

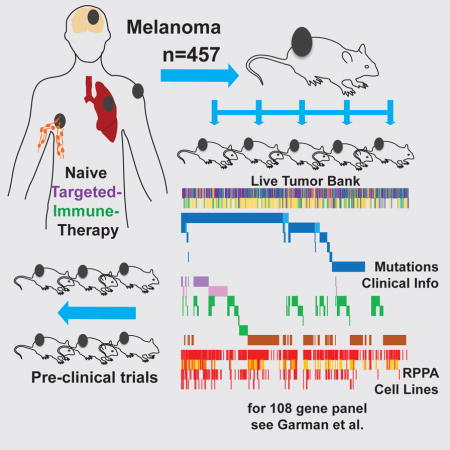
Summary
Therapy of advanced melanoma has been changing dramatically. Following mutational and biological sub-classification of this heterogeneous cancer, several targeted and immune therapies were approved and increased survival significantly. To facilitate further advancements through pre-clinical in vivo modeling, we have established 459 patient-derived xenografts (PDX) and live tissue samples from 384 patients representing the full spectrum of clinical, therapeutic, mutational, and biological heterogeneity of melanoma. PDX have been characterized using targeted sequencing and protein arrays, and are clinically annotated. This exhaustive live tissue resource includes PDX from 57 samples resistant to targeted therapy, 61 samples from responders and non-responders to immune checkpoint blockade, and 31 samples from brain metastasis. Uveal, mucosal, and acral subtypes are represented as well. We show examples of pre-clinical trials that highlight how the PDX collection can be used to develop and optimize precision therapies, biomarkers of response, and the targeting of rare genetic subgroups.
Keywords: melanoma, patient-derived xenografts, targeted therapy, immune checkpoint blockade, melanoma brain metastasis, in vivo models, BRAF inhibitor resistance, ERK inhibitor, MDM2 inhibitor, PI3K beta inhibitor
Graphical Abstract
INTRODUCTION
Advanced melanoma has gone from limited therapeutic options to approved kinase inhibitor and immune checkpoint therapy. Five-year survival rates have nearly doubled (Menzies et al., 2015; Schadendorf et al., 2015). Precision medicine and immune oncology are major areas of translational melanoma research. The complex melanoma landscape needs improved models reflecting all mutational and clinical subtypes. The UV carcinogenic etiology of melanoma makes it one of the most highly mutated cancers (Alexandrov et al., 2013). This high mutational burden may be the reason for the success of immune checkpoint blockade (Callahan et al., 2016), but makes rational “precision” therapies challenging (Krepler et al., 2016).
The Melanoma Cancer Genome Atlas (TCGA) includes comprehensive molecular characterization of 333 non-acral cutaneous melanomas and is an important resource. It confirmed the main mutational subgroups of BRAF, NRAS, NF1, and triple wild type, as well as highlighting the distinct heterogeneity and high mutational burden of melanoma (Cancer Genome Atlas, 2015). Subtypes not included in the TCGA but published elsewhere were uveal (Van Raamsdonk et al., 2010), acral cutaneous (Furney et al., 2014), and mucosal melanoma (Sheng et al., 2016).
Patient-derived xenografts (PDX) as xenotransplantation of human tumors into athymic nude mice were first described by (Rygaard and Povlsen, 1969). PDX are established directly from patient tumors in immune deficient mice and thus provide a source of tumor tissue closely resembling the clinical lesion (Hidalgo et al., 2014). Melanoma is uniquely suited to this approach as even single cells are tumorigenic in vivo (Quintana et al., 2008). Melanoma PDX were shown to accurately model the clinical disease and response to targeted therapy (Einarsdottir et al., 2014). We have shown recently that PDX derived from BRAF inhibitor relapsed patients and expanded on chronic therapy could be used to identify effective second line combination therapies based on genomic and proteomic profiling (Krepler et al., 2016). While these studies demonstrate the feasibility of the PDX approach, the melanoma TCGA and other studies (Arafeh et al., 2015; Cancer Genome Atlas, 2015; Krauthammer et al., 2015) highlight the pronounced heterogeneity of this cancer type. Both concepts are combined here in an unparalleled collection of 459 mutationally and clinically diverse melanoma PDX and live frozen tissues, providing an exhaustive and testable resource for the melanoma research field. This resource is highly clinically annotated, includes rare body sites and subtypes such as brain metastasis, uveal, mucosal, and acral melanoma, as well as pre- and post-therapy samples from targeted inhibitor and checkpoint blockade treated patients.
RESULTS
Establishment of Melanoma PDX
We have collected 694 melanoma samples for PDX generation from eight institutions (Figure 1A). Fresh tumor samples were either directly implantated within 24 hours subcutaneously (s.c.) in NOD/SCID/IL-2Rγnull (NSG) mice or banked as cryopreserved live tissue (Figure 1A). Keeping primary tissue in a live tumor bank was a cost-effective alternative to fresh implantation, but dependent on adequate amounts of tissue. Both approaches successfully established PDX and detailed methods are included in the experimental procedures section and in a standard operating procedures (SOP) handbook (Supplementary File S1).
Figure 1. Establishment and biology of PDX models.

(A) A total of 694 melanoma tissue samples from naïve, pre-, on-, or post- therapy time points receiving targeted kinase inhibitors (TT) or immune checkpoint inhibitors (IT) were used to generate PDX and/or banked as live tissue. (B) Success rate of establishing a tumor graft (green), banking of live tissue with the potential of establishing a PDX or establishment in progress (blue), no tumor growth at 6 to 12 months (orange), adverse events (gray) where we were not able to establish a PDX due to reasons other than tumor take (this analysis excludes uveal primary samples). (C) Take rate of cutaneous melanoma derived tissue. (D) Time to palpable for all FNA, core, and excisional biopsy patient samples. (E) Tumor growth rate comparison of FNA, core, and biopsies. Growth was calculated as tumor volume/weeks. (F) Fresh tumor biopsies (MP0) or PDX after MP1 from three patients were prepared as cell suspensions, leucocytes and endothelial cells excluded and injected s.c. into NSG mice at indicated cell numbers. (G) Single cell suspension was prepared as before and sorted for CD271 marker. CD271+ and negative cells were injected at indicated cell numbers.
Of the samples collected, 319 were established as PDX and 140 were banked as live primary tissue w (Figure 1B) totaling 459 models from 384 different patients. Failure to establish a PDX was due to sample contamination, unexpected death of a primary recipient animal, receipt of non-viable samples, or non-melanoma samples (Figure 1B). Thus, although the overall success rate for establishing melanoma PDX was 62%, the take rate corrected for these factors was 83% (Figure 1C). This excluded primary uveal samples whose take rate was 11%.
Time to Tumor Growth and Tumor Growth Rate
Tumor samples were obtained from either fine needle aspirates (FNA), core biopsies, or surgical excisions. We found no significant difference in latency (time from implantation to palpable tumor) and tumor growth rates (time to maximal tumor size) (Figure 1D,E).
Very Small Cell Numbers Are Needed to Establish a Melanoma PDX
Tissues from three patients were enzymatically digested and hematopoietic cells, red blood cells, and endothelial cells removed We observed consistent tumor engraftment in mice at 1000, 100, 10, and 1 cell(s)/mouse (Figure 1F). The latency period was extended by up to 4 months indicating that a follow up of 6 months is optimal to achieve maximum engraftment. Further, tumorigenicity did not significantly change when sorting the cells for the cancer stem cell marker CD271 (Boiko et al., 2010) (Figure 1G).
