, San Jose, CA) interfaced with a nanoACQUITY UPLC System (Waters, Milford, MA) at the front end. Samples were loaded into a trapping column (nanoACQUITY UPLC Symmetry C18 Trap Column, 180 μm × 20 mm, Product Number: 186006527) at a flowrate of 5 μl/min and separated with a C18 column (nanoACQUITY column Peptide BEH C18, 75 μm × 250 mm, Product number: 186003545). The peptides were eluted with buffer B (0.1% formic acid in acetonitrile) in a gradient from 6% to 35% in 150 min at a flowrate of 300 nL/min. LC-MS/MS data were acquired using 3 s, the top speed data-dependent acquisition mode. Details of the instrument settings can be found in Supporting Information S1.
Peptides and proteins were identified and quantified with the Sequest HT search engine using Proteome Discoverer v 2.1 (Thermo Scientific) software. A standardized SILAC 2plex (Arg10, Lys8) quantification workflow in the Proteome Discoverer was slightly modified as described below and used for analysis. Briefly, MS/MS data were searched against the SwissProt human database (downloaded in September 2015; number of protein entries = 20,193). In Peak Filters node, the S/N threshold was set to 1.5. The search criteria included 10 ppm precursor mass tolerance, 0.02 Da fragment mass tolerance, and a trypsin miscleavage setting of two. Static modification settings included carbamidomethylation (+57.021 Da) on cysteine, while dynamic modifications were set to include oxidation (+15.995 Da) on methionine and phosphorylation (+79.966 Da) on serine, threonine, and tyrosine. Peptide spectrum matches (PSMs) were verified based on q-values set to 1% false discovery rate (FDR) using Percolator. Precursor Ions Quantifier node was used in the processing step workflow and the Peptide and Protein Quantifier node was selected for the consensus workflow to calculate and quantify peptides, protein abundances, and ratios. PhosphoRS node (Taus et al., 2011) was used to obtain the localization probability of the phosphorylation sites in the peptides.
SILAC data analysis for identifying the preferred MELK amino acid context for phosphorylation
A cut-off of two-fold was used to define downregulation of phosphorylation level for validation experiments. To identify the MELK phosphorylation consensus site from the SILAC data, we used a prediction algorithm developed in house. The motifs were generated by R/Bioconductor package dagLogo (v.1.9.2). The background of the motifs was built from the human proteome retrieved via R/Bioconductor package UniProt.ws (v.2.11.9). The entire list of all quantified phosphopeptides are presented in Table S4 (for A375 cell line) and Table S5 (for M14 cell line). The SILAC proteomics data is submitted to PRIDE (https://www.ebi.ac.uk/pride/archive/). The accession number for this data is PXD007872.
Melanoma data analysis
The Talantov melanoma dataset (Talantov et al., 2005), Riker melanoma dataset (Riker et al., 2008), and Xu melanoma dataset (Xu et al., 2008) were analyzed for MELK expression using Oncomine (https://www.oncomine.org) and MELK expression across different samples was plotted as box plots. Additionally, three previously published melanoma gene expression datasets were analyzed for MELK expression and plotted as box plots (Eskiocak et al., 2016; Kabbarah et al., 2010; Scatolini et al., 2010).
Statistical analysis
All quantitative data were collected from experiments performed in at least triplicate and expressed as mean ± SD. Differences between groups were assayed using Student’s t test using using GraphPad Prism version 6.0h for Macintosh, GraphPad Software, San Diego California USA (www.graphpad.com). Significant differences were considered when p ≤ 0.05.
Supplementary Material
Heavy lysine and arginine (13C, 15N) peptide labelling efficacy for A375 cell line. M14 cell line heavy amino acids labeling was done using same protocol. Related to Figure 4.
A list proteins with significantly reduced phosphorylation of particular sites identified using SILAC in both A375 and M14 cell line. Proteins in yellow highlight represent the overlap between our study and Stuart et al., 2015. Phosphorylation sites highlighted in red bold represent sites identified in our study and Stuart et al., 2015. Related to Figure 4.
Primer sequences for RT-qPCR, ChIP, site-diected mutagenesis and cloning; clone ID and catalog numbers for shRNAs (Open Biosystems); antibodies used; source and concentration of chemical inhibitors used. Related to Experimental Methodology and Supplementary Experimental Methodology.
