

SUMMARY
Aerobic glycolysis, also known as the Warburg effect, is a hallmark of cancerous tissues. Despite its importance in cancer development, our understanding of mechanisms driving this form of metabolic reprogramming is incomplete. We report here an analysis of colorectal cancer cells engineered to carry a single point mutation in the active site of the Mediator-associated kinase CDK8, creating hypomorphic alleles sensitive to bulky ATP analogs. Transcriptome analysis revealed CDK8 kinase activity is required for expression of many components of the glycolytic cascade. CDK8 inhibition impairs glucose transporter expression, glucose uptake, glycolytic capacity and reserve, as well as cell proliferation and anchorage independent growth, both in normoxia and hypoxia. Importantly, CDK8 impairment sensitizes cells to pharmacological glycolysis inhibition, a result reproduced with Senexin A, a dual inhibitor of CDK8/CDK19. Altogether, these results contribute to our understanding of CDK8 as an oncogene and justify investigations to target CDK8 in highly glycolytic tumors.
Keywords: CDK8, CDK19, Mediator, glycolysis, Warburg effect, hypoxia, chemical genetics, HCT116, SW480, A549, H460
eTOC BLURB
Galbraith et al. use a chemical genetics approach to examine the role of CDK8 kinase activity in cancer cells. CDK8 activity is required for transcription of multiple genes encoding enzymes required for glucose metabolism. Impaired CDK8 activity reduces glucose uptake and glycolysis, and sensitizes cells to the glucose analog 2-deoxy-D-glucose.
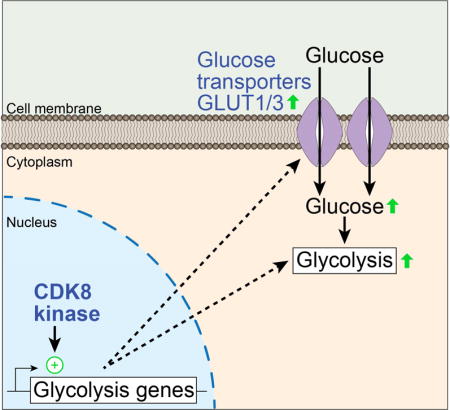
INTRODUCTION
Despite their diverse sites of origin and genetic heterogeneity, most tumors display a common set of hallmarks that support their continued ability to proliferate (Hanahan and Weinberg, 2011). One of these key features is reprograming of energy metabolism, and cancer cells exhibit high rates of glucose consumption and glycolysis relative to normal tissues (Hay, 2016). This is in part an adaptation to the intermittent hypoxia experienced by tumors as they outgrow their blood supply because glycolysis is necessary for continued ATP production during hypoxia. In fact, many genes encoding glycolytic enzymes are induced by the hypoxia inducible factor 1 alpha (HIF1A), a master transcriptional regulator of the hypoxic response (Gatenby and Gillies, 2004; Semenza, 2012). Increased glycolysis also helps to support proliferation by supplying intermediates for anabolic processes such as nucleotide and amino acid synthesis (Hay, 2016), and most cancer cells exhibit constitutive upregulation of glycolysis even under aerobic conditions, a phenomenon known as the Warburg effect (Koppenol et al., 2011; Warburg et al., 1927). Despite a wealth of evidence indicating that the Warburg effect could provide a therapeutic opportunity to target cancer cells, there are currently few drugs that inhibit this hallmark feature of cancer cells (Vander Heiden and DeBerardinis, 2017). Although HIF1A itself is recognized as a key regulator of the Warburg effect, its value as a pan-cancer therapeutic target remains unclear (Ioannou et al., 2015; Keith et al., 2011). Therefore, investigations aimed at identifying other master regulators of the Warburg effect could reveal potential therapeutic targets for a wide range of cancers.
CDK8 has been described as an oncogene in colorectal carcinoma (CRC) and several other cancer types including melanoma and breast cancer (Clark et al., 2015; Firestein et al., 2008; Firestein et al., 2009; Galbraith et al., 2010; Kapoor et al., 2010). However, the exact mechanisms by which CDK8 activity promotes cancer development remain to be elucidated. In fact, in certain contexts CDK8 has been described as a tumor suppressor (Clark et al., 2015; Gu et al., 2013; Li et al., 2014). We and others have shown that CDK8 is a positive regulator of transcriptional programs that drive the proliferation and survival of cancer cells, including those controlled by Wnt/β-catenin (Firestein et al., 2008), ERK/MAP kinase signaling (Donner et al., 2010), and HIF1A (Galbraith et al., 2013; Perez-Perri et al., 2016). Thus, it is possible that pharmacological inhibition of CDK8 could have therapeutic value by simultaneously decreasing the activity of multiple oncogenic pathways.
