

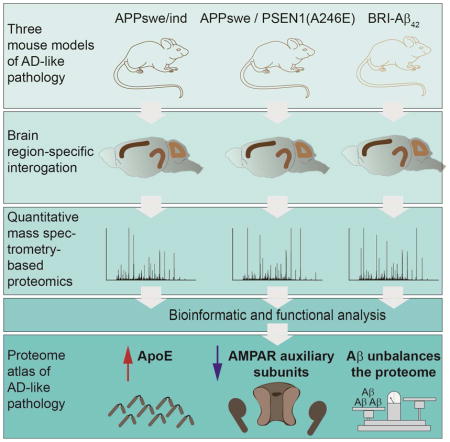
SUMMARY
Amyloid beta (Aβ) peptides impair multiple cellular pathways and play a causative role in Alzheimer’s disease (AD) pathology, but how the brain proteome is remodeled by this process is unknown. To identify protein networks associated with AD-like pathology, we performed global quantitative proteomic analysis in three mouse models at young and old ages. Our analysis revealed a robust increase in Apolipoprotein E (ApoE) levels in nearly all brain regions with increased Aβ levels. Taken together with prior findings on ApoE driving Aβ accumulation, this analysis points to a pathological dysregulation of the ApoE-Aβ axis. We also found dysregulation of protein networks involved in excitatory synaptic transmission. Analysis of the AMPA receptor (AMPAR) complex revealed specific loss of TARPγ-2, a key AMPAR trafficking protein. Expression of TARPγ-2 in hAPP transgenic mice restored of AMPA currents. This proteomic database represents a resource for the identification of protein alterations responsible for AD.
Keywords: AD, proteomics, mass spectrometry, synapses, proteostasis, WGCNA, amyloid beta, ApoE, AMPAr
Graphical Abstract
INTRODUCTION
AD is a progressive brain disorder that is the leading cause of dementia in adults, resulting in impaired memory and cognition. Pathological hallmarks include amyloid plaques, neurofibrillary tangles, astrogliosis, and changes in brain vasculature, which culminate in neurodegeneration. Abundant evidence shows AD brain pathology is complex and represents feedback and feed forward responses in multiple cell types (De Strooper and Karran, 2016). Many mechanisms have been proposed to explain AD, but a unifying and universally accepted description has yet to be achieved. There is near unanimous agreement that Aβ peptides generated by proteolytic processing of amyloid precursor protein (APP) represent a key toxic species. The “amyloid cascade hypothesis” has provided a useful theoretical framework for the basis of AD (Selkoe and Hardy, 2016), yet current evidence suggests a more complicated pathological etiology (Herrup, 2015). One major challenge impeding our understanding of AD pathology is that the molecular perturbations vary based on the stage of disease and brain region. Furthermore, even individuals with mutations in APP or Presenilin-1 (PS1) take decades to develop AD, suggesting that long incubation periods or aging is required. Several molecular pathways including impaired synaptic transmission, hampered protein degradation dynamics, defective mitochondrial function, and inflammatory responses all play important roles in AD pathogenesis (Akiyama et al., 2000; Lin and Beal, 2006; Mori et al., 1987; Sheng et al., 2012).
Dynamic changes in protein abundances represent key cellular responses to cope with cellular insults. However, broad toxicity, such as accumulation of Aβ peptides, likely affects many proteins and pathways. We hypothesized this must ultimately result in proteome remodeling (Powers et al., 2009). For example, proteins physically associated with Aβ plaques accumulate while proteins localized to synapses have reduced levels in AD (Lassmann et al., 1992). To understand the effects of Aβ peptides on the brain proteome, we performed quantitative proteomic analysis of frontal cortex (FC), hippocampal (HIP), and cerebellar (CB) extracts from transgenic mice expressing mutated hAPP or hAPP/PS1 (Borchelt et al., 1996; Mucke et al., 2000). While these models have provided insight into AD pathology, they are limited by over-expression of APP fragments outside Aβ in non-relevant cells (Saito et al., 2014). We also performed proteomic analysis of BRI-Aβ42 mouse brain extracts over-expressing Aβ42 without additional APP fragments (McGowan et al., 2005). To temporally resolve changes in protein abundance, we performed our analysis at ages before and after plaques form to find early and potentially reversible alterations.
We report an AD proteomic resource of 18,882 quantified proteins (10,288 genes) from mouse model brain region extracts. To delineate proximal effects of Aβ on the brain proteome, we determined the level of Aβ and amyloid plaques and performed our analyses at a- and symptomatic time points, based on previous reports (Dodart et al., 1999; Lalonde et al., 2005). We observed an age dependent increase in proteome remodeling, region specific patterns, and found many proteins genetically linked to late onset AD (LOAD), such as ApoE, as significantly altered. ApoE plays a key role in regulating Aβ metabolism and is co-deposited in plaques (Liu et al., 2013; Namba et al., 1991). ApoE also seems to play a role in Aβ aggregation (Bales et al., 1997). However, how ApoE levels are altered in AD remains unclear (Gupta et al., 2011; Hesse et al., 2000; Kuo et al., 2000; Liu et al., 2013). We found ApoE significantly increased in all three models and only in those datasets with increased Aβ levels Overall; we identified a small panel of proteins significantly altered in multiple models. Co-expression analysis revealed two distinct modules (ME) involved with synaptic transmission in HIP and two MEs related to mitochondria in FC. Biochemical purification of AMPARs, which represented core module proteins, showed a specific loss of TARPγ– 2 (known as stargazin). To assess if our proteomics can lead to important conclusions, we tested and found that correcting TARPγ–2 levels can restore glutamatergic synaptic transmission in vivo.
RESULTS
Remodeled proteomes in hAPP and hAPP/PS1 brains
To determine how Aβ peptide toxicity affects the brain proteome, we developed a quantitative proteomic strategy using nitrogen-15 (15N) labeled mice (McClatchy et al., 2007; Wu et al., 2004). The 15N “heavy” brains served as internal standards for proteome-wide quantitation of transgenic AD mouse models (Tg-AD) relative to non-transgenic (Non-Tg) littermates that were unlabeled and “light”. The mixed “light” and “heavy” proteins were digested to peptides, separated using multi-dimensional chromatography, and analyzed by tandem mass spectrometry (MS) (Figure 1A). We compared the FC, HIP, and CB between age-matched Tg-AD or Non-Tg littermates using the same whole brain “heavy” reference proteome and we performed our analysis at presumably a- (3 mo) and symptomatic (12 mo) time points to identify age dependent changes in protein abundance. In total, our MS analysis provided relative quantitation for > 3,800 different proteins in each dataset of three biological replicates per group (Figure 1B).
Figure 1.

Quantitative MS Analysis of Tg-AD Brains (A) Experimental workflow, 14N ratio 15N is calculated and normalized to controls. (B) Quantified protein counts for 12 datasets. (C) Brain region proteome remodeling of hAPP at 3 and 12 mo. (D) Brain region proteome remodeling of hAPP/PS1 at 3 and 12 mo. (E) Bar graphs of age dependent increase of the number of significantly altered proteins (FDR-adj p value < 0.05). (F–G) Venn diagrams of significantly altered proteins. hAPP black outline = CB lacking increased Aβ levels. (H) FC summary for both models, (I) HIP summary for both models. For (B and C) Solid bars are proteins with a > 50% change in ratio 2. See also Figure S1, S6 and Table S1–3, 6.