Patient Demographics Reflect the Clinical Spectrum of the Disease
Patients’ age ranged from 20–89 years with a peak between 60–69 years (Figure 2A)with a predominance of male patients, likely representing our sampling bias for advanced disease (Geller et al., 2002) (Figure 2B). More than 80% of patients had stage IV disease. The largest proportion of samples (68%) was metastases from patients with non-acral cutaneous primaries (Figure 2C), but we also included 59 unknown primary, 17 mucosal, 15 acral cutaneous, and 10 uveal melanomas. Approximately 44% were subcutaneous (Figure 2D) and 26% lymph node metastasis samples, since these are often excised for diagnostic or therapeutic reasons. Remarkably, 23% were distant organ metastates, including brain. Primary melanomas represented 5%, although these were thick primaries and the patients had often already developed stage III disease.
Figure 2. Demographics of patient samples used to generate PDX.

(A) Age of patients at time of biopsy in 10-year increments. (B) Gender of patients. (C) Primary tumor type. (D) Site of tissue biopsy; categorized into primary melanoma, subcutaneous metastasis (SQ), lymph node metastasis (LN), distant metastasis to organs (Distant met), and brain metastasis (Brain). (E) Targeted kinase or immune checkpoint inhibitor therapies the patient had received before or during the biopsy was taken. Samples without available data were excluded from the analysis.
Our collection spanned several years and the therapies for advanced melanoma have evolved during that period. Samples therefore reflect the standard of care and ongoing clinical trials at contributing centers, ranging from untreated through targeted therapy, to immune checkpoint blockade, and combination therapies (Figure 2E).
Genomic Characterization and Clinical Annotation
The majority (n=314, 68%) of PDX and tissues were analyzed for genomic alterations using massively parallel sequencing of a 108-gene targeted panel. The genes included in this panel were selected based on previously described mutations and copy number variations in melanoma. A full list of included genes and an in-depth analysis of mutational and copy number data of all PDX models as well as additional melanoma cell lines (n=488 total) are described in a companion resource article (Garman et al., 2017). An additional 90 patients were annotated by NGS targeted panels of 40–400 genes at their clinical institutions and we used these data to infer oncogenic driver mutation status of PDX. Both data sets were combined to classify a total of 372 PDX or banked tissues into major mutational subgroups.
Half (55%) of all samples analyzed were BRAF hotspot mutant, 20% NRAS mutant, 7% NF1 mutant, 2% KIT, 1.4% GNAQ/GNA11, and 18% wild-type (WT) (Figure 3A and Supplementary File S2). These results correlate with the melanoma TCGA data (Cancer Genome Atlas, 2015) and other published large scale sequencing studies (Arafeh et al., 2015; Hodis et al., 2012; Krauthammer et al., 2015).
Figure 3. Overview of PDX collection, immune therapy, targeted therapy resistant, and brain metastasis derived subsets.

(A) All PDX and live frozen tissue samples sorted by driver mutations and therapy received by the patients. Driver mutations are dark blue for hotspot and light blue for non-hotspot mutation. PDX from patients progressed on targeted therapies are shades of purple, patients treated with immune checkpoint inhibitors are green: sequential, combination CTLA4+PD-1 (IT combo), or combination with BRAF inhibition (TT/IT combo). Red indicates in vivo growth, presence of RPPA data, or a corresponding cell line. Samples that spontaneously metastasize to lungs in mice are red, yellow indicates no lung metastasis, white was not assessed. (B) Patients were treated with CTLA4 or PD-1 blocking therapy before, during, or after biopsy. Combination therapies are indicated. PDX are sorted by best response in the patients. Additional PDX with unknown response are not shown. (C) Genetic data of BRAF (−BR) and BRAF/MEK (−CR) inhibitor-resistant PDX. Deleterious and likely deleterious mutations, homozygous loss, and high copy number gains (>5) are shown. Numbering after dash (1–4) indicate additional PDX available from the same patient. Asterisks indicate resistant PDX with available patient matched pre-therapy derived PDX.(D) Patient matched pre- and post-therapy PDX models. Progression free survival of patients treated with BRAF or BRAF/MEK inhibitor (x-axis). Columns are labeled with putative resistance mechanisms. (E) Genetic profile and therapy received of 22 PDX with available sequencing data out of 31 total brain metastasis PDX. Deleterious and likely deleterious mutations, homozygous loss, and high copy number gains (>5) are shown. As an indication of PI3K pathway activation status RPPA levels of phosphorylated AKT are shown.
Thirty-seven of the BRAF hotspot mutation PDX were from patients progressed on a BRAF inhibitor (12 previously published in (Krepler et al., 2016) and 44 progressed on BRAF/MEK inhibitor combination therapy. We collected 190 samples from patients with immune checkpoint inhibitor therapy (anti CTLA4 and/or anti PD-1). These did not cluster to any mutational subgroup. We established PDX from patients progressed on both targeted and immune therapy (25 sequentially and 17 with BRAF inhibitor/PD-1 blockade combination therapy). (Figure 3A and Supplementary File S2)
The reverse phase protein array (RPPA) platform quantified ~300 proteins and phosphorylated proteins. These profiles are a useful complementary analysis to genetic sequencing (Krepler et al., 2016) and are available for 113 PDX models while others are in progress (Figure 3A and Supplementary File S3
PDX derived cell lines
We have established cell lines from 24 PDX tumors with a focus on targeted therapy resistant and brain metastasis samples (Figure 3A). These are added to the 112 cell lines of the “Wistar Melanoma” collection (https://www.wistar.org/lab/meenhard-herlyn-dvm-dsc/page/melanoma-cell-lines-0). As these PDX derived cell lines included 10 derived from targeted therapy resistant samples, the mutational distribution is biased for BRAF hotspot (71%). Further, the cell lines include seven from brain metastasis, two acral melanoma (WM4324: V600E, WM4235: Q61R) and one mucosal (WM4173: WT/WT).
PDX from Patients Treated with Checkpoint Inhibitors
We established 190 PDX from 140 immune checkpoint blockade therapy patients. Best response was complete response in 7 patients, partial response in 26, mixed response in 5, stable disease in 10, and progressive disease in 59 patients. Response data could not be obtained in 33 patients. Forty-three patients received only anti CTLA4, and 50 received only anti PD-1; 41 patients received both therapies sequentially and six as a combination therapy. All patient samples were collected either before, on-, or after immune therapy with 16 patients matched before and on or after therapy (Figure 3B).