CDK8 is one of the two kinases associated with the Mediator complex. Mediator is a conserved transcriptional coactivator required for transcription of most protein coding genes. Mediator is composed of ~30 subunits in vertebrates and many human subunits have been implicated in tumorigenesis (Clark et al., 2015; Poss et al., 2013), as well as other pathologies (Yin and Wang, 2014). CDK8, along with Cyclin C, MED12, and MED13 form a ‘CDK module’ that is reversibly associated with core Mediator, and that can regulate its transcriptional activity both positively and negatively (Galbraith et al., 2010; Poss et al., 2013). Interestingly, three of the four genes encoding CDK module subunits are duplicated in vertebrates, giving rise to the paralogous pairs CDK8/CDK19, MED12/MED12L, and MED13/13L. Each subunit variant can assemble with the rest of the CDK module in a mutually exclusive fashion with its paralog, thus allowing for up to eight variants of the CDK module (Daniels et al., 2013), which may be functionally specialized to enable regulatory diversity and specificity. Furthermore, the CDK module is likely to have additional functions and targets independent of core Mediator (Knuesel et al., 2009). Despite the obvious importance of Mediator in gene expression control, little is known about shared and unique functions of the CDK-module paralogs. Recently, a number of dual inhibitors of CDK8 and CDK19 have been developed, including Cortistatin A, Senexin A, CCT251921 and MSC2530818 (Cee et al., 2009; Clarke et al., 2016; Czodrowski et al., 2016; Dale et al., 2015; Koehler et al., 2016; Mallinger et al., 2015; Mallinger et al., 2016a; Mallinger et al., 2016b; Porter et al., 2012; Schiemann et al., 2016). While many of these compounds appear to have efficacy against cancer cell lines in vitro, it remains unclear whether the effects are due to inhibition of CDK8, CDK19 or both. Furthermore, while studies using mouse models demonstrated reduced tumor growth, testing in rats and dogs suggested that therapeutic application of dual CDK8/19 inhibitors could be limited due to adverse effects in a range of tissues (Clarke et al., 2016). Therefore, elucidating the functional specialization of CDK8 versus CDK19 and their relative value as therapeutic targets in cancer is a high priority.
To further investigate the oncogenic functions of CDK8, and to distinguish CDK8 action from that of CDK19, we have employed a chemical genetic strategy, enabled by CRC cells engineered to express an ATP analog-sensitive CDK8 variant (CDK8-AS). Using this approach, we identified widespread direct and indirect effects of impaired CDK8 activity on the transcriptome. This revealed a key role for CDK8 in expression of genes involved in glycolysis, and led to the discovery that impaired CDK8 activity reduces glucose uptake and sensitizes cells to the glycolysis inhibitor 2-deoxy-D-glucose (2DG). These effects of CDK8 are apparent in both hypoxia and normoxia, indicating that CDK8 activity promotes the Warburg effect. Altogether, these results suggest that CDK8-based combinatorial therapeutic strategies could be valuable in targeting cancer cells with altered glucose metabolism.
RESULTS
Generation of analog-sensitive CDK8 mutant cells
In vertebrates, the Mediator CDK module contains either of two paralogous protein kinases, CDK8 or CDK19 (Daniels et al, 2014). Previous work using genetic depletion, both in mice and human cells, indicates that CDK8 and CDK19 have specialized functions in development and gene expression control (e.g. Galbraith et al. (2013); Westerling et al. (2007)). However, it remains unclear to what degree their catalytic activities contribute to this functional specialization. To address this knowledge gap, we engineered HCT116 CRC cells to express CDK8-AS that is sensitive to inhibition by non-hydrolysable ATP analogs. Mutation of a conserved phenylalanine ‘gatekeeper’ residue in kinase active sites (F97 in CDK8) to glycine creates ‘analog-sensitive’ kinases able to accept bulky ATP analogs such as 3MB-PP1 (Figure 1A) (Blethrow et al., 2004), an approach that has been used to dissect the function of other transcriptional CDKs (Larochelle et al., 2006; Larochelle et al., 2007; Sanso et al., 2016). We utilized a two-round genome editing strategy that combined transcription activator-like effector nuclease (TALEN)-mediated double-strand break generation with a recombinant adeno-associated virus (rAAV)-based homology donor construct to sequentially introduce the F97G mutation at both CDK8 alleles (Figure 1B and S1A) (see also Experimental Procedures). We validated the resulting homozygous CDK8as/as HCT116 clones against wild-type (WT) and heterozygous cells using Southern blot hybridization (Figure 1C), and by restriction digestion and sequencing of PCR products (Figure S1B–C). These cells display unaltered levels of CDK8 protein, and mutant CDK8 retains its ability to interact with Cyclin C and the CDK module subunit MED12, as well as the core Mediator subunit MED14 (Figure 1D and S1D–E). In addition to autophosphorylation, CDK8 phosphorylates other members of the CDK module including MED12, MED13, and MED13L (Poss et al., 2016). Therefore, we next examined the sensitivity of CDK8-WT and CDK8-AS to inhibition by the ATP analogs 3MB-PP1 and 1NM-PP1 in an in vitro immunoprecipitation-kinase assay with γ-P32-ATP. While an obvious autophosphorylation signal is evident for both CDK8-WT and –AS (Figure 1E, lanes 1, 4, 7), the mutant kinase is less active in this context (Figure 1E, lanes 1–3). Importantly, only the mutant kinase is inhibited by either ATP analog (Figure 1E, lanes 4–9). As a further test, we treated WT and CDK8as/as cells with interferon gamma to stimulate phosphorylation of STAT1, a known substrate of CDK8 (Bancerek et al., 2013). This demonstrated that the mutant kinase is indeed less active against this target in vivo, and confirmed that it is sensitive to inhibition by 3MB-PP1 (Figure 1F).