By analyzing regional brain proteomes of two Tg-AD models with different degrees of pathology, we identified unknown AD pathological mechanisms and confirmed several previously reported. To identify proximal Aβ targets, we first analyzed mice over expressing APPswe/ind (hAPP) at moderate levels. The hAPP mice have hampered synaptic transmission at two to four mo, but do not develop plaques before 10 mo (Hsia et al., 1999; Mucke et al., 2000). We also analyzed a double Tg-AD mouse model over expressing APPswe/PSEN1(A246E) (hAPP/PS1) (Borchelt et al., 1996). hAPP/PS1 mice have plaques, dystrophic neurites, and behavioral deficits by 9 mo (Wang et al., 2003). To document the pathological state of our hAPP mice at 3 mo we performed Aβ1–42 ELISA and found a significant, ~50%, increase in the level of Aβ1–42 in the FC and HIP but not CB (Figure S1A). At 12 mo Aβ1–42 levels in the FC and HIP were more significantly increased compared to the Non-Tg littermates, while the CB did not possess significantly increased Aβ1–42 levels. The absence of a significant increase in Aβ1–42 levels in the CB of the hAPP provided a negative control for proteins sensitive to APP fragments outside Aβ. Aβ1–42 ELISA showed a large and significant increase in the Aβ1–42 level in all three brain regions at 3 and 12 mo in hAPP/PS1 (Figure S1B). We analyzed the amyloid plaque load in HIP by Congo red and Thioflavin S staining in both models and confirmed 3 mo hAPP mice lack plaques, the number of plaques increased with age in both models, and the plaque load even at 12 mo is moderate for both models (Figure S1C–P). Based on these results and previous behavioral analyses we consider the HIP and FC datasets from 3 mo hAPP mice to best represent early stages of pathology.
Our analysis revealed a global imbalance in protein levels, about 5% of the quantified proteins had a change of > 50% at 3 mo and an even larger percentage altered > 50% at 12 mo in 11 out of 12 of our brain region matched comparisons (Figures 1C–D). These measurements were made from, on average ≥ 30 different peptides per protein and > 28% of proteins were quantified in 50% of our samples (Figure S1Q–R). The total number of significantly altered proteins increased from 3 to 12 mo in all brain regions and models. Overall, the HIP possessed the greatest number of significantly (FDR-adjusted (adj) p value < 0.05) altered proteins (Figure 1E). We compared the levels of all the significantly altered proteins in the same Tg-AD model and brain region between the 3 and 12 mo datasets. For FC of hAPP but not hAPP/PS1 mice, we found that the difference between the average protein fold change at 3 to 12 mo was significantly increased (−0.0621 + 0.389 versus 0.214 + 0.320; mean + STD; p value = 1.34 E–8), consistent with an accumulation of protein in the context of more severe pathology (Figure S1S–T). For HIP, we found a significant increase between 3 and 12 mo in hAPP/PS1 (−0.266 ± 0.619 versus −0.155 ± 0.604 mean ± STD; p value = 0.0240). For CB we found a significant increase in the hAPP/PS1 model, (−0.00866 ± 0.554 versus 0.209 ± 0.489; mean ± STD; p value = 3.34 E–4). We determined the number of significantly altered proteins in each brain region and identified those altered in multiple brain regions. Each brain region in both models had 100s of significantly altered proteins (Figures 1F–G and Table S1–2). Bioinformatics to determine the cellular compartments that the altered proteins localize showed they reside in wide range of compartments (Figure S1U). We found 42 proteins significantly altered in FC of both models (Figure 1H and Table S3), and similarly, for HIP we found 184 (Figures 1I and Table S3). To determine how many of our altered proteins have yet to be reported, we performed a literature search, and found 17 FC and 92 HIP unreported in AD or AD models (Table S3). We next used hierarchical clustering to compare protein profiles and found region specific patterns (Figures S2A–C). Principal component analysis showed the 3 and 12 mo FC proteomes of both models are most similar to each other, CB both time points for hAPP or hAPP/PS1 were most similar, and HIP datasets were least similar between the models and time points (Figures S2D–F).
Connecting proteins to genes that predispose for AD
We tested the connection between genes linked to LOAD and those proteins we found with altered abundance (Bertram et al., 2007). Our screen identified twenty-three significantly altered proteins genetically linked to LOAD (Figure 2A). APOE and GOT1 were altered in six datasets, GSTM1 in five, and TF in four. We extracted the model, time point, and brain region where these alterations occurred. Interestingly, we found that APOE was significantly altered only in those datasets with significantly increased Aβ levels. Other proteins such BIN1 and CLU, were only altered in hAPP/PS1 mice (Figure 2B). These findings show that a large number of genes genetically linked to AD also have altered protein products in mouse model brains of AD-like pathology.
Figure 2.

Altered Protein Abundance of Genes Linked to LOAD (A) Significant (FDR-adj p value < 0.05) protein changes mapped to 153 genes previously linked to LOAD. - = not quantified. (B) Brain region(s) and age(s) with significant changes. Red = FC, orange = HIP, and teal = CB. # = hAPP CB that lacks Aβ accumulation. * = IDE1* is shown twice because of sequence variations, + = A2M+ was not identified - related alpha-2-macroglobulin protein Pzp is reported, # = CYP2D6 is not in the mouse genome, we measured related Cyp2d22.
We generated rank ordered summary “confidence plots” for the significant proteins (FDR-adj p value < 0.05). In hAPP mice, α-spectrin 2, which is known to be altered in AD, was the only protein identified as significantly altered in all four data sets with elevated Aβ (Figure 3A). We found 27 significantly altered proteins in three out of four hAPP datasets, 125 in two, and 691 in one. Similarly, only microtubule associated protein 2 (Map2) was significantly altered in all six hAPP/PS1 data sets, three proteins in five, 20 in four, 36 in three, 214 in two, and 469 in one (Figure S2G). Next, we compared the top 30 proteins (~4%) between the models and found nine proteins met this criterion in both models. These mostly-confidently altered proteins included ApoE, Glial fibrillary acidic protein (GFAP), Solute Carrier Family 12 Member 5 (Slc12a5), neurofilament medium peptide (Nefm), clatherin heavy chain 1, Ankyrin 2 (Ank2), spectrin β-III (Spnb3), aconitase 2 (Aco2), and dynein cytoplasmic 1 heavy chain 1 (Dync1h1). These results are consistent with previous findings that the structural proteins Nefm and Spnb3 are altered in AD brains (Ciavardelli et al., 2010; Masliah et al., 1990). Slc12a5, a major chloride extruder, shifts the effect of GABA, is linked to epilepsy, and may be relevant in AD (Lagostena et al., 2010; Palop and Mucke, 2009). Ank2, GFAP, Aco2, Cnp, and Dync1h1 have also been implicated in AD (Burbaeva et al., 2005; Kamphuis et al., 2014; Lazarov et al., 2002; Mangialasche et al., 2015; Silva et al., 2013; Soler-Lopez et al., 2011; Wang et al., 2003a).
Figure 3.