PDX from Targeted Therapy Resistant Patients
We collected 57 biopsies from 47 patients after progression on BRAF or BRAF and MEK combination targeted kinase inhibitor therapy (either still on or shortly after end of therapy) (Figure 3C). After initial establishment and expansion as PDX, the tumor graft bearing animals were continuously dosed with BRAF inhibitor (PLX4720) or BRAF/MEK inhibitor (PLX4720/PD-0325901) combination diet corresponding to the type of therapy received by the patient (Krepler et al., 2016). Targeted sequencing of resistant PDX tumors using our 108-gene panel (Garman et al., 2017) confirmed a BRAFV600 hotspot mutation in all but two of the models. These two PDX models were established from patients with clinical BRAFV600E positive tumors. However, the patient material tested for WM4323 was the primary cutaneous melanoma diagnostic biopsy accessioned 5 years prior to the specimen sent for PDX. This was done via pyrosequencing of codons 595 and 600 of exon 15 of the BRAF gene. The patient material tested for WM4352 was a metastatic lymph node accessioned 7 months prior to the specimen sent for PDX. This was done via NGS panel of 50 genes including, for BRAF, codons 439–473 of exon 11 and codons 581–611 of exon 15.
Several mechanisms of resistance were revealed by targeted sequencing. We found concomitant RAS (n=7/47 patients) and MAP2K1/2 (n=9/47 patients) mutations. These deleterious mutations were mutually exclusive and have been reported previously as activating mutations conferring resistance to BRAF inhibition (Emery et al., 2009; Nazarian et al., 2010). BRAF high level amplification (>5) in four patients and MET high level amplification (>5) in three patients were exclusive of each other and RAS and MAP2K activating mutations (Shi et al., 2013). PDX from 15 patients had alterations in the PI3K signaling pathway (13 PTEN deletion, 3 deleterious PTEN mutation, 5 likely deleterious PTEN mutation, 1 deleterious PIK3CA mutation) although these were not mutually exclusive with the other genomic changes observed.
Patient matched PDX from before start and after progression on targeted therapy were generated from seven patients. Of these, two (WM4298, WM4351) had acquired NRAS mutations on dabrafenib-trametinib combination therapy (D/T) and progressed after 406 and 161 days respectively. WM3901 was established from a solitary progressing (>10%) s.c. metastasis after 480 days on D/T and had acquired a BRAF amplification. WM4264 had PFS of 120 days and an acquired MEK2K61E heterozygous mutation in the relapse PDX. Although a variant of unknown significance per our algorithm (Garman et al., 2017), due to the location and glutamic acid change this might be a phosphomimetic activating mutation (Villanueva et al., 2013). WM4070 PDX were established from the patient with the shortest PFS (60 days) and we found a pre-existing MEK1 mutation in both pre-and post- therapy PDX. The remaining two models (WM4276, WM4237) had pre-existing loss of PTEN and amplification of MET respectively as possible contributors to resistance (Figure 3C).
Protein expression profiles
RPPA was performed on a total of 118 PDX models in triplicate divided on two batches. Set 102 (Supplementary File S3) had 184 profiles representing 60 models including one model with corresponding untreated and BRAF inhibitor treated samples. Set 119 ((Supplementary File S4) had 243 profiles containing 58 models, 23 of which have corresponding untreated and BRAFi and/or BRAFi/MEKi treated tumor samples. Set 102 assessed 279 phospho and total proteins, and set 119 assessed 299 phospho and total proteins.
PDX Derived from Brain Metastasis
We collected melanoma brain metastasis (MBM) tissue from 34 neurosurgeries of 28 patients to generate PDX models. Targeted sequencing data are currently available for 20 PDX and RPPA data for 12 (Figure 3D). Remarkably, four brain metastases were collected from the same patient (WM4237-1 to -4) at 2- to 4-month intervals. Although the patient had received dabrafenib/trametinib combination therapy (best response stable disease) after the first surgery, and had received anti PD-1 therapy during the last two surgeries, all four PDX had identical mutation profiles (BRAFV600E RB1N690fs TP53S241). PDX from seven patients had BRAF hotspot mutation and from six patients NRAS hotspot mutation. One of these had a co-occurring BRAF non-hotspot mutation. Another BRAF non-hotspot mutation was co-occurring with an NF1 mutation. Two patients were wild type for both BRAF and NRAS. Interestingly, the samples without BRAF hotspot mutation had significantly more concurrent deleterious and likely deleterious mutations overall. We found PTEN deletion or deleterious mutation in four of 7 patients with BRAF hotspot mutation which has been shown to be associated with MBM (Bucheit et al., 2014). On the protein expression level, both patients with deleterious PTEN mutations had evidence of PI3K pathway activation by relative increased phospho AKT compared to WT PTEN samples.
PDX from Primary or Metastatic Uveal Melanoma Samples
We implanted 45 uveal primary samples as tumor fragments s.c. in the interscapular fat pad of NSG mice with matrigel (Némati et al., 2010). After follow up of at least 12 months, we observed tumor growth in five models, albeit kinetics were slow. Three of these had mutations in GNAQ or GNA11, one was WT, and one failed genomic analysis. In contrast, the take rate for metastatic samples from uveal melanoma patients was comparable to cutaneous melanoma and we established four samples as PDX, one with a GNAQ mutation, and the others in process.
Availability of PDX models to the research community
A critical component of our PDX platform is its availability to the research community. Like cell line repositories, PDX tissue can be frozen and expanded as needed. Thus, we made a representative pre-selection of 26 “work horses” based on genetic, and clinical criteria available through www.horizondiscovery.com/patient-derived-xenograft/melanoma-pdx (Supplementary File S2). All other models are available upon request and tissue will be expanded either at Horizon Discovery Inc. (St. Louis, MO) or our laboratory.
Spontaneous Metastasis Rate is Associated with Mutational Group
When cells from a PDX model were inoculated into a human skin graft on NSG mice (Li et al., 2015), tumors formed within the human dermis. These then metastasized out of the human graft into the lungs of host mice as an indicator for distant organ metastasis (Figures 4A–C). This propensity to invade the mouse tissue and seed distant organs was reflected in the subsequently observed high rates of spontaneous metastasis in s.c. implanted PDX models. We analyzed lungs of mice at the time of tumor harvest (Figure 4D) and found that in 32% of PDX models assessed, more than 80% of the animals had micro- or macro-metastases (Figure 4E). There was a significant increase in metastatic ability of BRAF hotspot mutant PDX and a decreased metastatic rate in triple WT PDX (Figure 4F).
Figure 4. Melanoma PDX metastasize spontaneously.

(A) Animals were grafted with neonatal foreskin grafts and melanoma PDX cells were injected into established grafts. (B) Melanoma lesions formed in the human skin reconstructs. (C) Melanomas spontaneously metastasized to the mouse lungs from the human skin graft. H&E staining, and representative images. (D) Example of spontaneous micro-metastasis to lung. (E) Percentage of PDX that metastasize to lungs in more than 80% of animals from the subcutaneous tumor graft at the time point of maximal tumor volume. (F) Number of PDX with spontaneous lung metastasis compared to main mutational subgroups. (G) Luciferase transfected brain metastasis PDX injected s.c.. (H) Spontaneous metastases to the mouse brain were imaged ex vivo after a latency of 120 days after survival surgery. (I) Percentage of IGF1R positive cells in PDX from naïve patients, from patients progressed on BRAF inhibitor (−BR), on BRAF inhibitor or BRAF/MEKi combination diet.
Spontaneous brain metastasis model
An MBM derived PDX was established as a short-term culture, transfected with a luciferase reporter and implanted s.c. into NSG mice. To prolong survival of animals, primary tumor grafts were surgically removed once established (Figure 4G). We observed spontaneous metastasis to the mouse brain in 50% of animals after a latency of 120 days (Figure 4H). Additional such models are in development.