Figure 1. Engineering and validation of CDK8as/as HCT116 cells.
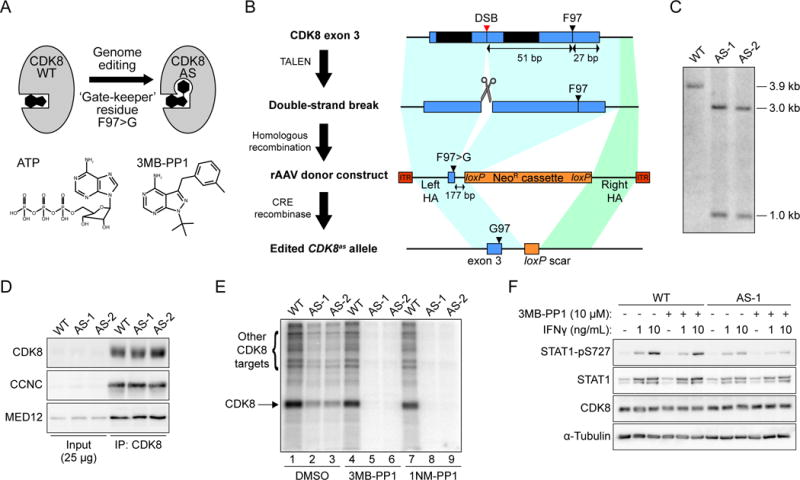
(A) Cartoon depicting the creation of ‘analog sensitive’ CDK8-AS by altering the ‘gate-keeper’ residue in the kinase active site. Structures of ATP and the analog 3MB-PP1 are shown for reference.
(B) Outline of genome editing strategy to generate CDK8as/as HCT116 cells. Each round entailed generation of a DNA double-strand break (DSB) in exon 3 of CDK8, using a transcription activator-like effector nuclease (TALEN) pair, followed by homologous recombination with a recombinant adeno-associated virus (rAAV)-based repair donor construct containing the F97G mutation and a loxP-flanked neomycin resistance (NeoR) cassette, selection for resistance, and finally removal of the NeoR cassette using transient expression of CRE recombinase. TAL-1 and TAL-2, TALEN binding sites; HA, homology arm; ITR, inverted terminal repeat.
(C) Southern blot hybridization analysis of AvrII-digested genomic DNA from WT and two independent homozygous CDK8as/as clones (AS-1 and AS-2), using a probe spanning the novel AvrII restriction site introduced along with the F97G mutation in CDK8 exon 3. Fragment sizes in kilo-base pairs are indicated at right.
(D) Western blot analysis of CDK8, Cyclin C (CCNC) and MED12 levels for inputs (2.5%) and CDK8 immunoprecipitations from WT and AS lysates.
(E) In vitro kinase assay with CDK8 immunoprecipitated material, as in D, showing labelling of proteins with 32P-ATP in the presence of vehicle (DMSO) or the ATP analogs 3MB-PP1 (10 μM) or 1NM-PP1 (10 μM). Arrows indicate bands representing phosphorylation of CDK8 itself, or additional proteins present in the immunoprecipitation.
(F) Western blot showing levels of S727-phosphorylated STAT1 (STAT1-pS727), total STAT1, and CDK8 in HCT116 WT or CDK8 AS-1 cell lysates following treatment with interferon gamma (IFNγ) and/or 10 μM 3MB-PP1.
CDK8 kinase activity is required for proliferation and tumorigenic properties of colorectal carcinoma cells
Given that the CDK8 protein regulates pro-proliferative and hypoxia-inducible genes (Firestein et al., 2008; Galbraith et al., 2013), and that its kinase activity was shown to be required for transformation of NIH-3T3 cells (Firestein et al., 2008), we examined the effect of impaired CDK8 activity on proliferation and tumorigenic properties of HCT116 cells. Vehicle (DMSO)-treated CDK8as/as cells displayed slower proliferation than WT cells in both normoxia and hypoxia (1% O2), and this was further exacerbated by treatment with 3MB-PP1 (Figure 2A–B). Clonogenic efficiency of single cells was reduced in CDK8as/as cells compared to WT, particularly in normoxia (Figure 2C–D), although 3MB-PP1 had little effect over that of the AS mutation alone. Notably, CDK8as/as cells formed smaller colonies, particularly in hypoxia (Figure 2E), in agreement with a proliferation defect. Another important property of transformed cells is the ability to escape anoikis and grow in an anchorage-independent fashion. CDK8as/as cells seeded into soft agar formed fewer colonies than WT cells in both normoxia and hypoxia, and these colonies were reduced in size (Figure S2A–C). Furthermore, while HCT116 cells readily formed three-dimensional spheroids when seeded into ultra-low attachment plates, the growth rates of these spheroids were reduced for CDK8as/as cells (Figure 2F–G). Finally, when injected into the flanks of nude mice, CDK8as/as xenograft tumors grew at a significantly lower rate than wild-type HCT116 (Figure 2H), while retaining normal CDK8 expression levels Figure S2D). Taken together, these results demonstrate that CDK8 kinase activity influences both proliferation and tumorigenic properties of HCT116 cells, consistent with its role as a positive regulator of several oncogenic signaling pathways in CRC.
Figure 2. CDK8 activity is required for proliferation and tumorigenic properties of HCT116 cells.