Most Significant Altered Proteins from Brain Regions with Elevated Aβ (A) Summary confidence index (see methods) plot for all significantly (FDR-adj p value < 0.05) altered proteins in the four datasets with significantly increased Aβ levels in hAPP. Proteins with small p values > 0 but < 3.42 E-09 graphed as 1E-13 (see Tables S1); proteins in the top 30 of both models are in bold. (B) APP levels in Tg-AD datasets revealed a significant increase compared to Non-Tg controls (n = 8 – 32 peptides). (C) Apo E levels in hAPP are significantly increased at 12 mo in FC and HIP but not CB. In hAPP/PS1 ApoE levels are significantly increased at 3 mo in the FC, and at 12 mo in all three brain regions (n = 17 – 39 peptides). (D) Apo E is increased in BRI-Aβ FC and HIP by WB. Median = colored, mean = grey or black, grey shading = datasets without increased Aβ levels. *p value < 0.05, ** < 0.01, *** < 0.001 by Student’s t test for (B–D). See also Figure S2–3 and Table S4.
Our finding that ApoE was among the most confidently altered proteins moved us to examine its level relative to APP and Aβ. In 3 mo hAPP mice, APP was increased ~2 fold in all brain regions and more so at 12 mo (Figure 3B). For hAPP/PS1, APP levels were slightly higher at both time points and in all brain regions. When we examined ApoE, which in mice is required for Aβ pathologies (Bales et al., 1997), we found its levels significantly increased in FC and HIP at 12 mo in hAPP. In hAPP/PS1, ApoE had significantly increased levels at both time points in FC and at 12 mo HIP and CB (Figure 3C). Interestingly, ApoE levels were higher exclusively in those brain regions with elevated Aβ levels, since it was unchanged at both time points in CB of hAPP (Figure S1A and Figure 3B). Altogether, ApoE was increased in five out of the five possible 12 mo datasets with the highest Aβ levels.
To specifically explore Aβ42 dependent proteome remodeling, we performed label-free quantitative proteomic analysis of FC, HIP, and CB extracts at 3 and 12 mo of BRI-Aβ42 mice (Figure S3A) (McGowan et al., 2005; Park et al., 2008). BRI-Aβ42 was only detected in mice carrying the transgene and overall levels increased over time (Figure S3B). Our BRI-Aβ42 proteomics quantified >4,000 proteins in each dataset and identified 100’s of proteins with significantly altered levels (Figure S3C and Table S4). As expected, we found no significant changes in APP levels but did detect a significant increase in ApoE. We confirmed increased ApoE levels in BRI-Aβ42 FC and HIP by WB (Figure 3D). Altogether, these results strongly suggest that Aβ42 accumulation leads to a concomitant increase in ApoE in vivo which likely has unexpected and important pathological consequences.
To test the confidence of our MS based protein measurements we performed semi-quantitative western blot (WB) analyses of six proteins. Indeed, these results unanimously confirmed proteins with both increased and decreased levels (Figure S3D–H). Moreover, BRI-Aβ42 proteomic analysis also provided additional independent confirmation of many proteins found altered in hAPP and hAPP/PS1. Altogether, these results show that our proteomic analysis possesses sufficient analytical power to reveal confident changes in protein levels relevant to AD pathology.
Protein co-expression network analysis of Tg-AD brains
We hypothesized consensus weighted gene/protein co-expression analysis (WGCNA) that identifies correlated patterns of protein levels in individual brain regions across the two time points and models, would allow us to delineate groups of co-regulated proteins (Langfelder and Horvath, 2007). We were able to generate consensus networks across genotypes and time points for both the FC and HIP datasets but not CB. We identified 25 FC and 10 HIP MEs across 30 FC and 34 HIP datasets (Figures 4A, S4A, and Table S5). WGCNA reduced 1000s of proteins measurements into 35 MEs that represent core proteome remodeling programs responding to Aβ (Zhang et al., 2013). Direct comparison of the two topological overlap matrices from the Tg-AD or Non-Tg datasets showed Aβ significantly (Z statistics p value < 1E-04) remodels several distinct molecular interaction networks (Figures 4B and S4B). To test if the MEs were enriched for distinct functions (Zhang et al., 2013), we subjected each ME to Gene Ontology cell component (GO:CC) enrichment analysis. Indeed, we identified significant gene ontologies assignments for seven of 10 HIP MEs. These included myelin sheath, regulation of actin polymerization, mitochondrion, synapse, adherens junction, and basal laminae (adj p values 6.41E-04 – 1.9E-17) (Table S5), consistent with previous findings (Bartzokis et al., 2007; Terry et al., 1991; Yamaguchi et al., 1992).
Figure 4.

Consensus Protein Coexpression Network Analysis of Tg-AD Brains (A) HIP protein clustering trees from both Tg-AD models and time points (top). Each module is in non-grey (middle). The two rows of heat maps below show the association of individual proteins with Q for Non-Tg or Tg-AD groups (bottom). Blue and red shading indicates proteins with reduced or increased expression, respectively, with increasing Q. Black bars show altered protein groups. (B) Individual topological overlap matrices of significantly (Z statistics p value < 1E-04) differentially connected modules in HIP between Tg-AD and Non-Tg datasets in both models. Module assignment based on most significant GO assignment (# for ME1 see Table S5). Shown are all modules with significant p values (< 1 E-4). (C) Summary expression value (eigenprotein) from the indicated HIP datasets, all Non-Tg/Tg-AD comparisons have p value < 0.05 calculated from Z statistics. See also Figure S4 and Table S5.
By decomposing our MEs into individual time points, we identified shared and unique protein expression patterns between the two models (Zhang and Horvath, 2005), and by using a meta-analysis of correlations of ME eigenproteins (summary expression profile) (Langfelder et al., 2016), we measured the relationship between MEs and each model at two time points. We found that six HIP and two FC MEs had significant (Z statistic and corresponding meta-analysis p value < 0.05), differences in abundance for each time point and model (Figures 4C and S4C–D). We found both synapse MEs (ME1 and ME3) in the HIP to have reduced protein levels. However, the myelin sheath (ME2) had only slightly altered expression patterns, except at 12 mo hAPP that showed a significant increase (Figure 4C). Bioinformatics of the FC MEs resulted in significant, but less dramatic eigenproteins (Figures S4D). Interestingly, only myelin sheath and mitochondrion MEs were found in both FC and HIP (Table S5). The strongest associations were in HIP. Noticeably, in hAPP mice, the synapse MEs (1 and 3) were significantly decreased at 3 mo (Figure 4C), which is before Aβ plaques form (Figure S1D and S1K), consistent with previous reports that synaptic deficits are caused by soluble Aβ peptides (Hsia et al., 1999). In contrast, the hAPP/PS1 synapse MEs were only slightly reduced at 3 mo (Figure 4C), but Aβ plaques were already formed (Figure S1E and S1L). The HIP myelin sheath (ME2) also provides a contrasting view of the two models (Figure 4C and S4D). In hAPP there were only slight differences in eigenprotein levels at both ages while in hAPP/PS1 we observed a substantial decrease at 3 mo and more dramatic reduction at 12 mo. Meta-analysis statistics allowed us to rank each ME in two Tg-AD MEs and highlighted related but distinct changes in the proteomes of the HIP and FC.