PDX tumors resistant to MAPK inhibitors have increased IGF1R expression
We assessed expression of a panel of melanoma surface receptors previously described as cancer stem cell markers including CD20 (Fang et al., 2005), CD271 (Boiko et al., 2010), and CD133 (Monzani et al., 2007) in two cohorts of therapy naïve and resistant PDX. There were no significant differences observed for any of the markers (data not shown). However, tumors derived from targeted therapy progressed patients had significantly higher levels of IGF-1R than tumors from therapy naïve patients (Figure 4I). IGF-1R/PI3K signaling has previously been implicated in conferring melanoma resistance to BRAF inhibitors (Villanueva et al., 2010). Interestingly, when the resistant tumor grafts were grown on continuous BRAFi or BRAF/MEKi combination diet the IGF-1R levels returned to baseline (Figure 4I). This phenomenon might indicate the transient nature of tyrosine kinase receptor upregulation and its modulation by MAPK pathway inhibitors.
Predictive Value of PDX for Response to Targeted Therapies
We selected a PDX from a 55-year-old female patient with metastatic melanoma and early relapse to vemurafenib after partial response using RECIST 1.1 criteria and progression free survival of 16 weeks. Lymph node lesions in her right and left axillary regions showed initial on-treatment regression: there was a 70.6% decrease in the target lesion (i.e., the right axillary node) and a partial response in the non-target lesion (the left axillary node) (Figure 5A). An FNA was taken from the left lymph node before therapy and used to generate a PDX. After in vivo expansion, tumor bearing animals were treated with the BRAF inhibitor PLX4720 alone and in combination with the MEK inhibitor PD-0325901. Tumors did not respond to BRAF inhibition, but regressed on BRAF/MEKi combination followed by relapse (Figure 5B). This was reflected in a reduced proliferation rate in the combination therapy tumor cells only (Figure 5C).
Figure 5. PDX models in pre-clinical trials.

(A) Computerized tomography scans of patient with early relapse on vemurafenib whose tumor was used to generated a PDX from a pre-therapy LN metastasis. Arrow indicates the lymph node metastasis biopsied, imaged before and 3 months on vemurafenib therapy. (B) The PDX bearing mice were fed a chemical additive diet containing PLX4720 200ppm as single agent or in combination with PD-0325901 7ppm (PLX+MEKi). The combination diet inhibited the PDX tumors’ growth, followed by early on-therapy relapse. (C) Ki67 staining indicating actively proliferating cells from tumor grafts on indicated treatments. (D) Two PDX models from patients relapsed on BRAF inhibition (n=10/group) were treated with chemical addictive diet containing the MEK inhibitor trametinib 2.1ppm (Tram), the PI3K beta inhibitor GSK231418 214.3ppm (GSK418) or the combination of both. (*) The combination significantly inhibited tumor growth over single agents in both models. (E) PDX model from a BRAF-V600E patient relapsed on vemurafenib (PFS 46 weeks, best response stable disease) that had an additional activating MEK mutation, TP53 WT, and a biomarker signature indicating sensitivity to p53 re-activation. PDX tumors (n=10/group) were treated with the ERK inhibitor BVD-523 50mg/kg twice daily oral gavage, the MDM2 inhibitor CGM097 100mg/kg once daily oral gavage, or the combination of both. (E, right panel) Single mouse growth curves of the BVD-523 + CGM treated group highlighting the heterogeneity of response in PDX models. While most tumors showed stable disease, two mice had early relapse and two mice had complete responses (CR). Dosing was stopped on day 38 (blue arrow) and the 2 CR mice showed regrowth of residual disease. (F) Twenty PDX of BRAFV600 mutant patients (naïve and BRAF inhibitor resistant), NRAS mutant, and BRAF-WT NRAS-WT (n=5 models each) were treated with the BET inhibitor BAY8097 10mg/kg once daily oral gavage (orange) or vehicle control (n=3/group, blue) in a rapid in vivo screen. Although variability within the PDX models was high, tumor growth velocity was decreased in a subset of models. Response was independent of mutation status. (G) IDH1 mutant PDX have increased 2-HG onco-metabolite levels in tumor tissue compared to IDH1 WT PDX.
MEK and PI3K Beta Inhibition as Second-line Therapy in BRAF Inhibitor Resistant Models
We selected three BRAF-V600E PDX models derived from patients relapsed on BRAF inhibitor. Two had homozygous PTEN deletion and one had an activating NRASQ61K mutation; all showed activation of both MAPK and PI3K pathways on the protein level (Krepler et al., 2016). The MEK inhibitor trametinib and the PI3K beta/delta isoform-specific inhibitor GSK418 (an analog of GSK2636771 (Rivero and Hardwicke) significantly decreased tumor growth in the two PDX models with PTEN deletion without evident toxicity (Figure 5D), but not in the PDX with concurrent BRAF and NRAS mutation.
ERK and MDM2 Inhibition Is Highly Effective in a BRAF Inhibitor-resistant PDX Model
WM3973 was derived from a patient progressed on vemurafenib with MAPK pathway reactivation via an activating MAP2K1 (MEK1) mutation as a potential resistance mechanism (Krepler et al., 2016). Accordingly, this PDX model did not respond to BRAF inhibition or even to the downstream targeting ERK inhibitor BVD-523. We then applied a previously published response biomarker signature for p53 re-activation (Jeay et al., 2015) to a cohort of nine TP53 wild type BRAF inhibitor resistant PDX models. The majority including WM3973, were predicted sensitive to MDM2 inhibition (data not shown). The MDM2 inhibitor CGM097 (Holzer et al., 2015) moderately inhibited WM3973 tumor growth as a single agent, but ERK and MDM2 inhibition synergized potently to induce stable disease over 6 weeks of dosing (Figure 5E, left panel).
Typical of the tumor growth heterogeneity seen in PDX experiments, single mice showed a variable response to the combination therapy (Figure 5E, right panel). Whereas most animals had stable disease, two tumors showed early relapse, and two tumors had complete responses at the end of dosing. Both regrew only after treatment was stopped, confirming that in PDX models small residual tumors can survive following several weeks of drug pressure. However, we did not observe any tumors acquiring resistance while on combination therapy, indicating that this approach could be explored further using additional models.
We analyzed protein expression profiles of tumor grafts at the end of dosing to investigate the heterogenous responses seen with this therapy. The clusters from unsupervised hierarchical clustering identified groups that were predominately based on proteins with a role in proliferation and correlated with tumor growth rates rather than dosing groups (Supplementary Figure S1). The BVD-523 single agent group whose tumors grew at the same rate as controls, clustered with the fastest growing tumors in the control group, indicating that ERK inhibition alone did not widely change the protein and phospho protein levels assessed in this array. Indeed, there was no inhibition of pERK on RPPA. However, the BVD-523 single agent group had the least tumor growth variability with all tumors progressing rapidly. All tumors with continued response to combination treatment clustered in one group, whereas the two tumors with early resistance to the combination therapy clustered with the CGM single agent samples.