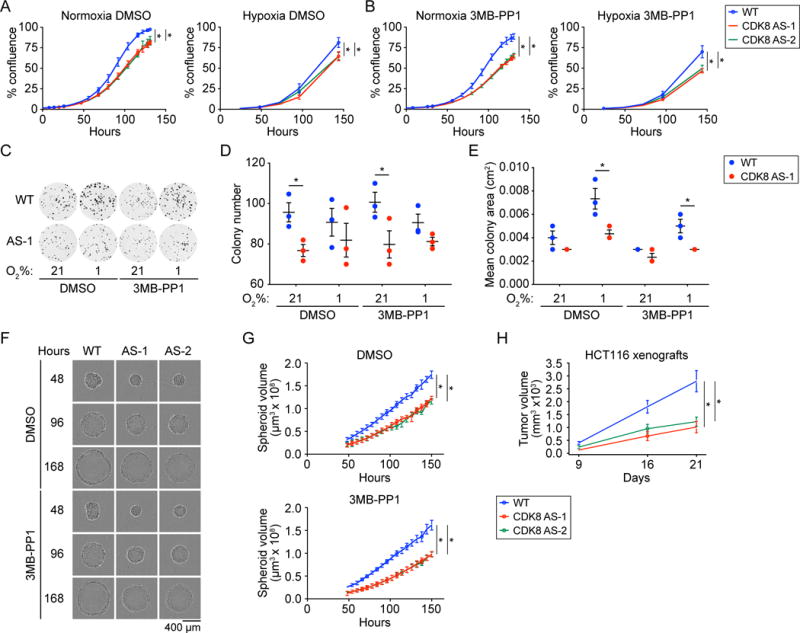
(A and B) Growth curves for WT and two independent homozygous CDK8as/as clones (AS-1 and AS-2) under normoxic and hypoxic (1% O2) conditions, and treated with (A) DMSO vehicle, or (B) 10 μM 3MB-PP1. Percentage confluence was monitored using an Incucyte imaging system. Data are represented as mean ± SEM from three independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(C) Clonogenic cell survival assay for WT and CDK8 AS-1 in normoxia and hypoxia (1% O2), and treated with vehicle (DMSO) or 10 μM 3MB-PP1. Representative images shown.
(D) Clonogenic colony number for cells treated as in C. Individual replicates are shown as circles. Lines and whiskers represent mean ± SEM from three independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(E) Clonogenic colony area for cells treated as in C. Individual replicates are shown as circles. Lines and whiskers represent mean ± SEM from three independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(F) Spheroid growth assay for WT and two independent homozygous CDK8as/as clones (AS-1 and AS-2) in normoxia, treated with vehicle (DMSO) or 10 μM 3MB-PP1. Representative images acquired using an Incucyte imaging system are shown.
(G) Growth curves for WT, AS-1, and AS-2 spheroids, treated as in G. Spheroid volume was calculated from area as monitored using an Incucyte imaging system. Data are represented as mean ± SEM from three independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(H) Xenograft tumor growth assay with wild-type and CDK8as/as HCT116 cells injected into the flanks of nude mice. Data are represented as mean ± SEM from 20 tumors per group. Asterisks indicate significant differences (unpaired t test, p < 0.05).
Inhibition of CDK8 kinase activity has widespread effects on gene expression
We and others have shown that CDK8 is a positive transcriptional coactivator of genes in pathways important for cancer development and progression, including MAPK signaling to serum-responsive genes, β-catenin activated transcription, and hypoxia (Donner et al., 2010; Firestein et al., 2008; Galbraith et al., 2013). However, none of these studies examined the role of the kinase activity in transcription per se. Therefore, we next sought to define the impact of impaired CDK8 kinase activity on the transcriptome of HCT116 cells in normoxia and hypoxia.
We first verified that stabilization of HIF1A during hypoxia is unaffected by CDK8-AS or treatment with 3MB-PP1 (Figure S3A). We then carried out RNA-seq analysis for WT and CDK8as/as HCT116 cells grown in normoxic or hypoxic conditions (1% O2, 24 hrs), and treated with vehicle (DMSO) or 3MB-PP1. Principle component analysis of the 500 most-variable mRNAs in these samples revealed large amounts of variation associated with hypoxia treatment (~58%), and CDK8 genotype (~35%) (Figure S3B). We next used DESeq2 to detect differentially expressed genes (Love et al., 2014). To identify changes caused by hypomorphic CDK8-AS, and to control for potential off-target effects of 3MB-PP1, we conducted separate comparisons of CDK8as/as to WT cells treated with DMSO or 3MB-PP1 (Figure S3C–D and Table S1). This revealed that mutation of the gatekeeper residue in CDK8 has profound effects on the transcriptome, even in the absence of the ATP analog (Figure S3C–D, DMSO). Addition of 3MB-PP1 affects a greater number of genes (Figure S3C–D, 3MB-PP1), including most of those observed with the AS mutation alone (Figure 3A–B). These global patterns confirm that CDK8-AS is a hypomorph, which can then be further inactivated by 3MB-PP1, leading to both down-regulation and upregulation of genes, which we define as CDK8 kinase-dependent and kinase-repressed, respectively (Figure 3A–B). This is most obvious under normoxic conditions (Figure 3A and S3C), where almost all genes affected by CDK8-AS alone are also altered in the presence of 3MB-PP1. Together, this indicates ~7.5% of all expressed genes require CDK8 for maximal expression during normoxia. In terms of absolute numbers, and as a fraction of all expressed genes, the impact of CKD8 inhibition is greater under hypoxic conditions, where ~17% of all expressed genes require CDK8 kinase activity (Figure 3A and S3D). Remarkably, ~23% of hypoxia-inducible genes require CDK8 activity for maximal expression (Figure 3A). For simplicity, we combined the genes affected in CDK8-AS-1 ± 3MB-PP1 for subsequent analyses (see Table S1).