Refining MEs to untangle pathology
Interpretation of affected protein pathway perturbations in Tg AD HIP and FC depends on confident ME characterizations. To identify the most confident groups of proteins in our datasets, we first considered the most significant HIP MEs. ME2 and ME4 were the among the most significant and highly populated MEs and GO:CC analysis showed these MEs enriched in myelin sheath proteins (adj p value = 2.22E-15; n = 213) and mitochondria respectively (adj p value = 1.57E-12; n = 117). Since these proteins are known to be impaired in AD, it confirms our analysis strategy (Bartzokis, 2004; Hirai et al., 2001). We focused on synapse related HIP ME1 and ME3, and used KEGG database to test if these MEs are significantly enriched in proteins with shared functional assignments (Kanehisa and Goto, 2000). Interestingly, for ME1, we found that long-term potentiation (LTP) to be the predominant assignment (Figure 5A). This result is consistent with previous reports showing that HIP LTP impairments may represent the basis of hampered memory formation in AD patients (Chapman et al., 1999), while ME3 was enriched for the synaptic vesicle cycle. Consistently, when we used the GO: biological processes (GO:BP) database (von Mering et al., 2005), to examine ME1 and ME3, we found these MEs were significantly enriched for synapse plasticity/actin organization and modulation of synaptic transmission/vesicle transport processes (Figure 5B). These results suggest that synaptic transmission may be altered in HIP by multiple mechanisms in AD-like pathology. To confirm this strategy, we next explored if our MEs relate to previously characterized AD pathology in the FC. We first used KEGG to test if our MEs are enriched for proteins assigned the AD pathway. We found that ME1, ME2, and ME4 significantly enriched in proteins previously associated with AD. To extend these findings, we subjected these three MEs to GO:BP analysis. This analysis showed that these BP terms, namely ME1 - excitatory postsynaptic potential, ME2 – Oxidative-reduction process, and ME4 – hydrogen ion transporter activity, were significantly enriched (Figure S4E) and all of these pathways are linked to AD pathology.
Figure 5.

Synaptic Proteins in ME1 and ME3 Have Specific Changes in Protein Levels (A) HIP ME1 and ME3 are enriched for proteins with distinct synaptic functions based on KEGG. Scatter plot showing enrichment FDR (y–axis) versus the number of proteins per KEGG pathway (x-axis). (B) HIP ME1 and ME3 are enriched in proteins with distinct synaptic functions based on GO:BP. (C–E) Protein expression matrix (ratio 2) for the indicated MEs for both Tg-AD models and time points. Black bars show datasets with contrasting protein expression patterns. Below bar graph based on hypergeometric enrichment p values, dotted line indicates an adj p value < 0.05. (F–G) Network of top 50 hub proteins in ME 1 and ME3 from the HIP (8 most connected = large, next 16 = medium, remaining 26 = small). (A and B) Most significant KEGG and GO:BP for ME1 and ME3 are underlined. See also Table S5.
We examined the expression pattern of the individual proteins of the most significant MEs which provided us with insight into the core protein network perturbations. First we inspected the hippocampal LTP module (ME1), which contained proteins such as GluA2 and GluN2A/B (Figure 5C). Overall, these proteins showed progressive and significant reduction in levels in hAPP but were mostly up at 3 and down regulated at 12 mo in hAPP/PS1. For FC and CB in both models, proteins in this module generally had increased levels. We obtained similar results for the ME1 proteins associated with actin organization, a key process for synapse remodeling (Figure 5D). In general, the proteins comprising the second HIP synapse related module (ME3) showed a more robust reduction in levels in hAPP compared to hAPP/PS1 (Figure 5E). ME2 and ME4, from the FC data sets, are involved with mitochondria and had mostly reduced expression in HIP, but increased expression in FC and CB (Figures S4F–G). Finally, we performed network analysis based on the continuous measure of membership and connectivity based on WGCNA to determine the top 50 hub proteins in HIP ME1 and ME3 (Langfelder et al., 2016). These hub protein alterations are similar to the eigenproteins of these MEs and largely reflected function of corresponding MEs. Several core hub proteins identified in ME1, including Wasf1 and Nckap1 (Figures 5F–G), play critical roles regulating spine structure and have been implicated in AD (Ceglia et al., 2015; Govek et al., 2005; Kim et al., 2006; Yamamoto et al., 2001).
AMPARs as core synaptic complexes altered in AD
WGCNA identified the core AMPAR subunit GluA2 as a top hub gene of ME1 in HIP. Additional GluA2 regulatory proteins such as Nsf and Camk2b (Braithwaite et al., 2002; Kristensen et al., 2011; Shanks et al., 2012), were also identified as hub genes (Figure 5F). This finding suggests GluA2 containing protein networks may represent key potential targets for AD therapeutic intervention. However, GluA2 plays essential synaptic functions in learning and memory and thus the possibility of directly manipulating it is problematic. Thus, we focused on finding key components of AMPAR complexes contributing in AD. To identify the earliest alterations we examined the levels of AMPAR subunits in HIP and FC datasets from hAPP at 3 and 12 mo. At 3 mo, the only significantly altered AMPAR proteins were TARPγ-2 in HIP (1.253 ± 0.162 versus 0.847 ± 0.231 mean ± STD, p value = 4.29 E-04) and TARPγ-3 in FC (1.165 ± 0.0354 versus 2.380 ± 0.139 mean ± STD, p value = 7.21 E-04) (Figure S5A). This result contrasts, results at 12 mo that showed both GluA1 and 2 significantly down regulated in HIP (Figure S5B). Based on the key effects of TARPs on AMPAR trafficking and synaptic responses, we tested if TARPγ-2/3 levels are altered in the context of intact AMPARs in AD model brains. Indeed, AMPARs immunopurified from hAPP brains with anti-GluA2 antibodies analyzed by WB and MS showed decreased levels of TARPγ-2/3, strongly suggesting AMPARs in AD model brains have altered interactions with TARP proteins (Figures 6A and S5C). To test if AMPAR complexes are remodeled in AD human cortex, we immunopurified AMPARs and analyzed the precipitates with MS and confirmed reduced TARPγ-2 levels in the context of human AD pathology (Figure 6B).
Figure 6.

AMPAR Complexes Are Hub Proteins Altered in AD (A) WBs of AMPAR complexes from hAPP or Non-Tg brains. Less TARP-γ2/3 was recovered in the hAPP brain compared to Non-Tg after normalizing to the recovery of GluA2: 1.0 verses 0.63. (B) Semi-quantitative MS analysis of AMPAR complexes from AD or healthy control brains show AMPARs in AD cortex have reduced levels of TARP-γ2 associated normalized to GluA2: healthy cortex = 0.082 ± 0.011 verses AD cortex = 0.035 ± 0.0058. Bar shows mean ± standard deviation, AD brains (n = 4 patients) and controls (n = 2). White bars = controls and solid bars = AD. (C) Strategy to test if injected LVs expressing Flag-TARP-γ2-IRES-GFP can rescue AMPA defect in hAPP mice. (D) Image of simultaneously recorded un- and infected CA1 cells in a hAPP HIP brain slices. (E) AMPA currents are not significantly different in Non-Tg mice (TARP-γ2: 70.248 ± 11.479; Uninf: 59.896 ± 11.020, n = 7, p value = 0.379). (F) AMPA currents are significantly increased in TARP-γ2 expressing cells in hAPP mice (TARP-γ2: 134.67 ± 27.812 pA; Uninf: 48.331 pA, n = 7, p value = 0.00385). (G) NMDA currents are not significantly different in Non-Tg mice (TARP-γ2: 114.2344 ± 22.5515, Uninf: 121.669 ± 29.973, n = 9, p value = 0.7089). (H) NMDA currents are not significantly different in hAPP mice (TARP-γ2: 112.148 ± 16.537; Uninf: 88.6866 ± 0.301, n = 5, p value = 0.3016). (E–H) Mean ± SEM. **p value < 0.01, *** < 0.001, by Student’s t test. See also Figure S5.