Rapid in Vivo Screen for BET Inhibitor Activity in a Broad PDX Panel
We used the novel BRD4 inhibitor BAY8097 to conduct a rapid in vivo screen on 20 PDX of diverse mutational profiles. To test feasibility, we reduced group size from 10 to 3 mice per group. Like the model in Figure 3E, we observed significant heterogeneity in tumor growth, a problem also encountered in a recently published study using only one tumor graft/PDX/therapy (Gao et al., 2015). We found that a subset of models not clustering into a mutational subgroup showed significant tumor growth inhibition using BAY8097 as a single agent (Figure 5G).
Validation of Increased Onco-metabolites in PDX with IDH1 Mutation
We identified eight PDX with the canonical IDH1 mutation R132C. Only one melanoma cell line with very slow growth kinetics has been described in the literature (Lopez et al., 2010). Indeed, we were unsuccessful in establishing cell lines from these patient samples (data not shown). We tested levels of the D-2-hydroxyglutarate (2-HG) onco-metabolite (Mondesir et al., 2016) and confirmed buildup to very high levels as compared to WT in PDX tissue (Figure 5H).
PDX Can Model Pathway Adaptation to Targeted Drugs Over Time
To assess the potential of PDX models to mimic acquired drug resistance, we performed a time course analysis of response and acquired resistance to a BRAF inhibitor in a targeted therapy-naïve BRAF-V600E PDX. The patient had received BRAF inhibitor therapy after the biopsy was taken and initially responded followed by relapse after 9 months. Although, the patient never received BRAF/MEK combination therapy, we followed up with this combination in our PDX model (Figure 6A). The PDX tumors initially responded to BRAF inhibition with almost complete tumor regression but relapsed after seven weeks; however, when the same animals were switched over to BRAF/MEK inhibitor combination they again responded continuously without relapse for up to 2.5 months. Tumors from each treatment were analyzed for protein expression by RPPA in a time course manner (Figure 6B, full dataset in Supplementary File S5). Protein expression only changed significantly with the onset of BRAF inhibitor resistance (Figure 6C), and the subsequent change to BRAF/MEK inhibitor combination therapy shut down cell proliferation, induced apoptosis and led to sustained tumor growth inhibition (Figure 6D). Thus, PDX models can be used to track changes in tumor cell signaling on the protein level over the course of therapy.
Figure 6. Protein pathway activation over time and in response to MAPK inhibition.

WM4007 was generated from a pre-BRAF inhibitor therapy biopsy. (A) PDX growth curves for mice treated with PLX4720 (BRAFi) or PLX4720+PD-0325901 (BRAF/MEKi) diet started at time points indicated by black data points. (B) Protein expression change patterns identified in RPPA data with K means clustering. All proteins within each cluster are averaged and standard deviation shown. Clusters in bold had variation above 0.1 and were analyzed further. (C) Hierarchical clustering of RPPA data normalized to controls depicting the significant K means clusters along each time point. (D) Ingenuity Pathway Analysis (IPA) was used to assign proteins within each cluster into distinct biological processes. The top five significant gene ontology terms within each cluster are displayed with bars, top axis. The percentage of each cluster’s proteins found within each biological functional category are displayed with orange dots, bottom axis.
DISCUSSION
Established melanoma cell lines have significant bias toward BRAF, TP53 mutations, and CDKN2A loss (Garman et al., 2017) since these adapt well to in vitro growth. The much higher success rate of PDX irrespective of mutational subgroup make PDX more clinically relevant (Byrne et al., 2017; Townsend et al., 2016). Several other research groups have established melanoma PDX models (Einarsdottir et al., 2014; Gao et al., 2015; Girotti et al., 2016; Kemper et al., 2016; Quintana et al., 2012). Quintana et al. established PDX from 25 stage IIIB/C patients and correlated spontaneous metastasis in the animals with patient outcome. Einarsdottir et al. established PDX from 23 patients and predicted targeted therapy responses in a subset. Gao et al. employed a 1×1×1 in vivo trial design in 277 PDX including 67 melanoma derived, demonstrating clinical translatability of this approach. Kemper et al. established 89 PDX, but focused on BRAF mutant patients with only 10 NRAS and 6 WT/WT samples. They then used this platform to identify a novel resistance mechanism to BRAF inhibition in the form of a duplicated kinase domain. Girotti et al. have built a collection of about 90 PDX models, of which they show 3 deeply characterized examples by following the development of resistance to targeted therapy over time using whole exome sequencing. Together, these studies show the promise and potential of PDX models in melanoma.
Multiple resistance mechanisms to targeted therapy have been described and these most often lead to re-activation of the MAPK pathway or activation of alternative pathways such as the PI3K signaling pathway (Rizos et al., 2014). Pre-clinical data by several groups have suggested that combining BRAF/MEK inhibitors with PI3K/mTOR inhibitors may overcome resistance in BRAF mutant melanomas (Atefi et al., 2011; Greger et al., 2012; Shannan et al., 2016; Villanueva et al., 2010). Phase I clinical trials using this combination demonstrated the safety of this combination approach and some early signs of clinical activity (Bedard et al., 2012; Juric et al., 2014), and further phase I/II trials are ongoing (NCT01449058, clinicaltrials.gov). On the other hand, a Phase I trial testing the combination of pan-PI3K/mTORC1/2 inhibitor GSK2126458 with trametinib was terminated due to a lack of tolerability and efficacy (NCT01248858), suggesting a narrower targeting profile might be advantageous. Thus, our pre-clinical PDX trial confirmed that combination of a beta isoform specific PI3K inhibitor retained synergistic potential with MEK inhibition but could potentially decrease toxicity.
We included PDX with diverse mutational backgrounds that were either naïve or progressed on targeted therapy in an in vivo screen of a novel BET inhibitor. Targeting the transcriptional activity of cancer cells has emerged recently as a novel strategy (Filippakopoulos et al., 2010). It is unclear however, which patients would benefit from these inhibitors and whether it would be a viable strategy in a clinical setting for melanoma (Segura et al., 2013). Our PDX collection is large enough to mirror the diversity of patients that would be studied in an early-stage clinical trial at a fraction of the cost and could be beneficial for early-stage drug screening as well as for the development of biomarkers. The activity of BET inhibition seen in a subset of PDX models, although hampered by high heterogeneity, still warrants further investigation into this class of compounds and use of the PDX data to identify response biomarkers.
Another strength of our large collection of PDX is the breadth of coverage including multiple samples with rare mutations, made possible by large-scale targeted sequencing of PDX (Garman et al., 2017). IDH1 is a rarely mutated oncogene in melanoma, representing about 6% of driver mutations (Cancer Genome Atlas, 2015) and has been described as a viable target in other cancers (Tateishi et al., 2015). Since PDX are a living resource we could functionally validate the mutation by assessing the accumulation of the onco-metabolite 2-HG in the tumor grafts. Thus, these models would be ideal to test inhibitors of IDH1.
MBM is a common event in late stage patients and has a poor prognosis of less than one year median survival (Staudt et al., 2010) even with modern systemic therapies (Forschner et al., 2017). Although current targeted and immune therapies have demonstrated activity in MBM, successful therapy is still a major challenge and an important area of current investigation (Glitza Oliva et al., 2017). MBM models are scarce and new therapies are needed urgently. Thus, we focused our collection efforts on samples derived from MBM and these will provide a valuable resource to study this challenging to treat and frequently lethal manifestation of late stage melanoma.