Figure 3. Inhibition of CDK8 has widespread effects on the transcriptome.
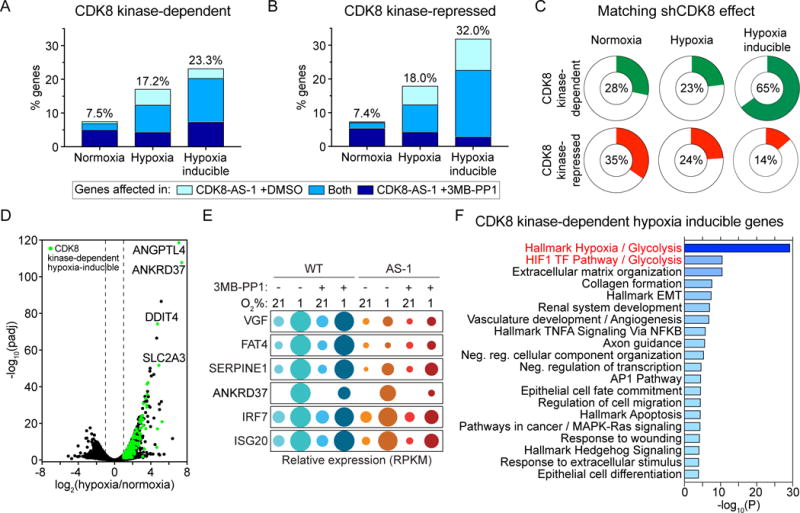
(A and B) Proportions of genes expressed during normoxia and hypoxia, or induced by hypoxia, that (A) depend on CDK8 kinase activity or (B) are repressed by CDK8 kinase activity. Colors indicate the relative numbers of genes affected in CDK8-AS-1 treated with vehicle (DMSO) (light blue) or 10 μM 3MB-PP1 (dark blue), or in both conditions (medium blue). All subsequent analyses combine genes affected in CDK8-AS-1 ±3MB-PP1 (see Table S1).
(C) Proportions of CDK8 kinase-dependent or –repressed genes for which CDK8 knockdown (shCDK8) produces an effect in the same direction.
(D) Volcano plot of log2 fold change (hypoxia/normoxia) against -log10(FDR adjusted p value) for WT HCT116 cells, with hypoxia inducible CDK8-dependent genes highlighted in green. Selected genes of interest are labelled.
(E) Bubble plots showing relative mRNA levels for example CDK8-dependent hypoxia inducible genes. Circle area corresponds to mean RPKM values relative to condition with maximum expression.
(F) Top 20 clusters from Metascape pathway enrichment analysis of the 292 CDK8-dependent hypoxia-inducible genes. Color and length of bars represents −log10(p value), based on the best-scoring term within each cluster.
See also Figure S3, Table S1, and Table S2.
We previously identified a role for CDK8 as a direct coactivator of hypoxia-inducible genes using shRNA-based knockdown in HCT116 cells (Galbraith et al., 2013). Interestingly, the overlap in gene expression changes observed when comparing data from CDK8-AS cells and shCDK8 cells was greatest for hypoxia-inducible genes (~65%, Figure 3C). This indicates that most hypoxia-inducible genes require the kinase activity of CDK8, rather than a scaffold function, for maximal expression, something that is not conserved among the other gene sets defined here (Figure 3C). Hence, we took a closer look at CDK8-dependent hypoxia-inducible genes. A volcano plot of transcriptome changes illustrates the widespread requirement for CDK8 kinase activity among hypoxia inducible genes (Figure 3D). Examples of hypoxia-inducible genes that require CDK8 kinase activity are shown in the bubble plots in Figure 3E. Whereas some genes are strongly affected by the AS mutation alone without further impact of the ATP analog 3MB-PP1 (e.g. VGF, FAT4), others are inhibited more strongly in the presence of the drug (e.g. ANKRD37, IRF7, ISG20). Notably, when we performed gene ontology and pathway enrichment analysis using Metascape (Tripathi et al., 2015) for the 292 hypoxia-inducible genes dependent on CDK8 kinase activity, the most significantly enriched gene sets were the Hallmark Hypoxia and Glycolysis gene sets (Liberzon et al., 2015), and the HIF1 TF and Glycolysis pathways (Figure 3F and Table S2). Of note, these two pathways were also enriched among CDK8-dependent genes in the normoxic condition, suggesting that the kinase activity is also required for basal expression of these genes (Figure S3E).
Altogether, these results indicate that CDK8 kinase activity could play an important role in the transcriptional programs required for the metabolic reprograming of cancer cells, under both basal and hypoxia-induced conditions.