Impaired excitatory synaptic transmission and AMPAR dysfunction have been implicated in AD HIP, however the details are unclear (Hsia et al., 1999; Hsieh et al., 2006). Consistently, we observed a significant reduction in the strength of evoked field potentials in hAPP HIP slices relative to control littermates (Figures S5D–F). We hypothesized that reduction of TARPγ-2 in AD could lead to destabilization of synaptic AMPARs since TARPγ-2 anchors AMPAR subunits to PSD-95, and this interaction can help deliver AMPARs to synapses e(Schnell et al., 2002). We wondered if TARPγ-2 might affect basal synaptic transmission in hAPP mice. To test this, we injected lentiviruses (LVs) that overexpress TARPγ-2 into CA1 in Tg-AD and Non-Tg littermates (Figures 6C–D and S5G). Consistent with previous findings (Schnell et al., 2002), we found overexpression of TARPγ-2 had no effect on basal AMPAR EPSCs in Non-Tg animals, but we found that TARPγ-2 overexpression resulted in a significant increase in AMPAR EPSCs in hAPP mice (Figures 6E–F). Additionally, we observed no effects on NMDAR EPSCs (Figures 6G–H). These results support TARPγ-2 as a potential target for treating in AD.
DISCUSSION
Proteomic characterization of AD model brains shows elevated Aβ levels cause proteome remodeling in multiple cell types and pathways. Most ApoE is produced by glia and is likely involved in multiple mechanisms important to AD (Holtzman et al., 2012). Our proteomics showed that ApoE levels are increased in brain regions with high levels of Aβ but not in the CB of hAPP, suggesting that it is primarily involved in Aβ clearance rather than production. This result may contrast previous results showing that the APP intracellular domain can drive ApoE gene transcription and may highlight differences between AD models and the complexity of ApoE biology (Liu et al., 2007). Recently, an allele specific role of ApoE in regulating complement C1q protein has an important role in regulating the rate of synaptic pruning (Chung et al., 2016). It is possible that the synaptic protein defects we identified could be due to altered ApoE levels at tripartite synapses.
Many previous reports have described impaired synaptic transmission and an overall reduction in the number of synapses in the HIP of AD models and patients before appearance of plaques (Shankar et al., 2008; Terry et al., 1991). We confirmed distinct components of excitatory synapses are altered prior to plaque formation. WGCNA revealed two synaptic MEs with reduced protein levels at early and late time points. ME1 is enriched in proteins involved with LTP and actin cyto-skeleton, both of which have been implicated as playing key roles in AD (Sheng et al., 2012). ME3 is enriched in proteins involved with pre-synaptic functions. Among the top 50 hub proteins in ME1 were AMPAR and NMDAR subunits. Examination of AMPAR components showed a pioneering reduction in protein abundance in the auxiliary subunits TARPγ-2/3. It was not until the later time point that GluA1 and 2 had significantly reduced levels in the HIP of hAPP mice. AMPAR density was shown reduced at perforated synapses and synapses onto parvalbumin (PV)-positive neurons in the CA1 region of TARPγ-2 KO mice interestingly we found significantly reduced levels of PV in the HIP of both models (Yamasaki et al., 2016). Furthermore, pore forming GluA3 and -4 subunits were not significantly altered. In other AD models they have been showed to be important Aβ substrates (Reinders et al., 2016). AMPAR downscaling and removal have been reported in multiple AD models, but the precise mechanism(s) have remained vague (Chang et al., 2006; Hsia et al., 1999; Hsieh et al., 2006). These findings motivated us to test whether restoring TARPγ-2 levels could rescue AMPA defects in hAPP mice. Indeed, over expression of TARPγ-2 caused a large increase in AMPAR- but not NMDAR-mediated currents. Our results suggest that GluA1, 2 containing AMPARs that are known to be enriched with TARPγ-2/3 may represent key Aβ targets due to their restricted expression patterns at the effected synapses (Schwenk et al., 2012). Finally, while both NMDARs and AMPARs play key roles in the establishment and maintenance of LTP, our results highlight a potential mechanism by which reduced expression of TARPγ-2 could result in impaired delivery of AMPARs to spines and thus compromise LTP in AD patients. TARP subtypes have been shown to differentially influence AMPAR gating (Milstein et al., 2007), and TARPγ-2 may specifically represent a therapeutic target to restore cognitive function in AD patients.
We determined protein substrates and mechanisms of Aβ toxicity to dissect the amyloid cascade hypothesis by using three complementary mouse models of AD-like pathology (Table S6). To maximize the accessibility our results, we have generated an online interactive AD model protein expression database (Proteomics INTegrator) as a resource for the entire AD research community: http://sealion.scripps.edu/pint/?project=3d7c1ac078930a798a07c6a397bd21ef. This resource allows users to query proteins of interest, visualize quantified peptides, and perform enrichment analyses (Figure S6). The importance of determining protein abundances is particularly relevant for the investigation of AD because altered proteostasis and reduced protein degradation capacities are hallmarks (Powers et al., 2009; Rubinsztein, 2006). However, our study is not without limitations since we failed to accurately quantify many proteins expressed at low levels, and our results are limited by protein abundance averaging between the multiple cell types present in the brain.
Our results show that Aβ causes a broad and progressive alteration in the level of many functionally and spatially linked components of the brain proteome. These findings, and those recently published from others (Seyfried et al., 2017), raise the possibility that studying groups of co-expressed proteins might be advantageous over the study of individual proteins due to the highly complex response of various cell types to toxic Aβ
EXPERIMENTAL PROCEDURES
Mice
Ten C57BL6 female mice were metabolically labelled with 15N-rich, Spirulina-based diet (Cambridge Isotopes) for 12 weeks starting at P21 (Wu et al., 2004). The 15N protein enrichment was calculated based on the shape of the shape of the peptide isotope envelope, and in the brain was determined to be 90–95% (MacCoss et al., 2005). For the hAPP model we used the less aggressive J9 line, except for the AMPAr immunoprecipitation – mass spectrometry/western blots and whole cell electrophysiology in which out of necessity we used J20 mice (RRID:IMSR_JAX:004662) (Mucke et al., 2000). All hAPP mice were on C3H/B6 background. The hAPP/PS1 mice used were (B6C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax– 129x1/SvJ) (RRID:MMRRC_037564-JAX) (Borchelt et al., 1996; Cohen et al., 2009). The BRI-Aβ42 mice were C57BL/6J (RRID:IMSR_JAX:007182) (McGowan et al., 2005). Institutional Animal Care and Use Committees of UCSD, Salk Institute, and the Scripps Research Institute, approved all animal procedures, and studies. Most of the Tg-AD mice used in this study were female, 3 mo = 3.0 – 3.6 mo and 12 mo = 12.2 – 15.0 mo. Brains were split into two hemispheres, one for proteomic analysis and ELISA, and the other for histology. Mice were from the Masliah, Koo, and Dillin lab colonies or JAX. See Supplemental Experimental Procedures for details.