Although patients can show long-lasting responses to immune checkpoint blockade, many patients do not respond or acquire resistance. Clinical studies point towards the importance of the immune infiltrate in tumors (Chen et al., 2016), however human tumor-infiltrating lymphocytes implanted with the initial patient tumor tissue are lost in PDX propagation. High mutational load is associated with increased response rates to immune therapies with neo-antigens the target of immune responses (Peng et al., 2016). Thus, PDX models from checkpoint inhibitor responders and non-responders could potentially be valuable tools to study the role of tumor biology in response to immune therapy and we are currently investigating neo-antigens. Our collection of PDX can be used to study checkpoint inhibitors or other immune therapies alone or in combination with targeted kinase inhibitors when employed in humanized mouse models (unpublished). In these models, human CD34+ hematopoietic stem cells are injected to reconstitute human B and T cells in NSG mice (Rongvaux et al., 2014). Thus, the current limitations of model could potentially be addressed using humanized mice and would allow PDX models to be at the forefront of immune and targeted therapy translational research (Sanmamed et al., 2016). These studies are ongoing.
In summary, we have built a unique and comprehensive melanoma PDX collection representing the entire spectrum of this cancer with multiple biological replicates even for rare subgroups. It is further enhanced through genetic and genomic analysis in our companion paper (Garman et al., 2017).
EXPERIMENTAL PROCEDURES
Detailed SOPs for all aspects of PDX generation and use are provided in Supplementary File S1.
Patient sample processing
Patient samples were collected under IRB approval. Tumor samples were processed within 24 hours of biopsy. Samples were mechanically dissociated and enzymatically digested if necessary. Tumor tissue was frozen in 10%DMSO 90%FBS, if sufficient quantities were available, or implanted directly into NSG mice. Mice were anesthetized, a small skin incision (~5mm) was made in the back of the animal, and a subcutaneous pocket created. Tumor fragments were implanted with 100 μL of matrigel, and the incision closed with a wound clip.
PDX Maintenance
All animal experiments were performed in accordance with institutional guidelines under Wistar IACUC approval. PDX were expanded in NSG mice. Tumor size was assessed once weekly by caliper measurements (lengthxwidth2/2). Animals were sacrificed when the tumors reached 1,000mm3 or when necessary for animal welfare. The larger part of the tumor was retained as a live tumor bank, the smaller part was re-implanted at 1:5 ratio. PDX tumors from patients progressed on BRAF or BRAF/MEK inhibitor therapy were expanded on continuous PLX4720 200ppm or PLX4720 200ppm + PD-0325901 7ppm chemical additive diet (Research Diets, New Brunswick, NJ).
Pre-clinical in vivo trials
When tumors reached 200mm3, mice were randomized into treatment groups. Groups in the efficacy studies were 10 animals each to account for variability among tumors, except for the BAY8097 rapid in vivo screen which was designed with three animals/group. Tumor sizes were assessed twice weekly per caliper measurement, and tumor volume was estimated using the formula (lengthxwidthxwidth/2). Mice were sacrificed after 2–3 weeks of treatment. If therapy groups showed tumor regression, dosing was prolonged.
Short Tandem Repeat (STR) Profiling
We performed STR profiling on one tumor per MP using AmpFlSTR® Identifiler® PCR Amplification Kit (Life Technologies, Carlsbad, CA) which uses loci consistent with all major worldwide STR standards. Genomic DNA was extracted from patient or xenograft tumor samples using DNeasy® Blood & Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. PCR amplification and STR allele separation and sizing was performed by the Wistar Genomics Facility. Profile interpretation was performed in our lab by interrogating the resulting DNA fingerprint to our internal database which includes over 1,000 fingerprints and is available on our website www.wistar.org/lab/meenhard-herlyn-dvm-dsc/page/melanoma-cell-str-profiles. DNA fingerprinting was matched to normal blood DNA if available to confirm identity of the samples.
Massively Parallel Sequencing
DNA from patients and/or PDX were characterized by massively parallel sequencing using a custom-designed 108 gene targeted panel. Results were annotated for mutations, insertions and deletions, and copy number changes. A detailed description of the methodology and analysis is provided in (Garman et al., 2017). Briefly, DNA was purified (DNeasy Blood & Tissue Kit), 500 ng of genomic DNA was sheared randomly into 200 bp fragments, and sheared DNA was A-tailed and ligated with adaptor-embedded indexes using the NEBNext® UltraTM DNA Library Prep Kit for Illumina® (New England BioLabs, Inc., Ipswich, MA). Samples were equimolarly pooled prior to capture with a 2.2 Mbp SureSelectXT Custom Target Enrichment Kit (Agilent Technologies, Santa Clara, CA) targeting 108 genes previously implicated in melanomagenesis. Paired-end (2X100 bp) sequencing was performed on the HiSeqTM 2000 sequencing system (Illumina, Inc., San Diego, CA).
To account for mouse DNA contamination, previously unreported variants with an allelic fraction of less than 0.15 were filtered out of the analysis.
Foreskin Grafting Procedure
Prepared rectangles of about 1.5 × 2 cm foreskin were placed on skin defects on the back of a mouse with the panniculus canosum remaining intact. The panniculus canosum was needed to help vascularize the graft. The foreskin graft was then secured in situ using Tegaderm (3M, St. Paul, MN). After 10 days, the dressing was removed and the graft was fully healed in 5–6 weeks.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections of xenograft tumors were cut into 4 μm sections, deparaffinized in xylene, rinsed in ethanol and rehydrated. Then, the tissues were stained with Ki-67 mouse clone MiB-1 (Dako, Carpinteria, CA; Catalog# M7240).
Flow Cytometry Staining
Tumors were analyzed after mechanical dissociation followed by filtration and red blood cell lysis. For surface staining, cells were incubated at 4°C for 30 minutes with anti-human PeCy7 CD146 (M-CAM), anti-mouse FITC- CD45, H2Kb and H2Kd and anti-human PE IGFR1 from BD Biosciences (San Jose, CA). Staining were performed in presence of LIVE/DEAD® Fixable Dead Cell Stains (Life Technologies). After dead cells and mouse cell exclusion, percentage of double positive CD146 and IGF1R cells were reported.
RPPA
The samples were prepared as previously described (Krepler et al., 2016). RPPA was performed by the MD Anderson Center RPPA core facility (Houston, TX) as previously described (Tibes et al., 2006). Unsupervised hierarchical clustering using centered correlation and complete linkage was performed on normalized log2 median-centered protein values using Cluster 3.0 software (http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm#ctv). Results were visualized using Java TreeView 3.0 software (http://jtreeview.sourceforge.net). For WM4007 time course analysis, normalizedLog2 values were median centered to the average of the untreated controls. The three tumors from each time point were averaged. K means clustering using Euclidean distance measure on 10 clusters (identified in unsupervised hierarchical clustering) run for 100 iterations was performed using Cluster 3.0 and visualized with Java TreeView. Clusters with variance greater than 0.10 across the time points were selected for Gene Ontology analysis using Ingenuity Pathway Analysis (Qiagen) for biological processes.
Statistical Analysis
The scatter plots with mean of multiple mice’s tumor growth rates were reported by FNA, core, and excisional biopsy patient samples, or by patient’s sample. Shapiro normality tests were used to examine the distribution of studied variables. Non-parametric Mann-Whitney tests were used for between specific gene mutant group comparison. Linear mixed-effect models were used to test the difference of the tumor growth trends among treatment groups.