Inhibition of CDK8 kinase activity impairs glucose uptake and glycolysis, and sensitizes HCT116 cells to 2-Deoxy-D-glucose
Having observed an enrichment of the glycolysis pathway among the CDK8 kinase-dependent genes in both normoxia and hypoxia, we decided to focus on a potential role for CDK8 in regulating glucose metabolism. During hypoxia, genes encoding enzymes at every step of the glycolytic cascade are induced in WT HCT116 cells (Figure 4A and Table S1), and half (11/22) require CDK8 kinase activity for their full expression (labeled red in Figure 4A). Of these, more than half (7/11) also depend on CDK8 kinase activity during normoxia (underlined in Figure 4A–B), suggesting that CDK8 may regulate glycolysis during both normoxia and hypoxia. Using our previously published ChIP-seq data for CDK8 in HCT116 cells (Galbraith et al., 2013), we observed CDK8 binding to all but one of the glycolytic genes affected by the CDK8-AS allele, indicative of direct CDK8 action (e.g. SLC2A3, HK1, ENO1, Figure 4C). We confirmed the effect of CDK8 inhibition on expression of several mRNAs by qRT-PCR (Figure 4D), with the most profound effect observed for SLC2A3, which encodes the glucose transporter GLUT3. The expression of these genes is also reduced in HCT116 CDK8as/as xenograft tumors (Figure S2D). Importantly, GLUT3 also displays reduced protein levels in the mutant cells with lower CDK8 activity (Figure 4E).
Figure 4. Inhibition of CDK8 impairs glucose uptake and glycolysis.
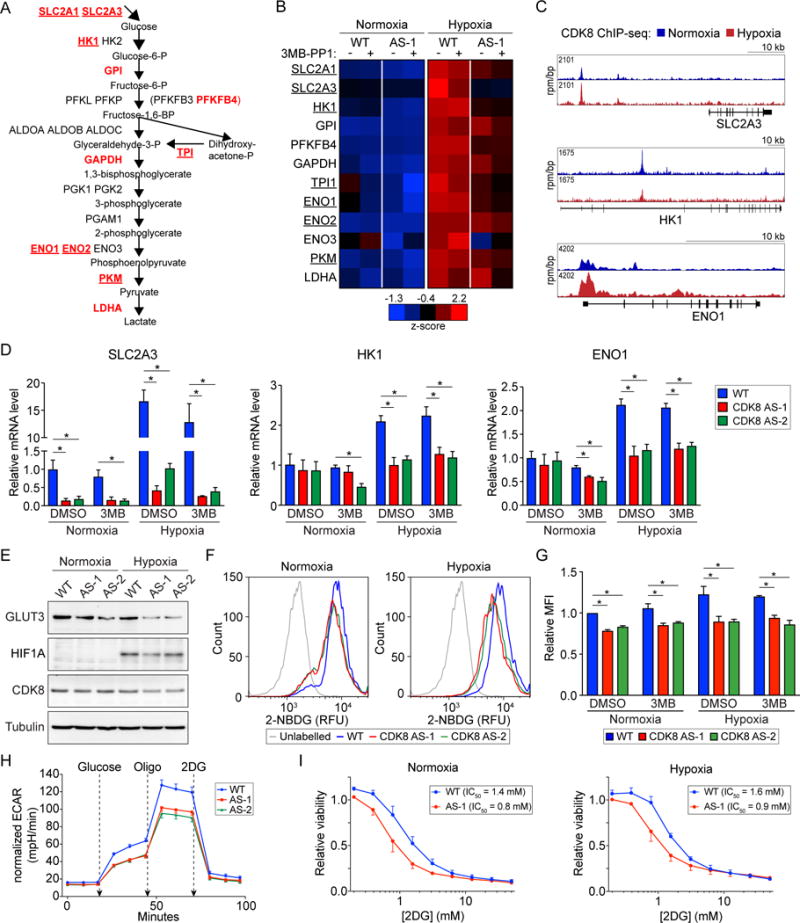
(A) Outline of the glycolysis pathway with metabolites in boxes. Glycolytic genes induced by hypoxia in HCT116 cells are listed at their corresponding step. Genes dependent on CDK8 kinase activity in hypoxia are listed in red, while those also dependent on CDK8 kinase activity in normoxia are underlined.
(B) Heat map showing relative expression of CDK8 kinase-dependent glycolytic genes in WT and CDK8-AS-1 cells across normoxic and hypoxic conditions ± treatment with 10 μM 3MB-PP1. Data are represented as z-scores of mean RNA-seq RPKM values.
(C) Genome browser views of CDK8 binding at selected CDK8 kinase-dependent glycolytic genes under normoxic (blue) and hypoxic (red) conditions.
(D) Relative mRNA levels for selected CDK8 kinase-dependent glycolytic genes, as assessed by qRT-PCR, under normoxic and hypoxic conditions ± treatment with 10 μM 3MB-PP1. Expression values were normalized to 18S ribosomal RNA and are expressed relative to the mean of DMSO treated wild-type samples. Data are represented as mean ± SEM from three independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(E) Western blot showing levels of GLUT3, HIF1A, and CDK8 in WT vs. CDK8-AS-1 and -AS-2 cells under normoxic and hypoxic conditions.