MS Analysis
Brain region homogenate corresponding to 100 μg, based on BCA assay was digested to peptides with trypsin overnight and was processed for MudPIT as previously described (Savas et al., 2015). The peptide loaded column was placed in line with an Agilent 1200 HPLC and analyzed with an 11 step method. MS analyses were completed with Thermo Orbitrap Velos or Orbitrap Fusion Tribrid mass spectrometers (see Table S6). Protein identification and quantification and analysis were done with ProLuCID, DTASelect2, Census, and QuantCompare within the Integrated Proteomics Pipeline (IP2) environment. Tandem mass spectra were matched to sequences using the ProLuCID algorithm with 50 ppm peptide mass tolerance for precursor ions and 400 ppm for fragment ions. Each dataset had a ~1% FDR rate at the protein level based on the target-decoy strategy. Since we analyzed brain tissue which has many cell types and mRNA alternative splicing we chose large databases to maximize our analysis potential. All the peptide identifications have been mapped to genes and the details can be found in our online database where all proteins were also mapped to UniprotKB accessions.
For MS1 based quantification “Census” first calculates the elemental compositions and corresponding isotopic distributions for both the unlabeled and labeled peptides. All isotopes with greater than 5 % of the calculated isotope cluster base peak abundance were used. MS1 files were used to generate chromatograms from the m/z range surrounding both the unlabeled and labeled precursor peptides. The core of Census is a linear least-squares correlation that is used to calculate the ratio (i.e., slope of the line) and closeness of fit [i.e., correlation coefficient (r)] between the data points of the unlabeled and labeled ion chromatograms. In this study, only peptide ratios with the coefficient correlation values (r2) greater than 0.5 were used for further analysis. The Grubbs test (p value < 0.01) was used to remove outlier peptides. Final protein ratios were generated with QuantCompare, which uses Log two-fold change on the biological replicates. The statistical significance labeled of the differential expression of all proteins was assessed using a two-tailed one-sample t-test on their corresponding peptide quantification ratios between both conditions. The obtained p values were FDR-adjusted for multiple hypothesis testing using the Benjamini–Hochberg correction (Benjamini and Hochberg, 1995). Proteins with FDR-adjusted p values < 0.05 and for which quantification measurements were obtained in at least two biological replicates in both conditions were considered for further analyses. We also performed label-free quantitative analysis using Census within IP2. See Supplemental Experimental Procedures for details.
Statistical and Bioinformatic Analyses
We compared our proteomic data to the previously described potential Alzheimer disease susceptibility genes (Bertram et al., 2007). For all heat maps, we used 3 biological replicates for each experimental group, required each protein to be quantified in a minimum of 57 (out of a total of 72) datasets and had to be quantified in each brain region. We used the Genes to Cognition protein database to identify synaptic proteins, MitoCarta2.0 for mitochondrial proteins, and InnateDB to identify the immunological proteins. Box plots (Figure 3) define 25th and 75th percentile and statistical analysis were performed with student’s t-test with 2 tails and un-equal variance. For quantification of APP levels, only peptide sequences with 100 % homology between human and mouse were used. Bar graphs (Figure S1) show mean ± SD with student’s t test with 1 tail and un-equal variance (A & B) with 2 tails and equal variance (I & P). Bar graphs for human AMPAR purifications with MS analysis, statistics were performed with Student’s t test with 1 tail and 2 sample equal variance (Figure 6B). Western blot quantification Bar graphs (Figure S3) show mean ± SEM and student’s t test with 1 tail and two-sample equal variance. We used the program ‘Venn Diagram Plotter’ to construct two and three circle Venn diagrams (aka Euler diagrams). Detailed description of each analysis, the tools used, and the specific criteria is in the Supplemental Experimental Procedures.
WGCNA
The consensus weighted correlation networks were constructed across both Non-Tg and Tg-AD groups for each brain regions with R (Langfelder and Horvath, 2008; Zhang and Horvath, 2005). Prior to analysis, protein IDs with 14 or more missing values were removed, resulting in three pairs of datasets: 1912 protein IDs for HIP, 2146 protein IDs for FC and 2169 protein IDs for CB. The specific datasets for each analysis is as follows (dataset numbers correspond to the numbering indicated in Table S6): FC = 137–9, 124–7, 89–92, 77–9, 112–6, 101–3, 65–7, 53–7. HIP = 140–4, 128–31, 93–5, 80–4, 117–9, 104–8, 68–72, 58–61. The soft power threshold was set to 12 for HIP groups, 6 for FC groups to arrive at the network adjacency. See Supplemental Experimental Procedures for details.
Affinity purification of AMPA receptor complexes
We used our previously reported method to purify native AMPARs (Nakagawa et al., 2005). Human brain (cortex) was obtained through the National Disease Research Interchange (NDRI), Researcher: Yates (code YAJ2), TSRI: IRB-11-5719. The antigen of the antibody against GluA2 is conserved in rat and human. See Supplemental Experimental Procedures for details.
Electrophysiology
Brains were cut into 300 μm sagittal sections on a vibratome and placed in ice-cold carbogenated artificial cerebrospinal fluid. Whole-cell recordings were made using 3–5 MΩ pipettes filled with an internal solution that contained 123 mM Cs-gluconate, 8 mM NaCl, 1 mM CaCl2, 10 mM EGTA, 10 mM HEPES, and 10 mM glucose, pH 7.3 with CsOH, 280–290 mOsm. Responses were evoked by stimulating axons in the stratum radiatum with a platinum 2-contact cluster electrode 100–200 microns lateral to the recording site. See Supplemental Experimental Procedures for details.
Supplementary Material
Table S1 (related to Figure 1). Significantly altered proteins in the frontal cortex, hippocampus, and cerebellum of hAPP mice at three and 12 months. Proteins identified as significantly altered in quantitative proteomic analyses from hAPP model, pre = 3 mo and post = 12 months of age. Each tab represents the indicated data set which are individually controlled with FDR-adjusted p values < 0.05. The number significant proteins identified included 111 for the frontal cortex at three months and 137 at 12 months., 212 for the hippocampus at three months and 386 at 12 mo, and 222 for the cerebellum at three months and 450 at 12 months. Experiment = specific data set, Locus = data base entry identifier, Gene = gene corresponding to identified protein, Ratio = Ratio log2 (Tg-AD/Non Tg), t test p value = t test p value, FDR-adjusted p value (see methods), Rank = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Total = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Description = protein description and identifier(s).
Table S2 (related to Figure 1). Significantly altered proteins in the frontal cortex, hippocampus, and cerebellum of hAPP/PS1 mice at three and 12 months. Proteins identified as significantly altered in quantitative proteomic analyses from hAPP model, pre = 3 mo and post = 12 mo of age. Each tab represents the indicated data set which are individually controlled with FDR-adjusted p values < 0.05. The number significant proteins identified included 61 for the frontal cortex at three months and 156 at 12 months., 214 for the hippocampus at three months and 450 at 12 months, and 123 for the cerebellum at three months and 153 at 12 months. Experiment = specific data set, Locus = data base entry identifier, Gene = gene corresponding to identified protein, Ratio = Ratio log2 (Tg-AD/Non Tg), t test p value = t test p value, FDR-adjusted p value (see methods), Rank = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Total = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Description = protein description and identifier(s).