Entire list of all quantified phosphopeptides from the SILAC analysis of A375 cells treated with MELK inhibitor OTSSP167. Related to Figure 4.
Entire list of all quantified phosphopeptides from the SILAC analysis of M14 cells treated with MELK inhibitor OTSSP167. Related to Figure 4.
HIGHLIGHTS.
MELK is upregulated in melanoma by the MAP kinase pathway via E2F1
MELK inhibition blocks melanoma growth
MELK phosphorylates a large number of BRAF and MEK substrates
MELK in part promotes melanoma by stimulating NF-κB pathway via SQSTM1
Acknowledgments
We gratefully acknowledge grants from the National Institutes of Health: R21CA197758-01 (N.W.), R21CA191364-01 (N.W.), R21CA195077-01A1 (NW), and R01CA200919-01 (NW). N.W. is also supported by Research Scholar Grant from American Cancer Society (128347-RSG-15-212-01-TBG) and grants from the Melanoma Research Alliance and the Melanoma Research Foundation. Authors declare no conflict of interest related to work presented in this manuscript.
Footnotes
AUTHORS CONTRIBUTIONS
R.J. and N.W. conceived and designed the experiments. R.J. performed all experiments. N.R. and T.L. performed SILAC and provided the list of identified proteins and altered phosphorylation sites. J.O. and J.L.Z. generated the program to predict consensus sites for MELK phosphorylation. M.R.G. provided reagents. R.J. and N.W. analyzed and interpreted the data. R.J. and N.W. co-prepared the figures and co-wrote the manuscript. All authors reviewed and commented on the manuscript.
ACCESSION NUMBERS
The SILAC proteomics data is submitted to PRIDE (https://www.ebi.ac.uk/pride/archive/). The accession number for this data is PXD007872.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 2013;14:249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- Badouel C, Korner R, Frank-Vaillant M, Couturier A, Nigg EA, Tassan JP. M-phase MELK activity is regulated by MPF and MAPK. Cell Cycle. 2006;5:883–889. doi: 10.4161/cc.5.8.2683. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Suzuki H, Miyamoto T, Takamatsu N, Tatsuguchi A, Ueda K, Kijima K, Nakamura Y, Matsuo Y. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget. 2012;3:1629–1640. doi: 10.18632/oncotarget.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davezac N, Baldin V, Blot J, Ducommun B, Tassan JP. Human pEg3 kinase associates with and phosphorylates CDC25B phosphatase: a potential role for pEg3 in cell cycle regulation. Oncogene. 2002;21:7630–7641. doi: 10.1038/sj.onc.1205870. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Dhawan P, Richmond A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J Biol Chem. 2002;277:7920–7928. doi: 10.1074/jbc.M112210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- Erstad DJ, Cusack JC., Jr Targeting the NF-kappaB pathway in cancer therapy. Surg Oncol Clin N Am. 2013;22:705–746. doi: 10.1016/j.soc.2013.06.011. [DOI] [PubMed] [Google Scholar]
- Eskiocak U, Ramesh V, Gill JG, Zhao Z, Yuan SW, Wang M, Vandergriff T, Shackleton M, Quintana E, Johnson TM, et al. Synergistic effects of ion transporter and MAP kinase pathway inhibitors in melanoma. Nat Commun. 2016;7:12336. doi: 10.1038/ncomms12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JA, Geraghty KM, Lavoie G, Kanshin E, Tcherkezian J, Calabrese V, Jeschke GR, Turk BE, Ballif BA, Blenis J, et al. Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14-3-3. Proc Natl Acad Sci U S A. 2014;111:E2918–2927. doi: 10.1073/pnas.1405601111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray D, Jubb AM, Hogue D, Dowd P, Kljavin N, Yi S, Bai W, Frantz G, Zhang Z, Koeppen H, et al. Maternal embryonic leucine zipper kinase/murine protein serine-threonine kinase 38 is a promising therapeutic target for multiple cancers. Cancer Res. 2005;65:9751–9761. doi: 10.1158/0008-5472.CAN-04-4531. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Kato T, Olugbile S, Tamura K, Chung S, Miyamoto T, Matsuo Y, Salgia R, Nakamura Y, Park JH. Effective growth-suppressive activity of maternal embryonic leucine-zipper kinase (MELK) inhibitor against small cell lung cancer. Oncotarget. 2016;7:13621–13633. doi: 10.18632/oncotarget.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Sosman JA. Therapeutic Advances and Treatment Options in Metastatic Melanoma. JAMA Oncol. 2015;1:380–386. doi: 10.1001/jamaoncol.2015.0565. [DOI] [PubMed] [Google Scholar]
- Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, Nardini D, Sobol RW, Chow LM, Kornblum HI, et al. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells. 2013;31:1051–1063. doi: 10.1002/stem.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Seong HA, Ha H. Murine protein serine/threonine kinase 38 activates apoptosis signal-regulating kinase 1 via Thr 838 phosphorylation. J Biol Chem. 2008;283:34541–34553. doi: 10.1074/jbc.M807219200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbarah O, Nogueira C, Feng B, Nazarian RM, Bosenberg M, Wu M, Scott KL, Kwong LN, Xiao Y, Cordon-Cardo C, et al. Integrative genome comparison of primary and metastatic melanomas. PLoS One. 2010;5:e10770. doi: 10.1371/journal.pone.0010770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Inoue H, Imoto S, Tamada Y, Miyamoto T, Matsuo Y, Nakamura Y, Park JH. Oncogenic roles of TOPK and MELK, and effective growth suppression by small molecular inhibitors in kidney cancer cells. Oncotarget. 2016;7:17652–17664. doi: 10.18632/oncotarget.7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA, Fecher LA, Millward M, McArthur GA, Hwu P, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–489. doi: 10.1200/JCO.2012.43.5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler RS, Kettelhack H, Knipprath-Meszaros AM, Fedier A, Schoetzau A, Jacob F, Heinzelmann-Schwarz V. MELK expression in ovarian cancer correlates with poor outcome and its inhibition by OTSSP167 abrogates proliferation and viability of ovarian cancer cells. Gynecol Oncol. 2017;145:159–166. doi: 10.1016/j.ygyno.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Lin ML, Park JH, Nishidate T, Nakamura Y, Katagiri T. Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 2007;9:R17. doi: 10.1186/bcr1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY, Lai F, Yan XG, Jiang CC, Guo ST, Wang CY, Croft A, Tseng HY, Wilmott JS, Scolyer RA, et al. RIP1 Kinase Is an Oncogenic Driver in Melanoma. Cancer Res. 2015;75:1736–1748. doi: 10.1158/0008-5472.CAN-14-2199. [DOI] [PubMed] [Google Scholar]
- Long J, Garner TP, Pandya MJ, Craven CJ, Chen P, Shaw B, Williamson MP, Layfield R, Searle MS. Dimerisation of the UBA domain of p62 inhibits ubiquitin binding and regulates NF-kappaB signalling. J Mol Biol. 2010;396:178–194. doi: 10.1016/j.jmb.2009.11.032. [DOI] [PubMed] [Google Scholar]
- Madonna G, Ullman CD, Gentilcore G, Palmieri G, Ascierto PA. NF-kappaB as potential target in the treatment of melanoma. J Transl Med. 2012;10:53. doi: 10.1186/1479-5876-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AJ, Mihm MC., Jr Melanoma. N Engl J Med. 2006;355:51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- Mirey G, Chartrain I, Froment C, Quaranta M, Bouche JP, Monsarrat B, Tassan JP, Ducommun B. CDC25B phosphorylated by pEg3 localizes to the centrosome and the spindle poles at mitosis. Cell Cycle. 2005;4:806–811. doi: 10.4161/cc.4.6.1716. [DOI] [PubMed] [Google Scholar]
- Nakano I, Paucar AA, Bajpai R, Dougherty JD, Zewail A, Kelly TK, Kim KJ, Ou J, Groszer M, Imura T, et al. Maternal embryonic leucine zipper kinase (MELK) regulates multipotent neural progenitor proliferation. J Cell Biol. 2005;170:413–427. doi: 10.1083/jcb.200412115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E) Nature. 2011;480:387–390. doi: 10.1038/nature10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riker AI, Enkemann SA, Fodstad O, Liu S, Ren S, Morris C, Xi Y, Howell P, Metge B, Samant RS, et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med Genomics. 2008;1:13. doi: 10.1186/1755-8794-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- Ryu B, Kim DS, Deluca AM, Alani RM. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS One. 2007;2:e594. doi: 10.1371/journal.pone.0000594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandru A, Voinea S, Panaitescu E, Blidaru A. Survival rates of patients with metastatic malignant melanoma. J Med Life. 2014;7:572–576. [PMC free article] [PubMed] [Google Scholar]