Supplementary Material
SOP handbook detailing all procedures and protocols for PDX generation, expansion, and banking.
Excel sheet of all PDX and banked tissue containing pertinent clinical information and main mutations.
Set 102 PDX RPPA data.
Set 119 PDX RPPA data
WM4007 RPPA data.
Acknowledgments
We thank the animal, genomics, histology, flow cytometry, and imaging core facilities at the Wistar Institute. We thank the University of Pennsylvania Perelman School of Medicine Biobank (Federico Valdivieso, Caitlin Feltcher, Amber McKeown, Emma Gasper) with support from the Perelman School of Medicine and Abramson Cancer Center (P30 CA016520-40). We thank Lori E. Huelsenbeck-Dill and Patricia L. Swanson at the Helen F. Graham Cancer Center. We thank the tissue collection core facilities at the MD Anderson Cancer Center, Massachusetts General Hospital, and John Wayne Cancer Institute. We thank Drew A. Torigian for CT image analysis. We thank G. Bollag at Plexxikon for supplying PLX4720.
Support for Shared Resources utilized in this study was provided by Cancer Center Support Grant (CCSG) P30CA010815 to the Wistar Institute; by Cancer Center Support Grant (CCSG) CA016672 to MDACC and the MDACC Melanoma SPORE (P50 CA093459). This work was supported by NIH grants P01 CA114046, P01 CA025874, R01 CA047159 to M.H.; the P50 CA174523-02 SPORE on Skin Cancer to the Wistar Institute and the University of Pennsylvania; R01 CA182635 to A.E.A.; R01CA198015, 5P30 CA016520 to R.A.; the Margaretta and R.R.M Carpenter, SR Cancer Stem Cell Research Program to HFGCC; the University of North Carolina Cancer Research Fund to D.D.; philanthropic contributions to The University of Texas MD Anderson Cancer Center Melanoma Moon Shot Program; and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was supported in part by grants from GSK, Novartis, and Bayer.
Footnotes
AUTHOR CONTRIBUTIONS
C. K., K. S., M. B., M. X., B. S., A. W., A. V. participated in PDX establishment, expansion, banking, and in vivo experiments. M. P. performed and analyzed flow cytometry experiments. G. Z. performed and analyzed IHC staining of tumor grafts. P. B. developed, performed, and analyzed quality control procedures. P.B analyzed RPPA data. K.S. and C.K. developed experimental procedures. X. Y., Q. L. performed statistical analysis. B. G., I. N. A., B. W., M. A. W., K. L. N. developed, performed, and analyzed targeted sequencing. W. X., G. K., M.F., X. X., R. A., T. C. G., D. E. E., D. A. T., L. S., L. H., J. A. W., M. D., Y. L., G. M., D. T. F., M. B., K. T. F., D. S. H., M. G., J. B., N. J. P., C. L. S., T. S., A. A., A. R., performed tissue and clinical data collection. D. D., S. A., R. K., E. H., G. C., S. J., J. W., A. W., M. O., M. B. B. participated in planning of in vivo experiments and data analysis. C.K., K. L. N., L. S., M. H. participated in conception and design of the project. C. K., P. B., M. H. wrote the manuscript. C.K. and M.H. supervised the work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arafeh R, Qutob N, Emmanuel R, Keren-Paz A, Madore J, Elkahloun A, Wilmott JS, Gartner JJ, Di Pizio A, Winograd-Katz S, et al. Recurrent inactivating RASA2 mutations in melanoma. Nat Genet. 2015;47:1408–1410. doi: 10.1038/ng.3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS ONE. 2011;6:e28973. doi: 10.1371/journal.pone.0028973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard P, Tabernero J, Kurzrock R, Britten CD, Stathis A, Perez-Garcia JM, Zubel A, Le NT, Carter K, Bellew KM, et al. A phase lb, open-label, multicenter, dose-escalation study of the oral pan-PI3K inhibitor BKM120 in combination with the oral MEK1/2 inhibitor GSK1120212 in patients (pts) with selected advanced solid tumors. ASCO Meeting Abstracts. 2012;30:3003. [Google Scholar]
- Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, Butler PD, Yang GP, Joshua B, Kaplan MJ, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–137. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucheit AD, Chen G, Siroy A, Tetzlaff M, Broaddus R, Milton D, Fox P, Bassett R, Hwu P, Gershenwald JE, et al. Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin Cancer Res. 2014;20:5527–5536. doi: 10.1158/1078-0432.CCR-14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinska E, Caldas C, Chang DK, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017;17:254–268. doi: 10.1038/nrc.2016.140. [DOI] [PubMed] [Google Scholar]
- Callahan MK, Postow MA, Wolchok JD. Targeting T Cell Co-receptors for Cancer Therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016;6:827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einarsdottir BO, Bagge RO, Bhadury J, Jespersen H, Mattsson J, Nilsson LM, Truve K, Lopez MD, Naredi P, Nilsson O, et al. Melanoma patient-derived xenografts accurately model the disease and develop fast enough to guide treatment decisions. Oncotarget. 2014;5:9609–9618. doi: 10.18632/oncotarget.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20411–20416. doi: 10.1073/pnas.0905833106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer research. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forschner A, Eichner F, Amaral T, Keim U, Garbe C, Eigentler TK. Improvement of overall survival in stage IV melanoma patients during 2011–2014: analysis of real-world data in 441 patients of the German Central Malignant Melanoma Registry (CMMR) Journal of cancer research and clinical oncology. 2017;143:533–540. doi: 10.1007/s00432-016-2309-y. [DOI] [PubMed] [Google Scholar]
- Furney SJ, Turajlic S, Stamp G, Thomas JM, Hayes A, Strauss D, Gavrielides M, Xing W, Gore M, Larkin J, et al. The mutational burden of acral melanoma revealed by whole-genome sequencing and comparative analysis. Pigment Cell Melanoma Res. 2014;27:835–838. doi: 10.1111/pcmr.12279. [DOI] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang YZ, Singh M, Zhang C, Schnell C, Yang GZ, Zhang Y, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature Medicine. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Garman B, Anastopoulos IA, Krepler C, Sproesser K, Brafford P, Wilson M, Wubbenhorst B, Amaravadi R, Bennett J, Beqiri M, et al. Genetic and genomic characterization of 462 melanoma patient-derived xenografts, tumor biopsies and cell lines. 2017 doi: 10.1016/j.celrep.2017.10.052. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geller AC, Miller DR, Annas G, Demierre M, Gilchrest BA, Koh HK. Melanoma incidence and mortality among us whites, 1969–1999. JAMA. 2002;288:1719–1720. doi: 10.1001/jama.288.14.1719. [DOI] [PubMed] [Google Scholar]
- Girotti MR, Gremel G, Lee R, Galvani E, Rothwell D, Viros A, Mandal AK, Lim KH, Saturno G, Furney SJ, et al. Application of Sequencing, Liquid Biopsies, and Patient-Derived Xenografts for Personalized Medicine in Melanoma. Cancer Discov. 2016;6:286–299. doi: 10.1158/2159-8290.CD-15-1336. [DOI] [PubMed] [Google Scholar]