Table S3 (related to Figure 1). Significantly altered proteins in the frontal cortex or hippocampus in hAPP and hAPP/PS1 mice at 3 or 12 months. This table represents an intersection of proteins (based on being from the same gene) with significantly altered levels at either time point from the frontal cortex or hippocampus in both hAPP and hAPP/PS1 models. Results from literature search to determine if each confirmed protein has been reported as being altered at the protein, mRNA, or as having a genetic association in AD are included. The first reference to establish the connection to AD is highlighted in red. Subsequent references are in black. If no literature could be found including the protein and a reference to AD, it is indicated by (*No Prior Reference Found). If literature could be found that reference both the protein and AD, but merely address related metabolites and larger protein complexes, it is indicated by (*No Specific Reference Linking [protein] to AD Found).
Table S4 (related to Figure 3). Summary of significantly altered proteins in BRI-Aβ42 frontal cortex, hippocampus, and cerebellum at 3 or 12 months. All proteins quantified by MS-based label free analysis with a FDR-adjusted p value < 0.05 are included
Table S5 (related to Figure 4). Summary of WGCNA analysis. Sheets ‘Hip (or FC) module membership’ show the assigned module label, color, meta-analysis Z statistics for module membership in all modules for each quantified protein IDs in all modules at Non-Tg and Tg-AD groups (both models). As resources, these tables can be used to evaluate the hub proteins in a module (protein IDs with highest module membership Z statistics) for the follow-up functional studies.
Table S6 (related to Figure 1). Complete summary of proteomic datasets reported in this study. This table lists each dataset, the number or protein identifications, protein and peptide FDRs based on target-decoy strategy, peptide quantification efficiency, DTAselect filtering parameters, instrument (MS), and liquid chromatography (LC) strategy for each analysis. RP = reversed phase, MuDPIT = multidimensional protein identification technology, and “T2” indicates technical replicate.
Acknowledgments
We thank L. Bertram, F. Sarsoza, I. Morantte, J. De Wit, C. Delahunty, E. Rockenstein, J. Choi, and A. Dillin for their assistance. This work was supported by NIH awards F32AG039127, R00DC013805 (JNS); R01MH067880, P41GM103533 (JRY); P30CA060553 (NU CAM and MHPL). Fonds de Recherche du Québec—Nature et Technologies fellowship (MLA).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and six figures and six tables can be found with this article online at:
AUTHOR CONTRIBUTIONS
J.N.S, Y.Z.W., D.B.m.C., L.A.D., E.H.K., T.N., A.G., and J.R.Y. designed research; J.N.S., L.A.D., D.B.m.C., N.F.S., T.J.H., K.A.C., and S.N.S., performed research; J.N.S., Y.Z.W., S.M.B., S.K.P., M.L.A., E.H.K., E.M., and J.R.Y. contributed reagents/analytical tools; all authors contributed to analyzing the data; J.N.S. and J.R.Y. wrote the paper.
ACCESSION NUMBERS
All the raw MS data is publically available through ProteomeXchange (PXD005595) and MassIVE (MSV000080431).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, et al. Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. author reply 49–62. [DOI] [PubMed] [Google Scholar]
- Bartzokis G, Lu PH, Mintz J. Human brain myelination and amyloid beta deposition in Alzheimer’s disease. Alzheimers Dement. 2007;3:122–125. doi: 10.1016/j.jalz.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1- 42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Xia H, Malenka RC. Differential roles for NSF and GRIP/ABP in AMPA receptor cycling. Proc Natl Acad Sci U S A. 2002;99:7096–7101. doi: 10.1073/pnas.102156099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceglia I, Reitz C, Gresack J, Ahn JH, Bustos V, Bleck M, Zhang X, Martin G, Simon SM, Nairn AC, et al. APP intracellular domain-WAVE1 pathway reduces amyloid-beta production. Nat Med. 2015;21:1054–1059. doi: 10.1038/nm.3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Chung WS, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, Holtzman DM, Barres BA. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A. 2016;113:10186–10191. doi: 10.1073/pnas.1609896113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Karran E. The Cellular Phase of Alzheimer’s Disease. Cell. 2016;164:603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Meziane H, Mathis C, Bales KR, Paul SM, Ungerer A. Behavioral disturbances in transgenic mice overexpressing the V717F beta-amyloid precursor protein. Behav Neurosci. 1999;113:982–990. doi: 10.1037//0735-7044.113.5.982. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Gupta VB, Laws SM, Villemagne VL, Ames D, Bush AI, Ellis KA, Lui JK, Masters C, Rowe CC, Szoeke C, et al. Plasma apolipoprotein E and Alzheimer disease risk: the AIBL study of aging. Neurology. 2011;76:1091–1098. doi: 10.1212/WNL.0b013e318211c352. [DOI] [PubMed] [Google Scholar]
- Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat Neurosci. 2015;18:794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- Hesse C, Larsson H, Fredman P, Minthon L, Andreasen N, Davidsson P, Blennow K. Measurement of apolipoprotein E (apoE) in cerebrospinal fluid. Neurochem Res. 2000;25:511–517. doi: 10.1023/a:1007516210548. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci U S A. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies A beta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, et al. Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature. 2006;442:814–817. doi: 10.1038/nature04976. [DOI] [PubMed] [Google Scholar]
- Kristensen AS, Jenkins MA, Banke TG, Schousboe A, Makino Y, Johnson RC, Huganir R, Traynelis SF. Mechanism of Ca2+/calmodulin-dependent kinase II regulation of AMPA receptor gating. Nat Neurosci. 2011;14:727–735. doi: 10.1038/nn.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Crawford F, Mullan M, Kokjohn TA, Emmerling MR, Weller RO, Roher AE. Elevated A beta and apolipoprotein E in A betaPP transgenic mice and its relationship to amyloid accumulation in Alzheimer’s disease. Mol Med. 2000;6:430–439. [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Kim HD, Maxwell JA, Fukuchi K. Exploratory activity and spatial learning in 12- month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci Lett. 2005;390:87–92. doi: 10.1016/j.neulet.2005.08.028. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Cantle JP, Chatzopoulou D, Wang N, Gao F, Al-Ramahi I, Lu XH, Ramos EM, El-Zein K, Zhao Y, et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat Neurosci. 2016;19:623–633. doi: 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann H, Weiler R, Fischer P, Bancher C, Jellinger K, Floor E, Danielczyk W, Seitelberger F, Winkler H. Synaptic pathology in Alzheimer’s disease: immunological data for markers of synaptic and large dense-core vesicles. Neuroscience. 1992;46:1–8. doi: 10.1016/0306-4522(92)90003-k. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007;56:66–78. doi: 10.1016/j.neuron.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCoss MJ, Wu CC, Matthews DE, Yates JR., 3rd Measurement of the isotope enrichment of stable isotope-labeled proteins using high-resolution mass spectra of peptides. Anal Chem. 2005;77:7646–7653. doi: 10.1021/ac0508393. [DOI] [PubMed] [Google Scholar]