(F) Representative histograms of relative fluorescence for unlabeled vs. 2-NBDG-labelled WT and CDK8-AS-1 and -AS-2 cells in normoxic and hypoxic conditions, treated with vehicle (DMSO).
(G) Relative uptake of 2-NBDG by WT and CDK8-AS-1 and -AS-2 cells in normoxic and hypoxic conditions, and treated with vehicle (DMSO) or 10 μM 3MB-PP1. Data are represented as mean ± SEM from three independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(H) Glycolytic function of WT vs. CDK8-AS-1 and -AS-2 cells, as measured by extracellular acidification rate (ECAR), when starved of glucose for 1 hr, and after addition of glucose followed by oligomycin (oligo) and 2DG. Data are represented as mean ± SEM from five independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(I) 2DG dose-response curves for WT and CDK8-AS-1 cells under normoxic and hypoxic conditions. Data are represented as mean ± SEM from three independent replicates.
See also Figure S4 and Table S1.
We previously reported that CDK8, but not CDK19, is a coactivator of HIF1A target genes (Galbraith et al., 2013). We therefore asked if the effect on glycolytic genes is also exclusive to CDK8. Analysis of our published microarray data for stable knockdown of CDK8 or CDK19 (Galbraith et al., 2013) indicates that while the majority of CDK8 kinase-dependent glycolytic genes, including SLC2A3, HK1, ENO1, have reduced expression with CDK8 knockdown, depletion of CDK19 does not affect their expression (Figure S4A). To explore if the regulation of glycolytic genes by CDK8 might be a common feature across cancers, we examined the association between expression of CDK8 or CDK19 and genes in the glycolysis pathway using TCGA data for 25 cancer types (https://cancergenome.nih.gov/). In a pan-cancer analysis, many glycolytic genes display strong positive correlations with CDK8 expression (i.e. SLC2A1, HK1, HK2, Figure S4B). Of note, this pattern is not conserved for CDK19 (Figure S4B). At the level of individual cancer types, there is clearly a more prevalent positive association between expression of CDK8 and glycolytic genes than for CDK19 (Figure S4C). These correlations were stronger for specific cancer types, such as lung adenocarcinoma (LUAD).
To test whether CDK8 activity influences glucose uptake, we used the fluorescent glucose analog 2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose (2-NBDG). WT HCT116 cells grown in normoxic conditions exhibited strong labelling after 1 hr incubation with 2-NBDG, with a slight increase when grown in hypoxic conditions (Figure 4F–G and S4D). However, 2-NBDG uptake by CDK8as/as cells was significantly reduced in both normoxia and hypoxia. We next assessed the performance of WT and CDK8as/as cells during a glycolysis stress test using extracellular acidification (ECAR) as a measure of glycolytic activity (Figure 4H). Compared to WT, the CDK8as/as cells have a reduction in both their basal and maximal ECAR, indicating that they have significant defects in glycolysis, glycolytic capacity, and glycolytic reserve (Figure S4E). Additionally, the differences in glycolytic activity were accompanied by a decrease in basal oxygen consumption rate (OCR, Figure S4F), although this was not significant.
Finally, we hypothesized that, due to impaired glucose uptake and glycolysis, CDK8as/as cells would be more sensitive to inhibition of glycolysis than WT HCT116 cells. To test this, we treated cells with 2-Deoxy-D-glucose (2DG) under both normoxic and hypoxic conditions (Figure 4I). 2DG is phosphorylated by hexokinases to form 2-Deoxy-D-glucose-6-phosphate which cannot be metabolized further and in turn inhibits the hexokinases, leading to inhibition of glycolysis (Pelicano et al., 2006). While at higher 2DG concentrations we observed reduced viability of WT cells (IC50 of ~1.4 mM and ~1.6 mM in normoxia and hypoxia), the CDK8as/as cells displayed a clear increase in sensitivity in both normoxia and hypoxia (IC50 of ~0.8 mM and ~0.9 mM, respectively). Taken together, these results demonstrate that CDK8 activity is required for HCT116 cells to maintain maximal rates of glucose uptake and glycolysis. This function is conserved in both normoxia and hypoxia, suggesting that not only is CDK8 important for the cellular adaptation to hypoxia, but that it also promotes aerobic glycolysis i.e. the Warburg effect that is exhibited by many cancer types.
The dual CDK8/19 inhibitor Senexin A impairs glucose uptake and sensitizes cancer cells to 2DG
Given the importance of hypoxic metabolism and the Warburg effect in solid tumors such as CRC, we next looked to test if the effects of CDK8 inhibition demonstrated using our chemical genetics approach could be recapitulated using a potential therapeutic compound in HCT116 and additional cancer cell lines. SW480 CRC cells exhibit amplification of CDK8 locus, and are more sensitive to depletion of CDK8 than HCT116 (Firestein et al., 2008), and Senexin A is one of several recently developed dual CDK8/CDK19 inhibitors (Porter et al., 2012). We first confirmed that 10 μM Senexin A treatment is able to reduce IFN gamma-stimulated phosphorylation of STAT1 (Figure S5A). We next examined the effect of Senexin A on expression of several glycolytic enzymes in the two CRC cell lines (Figure 5A). Surprisingly, in HCT116 cells, GLUT3 expression was increased by Senexin A treatment, potentially due to its dual effects on CDK8 and CDK19. However, expression of GLUT1 and HK1 was reduced by Senexin A treatment, consistent with our results for CDK8as/as cells. In SW480 cells, expression of both glucose transporters and HK1 was reduced by Senexin A. Accordingly, Senexin A had a more significant impact on 2-NBDG uptake in SW480 cells (Figure 5B–C and S5B).