- Glitza Oliva I, Tawbi H, Davies MA. Melanoma Brain Metastases: Current Areas of Investigation and Future Directions. Cancer J. 2017;23:68–74. doi: 10.1097/PPO.0000000000000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger JG, Eastman SD, Zhang V, Bleam MR, Hughes AM, Smitheman KN, Dickerson SH, Laquerre SG, Liu L, Gilmer TM. Combinations of BRAF, MEK, and PI3K/mTOR Inhibitors Overcome Acquired Resistance to the BRAF Inhibitor GSK2118436 Dabrafenib, Mediated by NRAS or MEK Mutations. Molecular Cancer Therapeutics. 2012;11:909–920. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, Berghausen J, Bouisset-Leonard M, Buschmann N, Pissot-Soldermann C, et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J Med Chem. 2015;58:6348–6358. doi: 10.1021/acs.jmedchem.5b00810. [DOI] [PubMed] [Google Scholar]
- Jeay S, Gaulis S, Ferretti S, Bitter H, Ito M, Valat T, Murakami M, Ruetz S, Guthy DA, Rynn C, et al. A distinct p53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. Elife. 2015:4. doi: 10.7554/eLife.06498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric D, Soria JC, Sharma S, Banerji U, Azaro A, Desai J, Ringeisen FP, Kaag A, Radhakrishnan R, Hourcade-Potelleret F, et al. A phase 1b dose-escalation study of BYL719 plus binimetinib (MEK162) in patients with selected advanced solid tumors. ASCO Meeting Abstracts. 2014;32:9051. [Google Scholar]
- Kemper K, Krijgsman O, Kong X, Cornelissen-Steijger P, Shahrabi A, Weeber F, van der Velden Daphne L, Bleijerveld Onno B, Kuilman T, Kluin Roel JC, et al. BRAFV600E Kinase Domain Duplication Identified in Therapy-Refractory Melanoma Patient-Derived Xenografts. Cell Reports. 2016;16:263–277. doi: 10.1016/j.celrep.2016.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, McCusker JP, Ma S, Cheng E, Straub R, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nat Genet. 2015;47:996–1002. doi: 10.1038/ng.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Xiao M, Sproesser K, Brafford PA, Shannan B, Beqiri M, Liu Q, Xu W, Garman B, Nathanson KL, et al. Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin Cancer Res. 2016;22:1592–1602. doi: 10.1158/1078-0432.CCR-15-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Fukunaga-Kalabis M, Herlyn M. Establishing Human Skin Grafts in Mice as Model for Melanoma Progression. Methods Mol Biol. 2015 doi: 10.1007/7651_2015_301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez GY, Reitman ZJ, Solomon D, Waldman T, Bigner DD, McLendon RE, Rosenberg SA, Samuels Y, Yan H. IDH1(R132) mutation identified in one human melanoma metastasis, but not correlated with metastases to the brain. Biochem Biophys Res Commun. 2010;398:585–587. doi: 10.1016/j.bbrc.2010.06.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies AM, Wilmott JS, Drummond M, Lo S, Lyle M, Chan MM, Thompson JF, Guminski A, Carlino MS, Scolyer RA, et al. Clinicopathologic features associated with efficacy and long-term survival in metastatic melanoma patients treated with BRAF or combined BRAF and MEK inhibitors. Cancer. 2015;121:3826–3835. doi: 10.1002/cncr.29586. [DOI] [PubMed] [Google Scholar]
- Mondesir J, Willekens C, Touat M, de Botton S. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. Journal of blood medicine. 2016;7:171–180. doi: 10.2147/JBM.S70716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, Gritti A, Piccinini A, Porro D, Santinami M, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–946. doi: 10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MKK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Némati F, Sastre-Garau X, Laurent C, Couturier J, Mariani P, Desjardins L, Piperno-Neumann S, Lantz O, Asselain B, Plancher C, et al. Establishment and Characterization of a Panel of Human Uveal Melanoma Xenografts Derived from Primary and/or Metastatic Tumors. Clinical Cancer Research. 2010;16:2352–2362. doi: 10.1158/1078-0432.CCR-09-3066. [DOI] [PubMed] [Google Scholar]
- Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Piskounova E, Shackleton M, Weinberg D, Eskiocak U, Fullen DR, Johnson TM, Morrison SJ. Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Sci Transl Med. 2012;4:159ra149. doi: 10.1126/scitranslmed.3004599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456:593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero RA, Hardwicke MA. Abstract 2913: Identification of GSK2636771, a potent and selective, orally bioavailable inhibitor of phosphatidylinositol 3-kinase-beta (PI3Kα) for the treatment of PTEN deficient tumors. Cancer Research. 2014;72:2913–2913. [Google Scholar]
- Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res. 2014;20:1965–1977. doi: 10.1158/1078-0432.CCR-13-3122. [DOI] [PubMed] [Google Scholar]
- Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, et al. Development and function of human innate immune cells in a humanized mouse model. Nat Biotechnol. 2014;32:364–372. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to “Nude” mice. Acta pathologica et microbiologica Scandinavica. 1969;77:758–760. doi: 10.1111/j.1699-0463.1969.tb04520.x. [DOI] [PubMed] [Google Scholar]
- Sanmamed MF, Chester C, Melero I, Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2016;27:1190–1198. doi: 10.1093/annonc/mdw041. [DOI] [PubMed] [Google Scholar]
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TTT, Berman DM, Wolchok JD. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2015;33:1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, Gonzalez-Gomez P, Morante M, Jubierre L, Zhang W, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264–6276. doi: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannan B, Chen Q, Watters A, Perego M, Krepler C, Thombre R, Li L, Rajan G, Peterson S, Gimotty PA, et al. Enhancing the evaluation of PI3K inhibitors through 3D melanoma models. Pigment Cell Melanoma Res. 2016;29:317–328. doi: 10.1111/pcmr.12465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X, Kong Y, Li Y, Zhang Q, Si L, Cui C, Chi Z, Tang B, Mao L, Lian B, et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur J Cancer. 2016;65:156–163. doi: 10.1016/j.ejca.2016.06.019. [DOI] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer discovery. 2013;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudt M, Lasithiotakis K, Leiter U, Meier F, Eigentler T, Bamberg M, Tatagiba M, Brossart P, Garbe C. Determinants of survival in patients with brain metastases from cutaneous melanoma. Br J Cancer. 2010;102:1213–1218. doi: 10.1038/sj.bjc.6605622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi K, Wakimoto H, Iafrate AJ, Tanaka S, Loebel F, Lelic N, Wiederschain D, Bedel O, Deng G, Zhang B, et al. Extreme Vulnerability of IDH1 Mutant Cancers to NAD+ Depletion. Cancer Cell. 2015;28:773–784. doi: 10.1016/j.ccell.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell. 2016;29:574–586. doi: 10.1016/j.ccell.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Infante JR, Krepler C, Reyes-Uribe P, Samanta M, Chen HY, Li B, Swoboda RK, Wilson M, Vultur A, et al. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–1099. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SOP handbook detailing all procedures and protocols for PDX generation, expansion, and banking.
Excel sheet of all PDX and banked tissue containing pertinent clinical information and main mutations.
Set 102 PDX RPPA data.
Set 119 PDX RPPA data
WM4007 RPPA data.