- McClatchy DB, Dong MQ, Wu CC, Venable JD, Yates JR., 3rd 15N metabolic labeling of mammalian tissue with slow protein turnover. J Proteome Res. 2007;6:2005–2010. doi: 10.1021/pr060599n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein AD, Zhou W, Karimzadegan S, Bredt DS, Nicoll RA. TARP subtypes differentially and dose-dependently control synaptic AMPA receptor gating. Neuron. 2007;55:905–918. doi: 10.1016/j.neuron.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science. 1987;235:1641–1644. doi: 10.1126/science.3029875. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Cheng Y, Ramm E, Sheng M, Walz T. Structure and different conformational states of native AMPA receptor complexes. Nature. 2005;433:545–549. doi: 10.1038/nature03328. [DOI] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- Park SK, Venable JD, Xu T, Yates JR., 3rd A quantitative analysis software tool for mass spectrometry-based proteomics. Nat Methods. 2008;5:319–322. doi: 10.1038/nmeth.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Reinders NR, Pao Y, Renner MC, da Silva-Matos CM, Lodder TR, Malinow R, Kessels HW. Amyloid-beta effects on synapses and memory require AMPA receptor subunit GluA3. Proc Natl Acad Sci U S A. 2016;113:E6526–E6534. doi: 10.1073/pnas.1614249113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–786. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17:661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- Savas JN, Ribeiro LF, Wierda KD, Wright R, DeNardo-Wilke LA, Rice HC, Chamma I, Wang YZ, Zemla R, Lavallee-Adam M, et al. The Sorting Receptor SorCS1 Regulates Trafficking of Neurexin and AMPA Receptors. Neuron. 2015;87:764–780. doi: 10.1016/j.neuron.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci U S A. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Brechet A, Zolles G, Berkefeld H, Muller CS, Bildl W, Baehrens D, Huber B, Kulik A, et al. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron. 2012;74:621–633. doi: 10.1016/j.neuron.2012.03.034. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried NT, Dammer EB, Swarup V, Nandakumar D, Duong DM, Yin L, Deng Q, Nguyen T, Hales CM, Wingo T, et al. A Multi-network Approach Identifies Protein-Specific Co-expression in Asymptomatic and Symptomatic Alzheimer’s Disease. Cell Syst. 2017;4:60–72. e64. doi: 10.1016/j.cels.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks NF, Savas JN, Maruo T, Cais O, Hirao A, Oe S, Ghosh A, Noda Y, Greger IH, Yates JR, 3rd, et al. Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep. 2012;1:590–598. doi: 10.1016/j.celrep.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012;4 doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Wang J, Tanila H, Puolivali J, Kadish I, van Groen T. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis. 2003;14:318–327. doi: 10.1016/j.nbd.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Wu CC, MacCoss MJ, Howell KE, Matthews DE, Yates JR., 3rd Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal Chem. 2004;76:4951–4959. doi: 10.1021/ac049208j. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Yamazaki T, Ishiguro K, Shoji M, Nakazato Y, Hirai S. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol. 1992;85:15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Suzuki T, Sakaki Y. Isolation of hNap1BP which interacts with human Nap1 (NCKAP1) whose expression is down-regulated in Alzheimer’s disease. Gene. 2001;271:159–169. doi: 10.1016/s0378-1119(01)00521-2. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Fukaya M, Yamazaki M, Azechi H, Natsume R, Abe M, Sakimura K, Watanabe M. TARP gamma-2 and gamma-8 Differentially Control AMPAR Density Across Schaffer Collateral/Commissural Synapses in the Hippocampal CA1 Area. J Neurosci. 2016;36:4296–4312. doi: 10.1523/JNEUROSCI.4178-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (related to Figure 1). Significantly altered proteins in the frontal cortex, hippocampus, and cerebellum of hAPP mice at three and 12 months. Proteins identified as significantly altered in quantitative proteomic analyses from hAPP model, pre = 3 mo and post = 12 months of age. Each tab represents the indicated data set which are individually controlled with FDR-adjusted p values < 0.05. The number significant proteins identified included 111 for the frontal cortex at three months and 137 at 12 months., 212 for the hippocampus at three months and 386 at 12 mo, and 222 for the cerebellum at three months and 450 at 12 months. Experiment = specific data set, Locus = data base entry identifier, Gene = gene corresponding to identified protein, Ratio = Ratio log2 (Tg-AD/Non Tg), t test p value = t test p value, FDR-adjusted p value (see methods), Rank = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Total = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Description = protein description and identifier(s).
Table S2 (related to Figure 1). Significantly altered proteins in the frontal cortex, hippocampus, and cerebellum of hAPP/PS1 mice at three and 12 months. Proteins identified as significantly altered in quantitative proteomic analyses from hAPP model, pre = 3 mo and post = 12 mo of age. Each tab represents the indicated data set which are individually controlled with FDR-adjusted p values < 0.05. The number significant proteins identified included 61 for the frontal cortex at three months and 156 at 12 months., 214 for the hippocampus at three months and 450 at 12 months, and 123 for the cerebellum at three months and 153 at 12 months. Experiment = specific data set, Locus = data base entry identifier, Gene = gene corresponding to identified protein, Ratio = Ratio log2 (Tg-AD/Non Tg), t test p value = t test p value, FDR-adjusted p value (see methods), Rank = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Total = rank ordered proteins based on p value (if ratio value is identical the redundant protein is not considered), Description = protein description and identifier(s).
Table S3 (related to Figure 1). Significantly altered proteins in the frontal cortex or hippocampus in hAPP and hAPP/PS1 mice at 3 or 12 months. This table represents an intersection of proteins (based on being from the same gene) with significantly altered levels at either time point from the frontal cortex or hippocampus in both hAPP and hAPP/PS1 models. Results from literature search to determine if each confirmed protein has been reported as being altered at the protein, mRNA, or as having a genetic association in AD are included. The first reference to establish the connection to AD is highlighted in red. Subsequent references are in black. If no literature could be found including the protein and a reference to AD, it is indicated by (*No Prior Reference Found). If literature could be found that reference both the protein and AD, but merely address related metabolites and larger protein complexes, it is indicated by (*No Specific Reference Linking [protein] to AD Found).
Table S4 (related to Figure 3). Summary of significantly altered proteins in BRI-Aβ42 frontal cortex, hippocampus, and cerebellum at 3 or 12 months. All proteins quantified by MS-based label free analysis with a FDR-adjusted p value < 0.05 are included
Table S5 (related to Figure 4). Summary of WGCNA analysis. Sheets ‘Hip (or FC) module membership’ show the assigned module label, color, meta-analysis Z statistics for module membership in all modules for each quantified protein IDs in all modules at Non-Tg and Tg-AD groups (both models). As resources, these tables can be used to evaluate the hub proteins in a module (protein IDs with highest module membership Z statistics) for the follow-up functional studies.
Table S6 (related to Figure 1). Complete summary of proteomic datasets reported in this study. This table lists each dataset, the number or protein identifications, protein and peptide FDRs based on target-decoy strategy, peptide quantification efficiency, DTAselect filtering parameters, instrument (MS), and liquid chromatography (LC) strategy for each analysis. RP = reversed phase, MuDPIT = multidimensional protein identification technology, and “T2” indicates technical replicate.