- Scatolini M, Grand MM, Grosso E, Venesio T, Pisacane A, Balsamo A, Sirovich R, Risio M, Chiorino G. Altered molecular pathways in melanocytic lesions. Int J Cancer. 2010;126:1869–1881. doi: 10.1002/ijc.24899. [DOI] [PubMed] [Google Scholar]
- Seong HA, Gil M, Kim KT, Kim SJ, Ha H. Phosphorylation of a novel zinc-finger-like protein, ZPR9, by murine protein serine/threonine kinase 38 (MPK38) Biochem J. 2002;361:597–604. doi: 10.1042/0264-6021:3610597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart SA, Houel S, Lee T, Wang N, Old WM, Ahn NG. A Phosphoproteomic Comparison of B-RAFV600E and MKK1/2 Inhibitors in Melanoma Cells. Mol Cell Proteomics. 2015;14:1599–1615. doi: 10.1074/mcp.M114.047233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- Toure BB, Giraldes J, Smith T, Sprague ER, Wang Y, Mathieu S, Chen Z, Mishina Y, Feng Y, Yan-Neale Y, et al. Toward the Validation of Maternal Embryonic Leucine Zipper Kinase: Discovery, Optimization of Highly Potent and Selective Inhibitors, and Preliminary Biology Insight. J Med Chem. 2016;59:4711–4723. doi: 10.1021/acs.jmedchem.6b00052. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Richmond A. NF-kappaB activation in melanoma. Pigment Cell Res. 2006;19:112–124. doi: 10.1111/j.1600-0749.2006.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Begley M, Li Q, Huang HT, Lako A, Eck MJ, Gray NS, Mitchison TJ, Cantley LC, Zhao JJ. Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proc Natl Acad Sci U S A. 2016;113:9810–9815. doi: 10.1073/pnas.1606862113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lee YM, Baitsch L, Huang A, Xiang Y, Tong H, Lako A, Von T, Choi C, Lim E, et al. MELK is an oncogenic kinase essential for mitotic progression in basal-like breast cancer cells. Elife. 2014;3:e01763. doi: 10.7554/eLife.01763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004a;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D, Springer CJ, Marais R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004b;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- Whittaker SR, Theurillat JP, Van Allen E, Wagle N, Hsiao J, Cowley GS, Schadendorf D, Root DE, Garraway LA. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3:350–362. doi: 10.1158/2159-8290.CD-12-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280:35625–35629. doi: 10.1074/jbc.C500237200. [DOI] [PubMed] [Google Scholar]
- Xu L, Shen SS, Hoshida Y, Subramanian A, Ross K, Brunet JP, Wagner SN, Ramaswamy S, Mesirov JP, Hynes RO. Gene expression changes in an animal melanoma model correlate with aggressiveness of human melanoma metastases. Mol Cancer Res. 2008;6:760–769. doi: 10.1158/1541-7786.MCR-07-0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotti T, Scudiero I, Settembre P, Ferravante A, Mazzone P, D’Andrea L, Reale C, Vito P, Stilo R. TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Mol Immunol. 2014;58:27–31. doi: 10.1016/j.molimm.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Heavy lysine and arginine (13C, 15N) peptide labelling efficacy for A375 cell line. M14 cell line heavy amino acids labeling was done using same protocol. Related to Figure 4.
A list proteins with significantly reduced phosphorylation of particular sites identified using SILAC in both A375 and M14 cell line. Proteins in yellow highlight represent the overlap between our study and Stuart et al., 2015. Phosphorylation sites highlighted in red bold represent sites identified in our study and Stuart et al., 2015. Related to Figure 4.
Primer sequences for RT-qPCR, ChIP, site-diected mutagenesis and cloning; clone ID and catalog numbers for shRNAs (Open Biosystems); antibodies used; source and concentration of chemical inhibitors used. Related to Experimental Methodology and Supplementary Experimental Methodology.
Entire list of all quantified phosphopeptides from the SILAC analysis of A375 cells treated with MELK inhibitor OTSSP167. Related to Figure 4.
Entire list of all quantified phosphopeptides from the SILAC analysis of M14 cells treated with MELK inhibitor OTSSP167. Related to Figure 4.