Figure 5. Senexin A reduces glucose uptake and sensitizes CRC cells to 2DG.
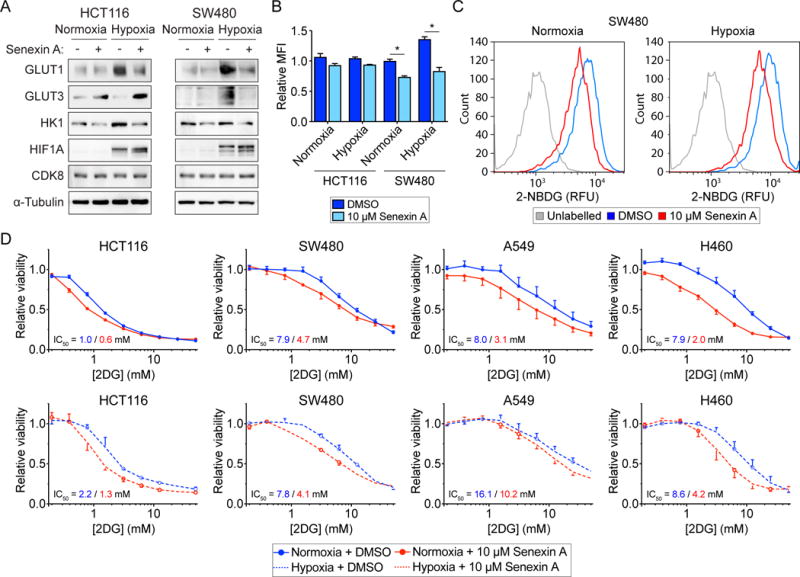
(A) Western blots showing levels of GLUT1, GLUT3, HK1, HIF1A, and CDK8 in HCT116 and SW480 cells under normoxic conditions, and treated with vehicle (DMSO) or 10 μM Senexin A.
(B) Relative uptake of 2-NBDG by HCT116 and SW480 cells in normoxic and hypoxic conditions, and treated with vehicle (DMSO) or 10 μM Senexin A. Data are represented as mean ± SEM from three independent replicates. Asterisks indicate significant differences (unpaired t test, p < 0.05).
(C) Representative histograms of relative fluorescence for unlabeled vs. 2-NBDG-labelled SW480 cells in normoxic and hypoxic conditions, treated with vehicle (DMSO) or 10 μM Senexin A.
(D) 2DG dose-response curves for the indicated cells under normoxic and hypoxic conditions, and treated with vehicle (DMSO) or 10 μM Senexin A. Data are represented as mean ± SEM from three independent replicates.
See also Figure S5.
To serve as a proof of principle for potential therapeutic strategies, we next tested the effect of Senexin A on viability of the HCT116 and SW480 CRC cell lines, as well two non-small cell lung cancer lines, A549 and H460. Note that all four lines express readily detectable amounts of both CDK8 and CDK19 at the protein and RNA levels (Figure S5C–D), with SW480 expressing the highest level of CDK8, consistent with the known amplification of the CDK8 locus in this cell line (Firestein et al., 2008). Treatment with 10 μM Senexin A alone led to modest reductions in viability of all four cell lines during normoxia, but not hypoxia (Figure S5C). Combination treatment with 2DG further decreased viability in both normoxia and hypoxia (Figure 5D), confirming that CDK8/19 inhibition can enhance the effects of 2DG on cancer cells. Finally, while the growth of HCT116 spheroids is essentially unaffected by treatment with either compound alone, the combination treatment causes a significant defect in spheroid growth (Figure S5D). These results suggest that pharmacological inhibition of both CDK8 and glycolysis may be an effective strategy for targeting CRC and other tumor types.
DISCUSSION
Upregulation of glycolysis is important for the proliferation and survival of cancerous cells in both aerobic and hypoxic conditions, with most cancer cells displaying constitutively high rates of glucose consumption regardless of oxygen availability. This glycolytic switch may initially be driven by adaptation to the intermittent hypoxia experienced by proliferating cells early in the transformation process prior to angiogenesis and metastasis (Gatenby and Gillies, 2004). Furthermore, there is also evidence that elevated glycolysis confers a proliferative advantage upon cancer cells over and above maintaining ATP production (Hay, 2016). Both hypoxia and high levels of glucose uptake are associated with poor prognosis (Chen et al., 2017; Kunkel et al., 2003). However, high rates of glycolysis are also a feature of normal cells with high rates of proliferation which may lead to on-target toxicities with prolonged high-dose treatment with glycolysis inhibitors such as 2DG.
Control of glycolytic flux in cancer appears to be mainly at the level of glucose import and phosphorylation, by the SLC/GLUT family of transporters and the hexokinases, respectively (Gatenby and Gillies, 2004). These enzymes can be upregulated by HIFs in response to hypoxia, or constitutively by oncogenic pathways and transcription factors, such as oncogenic KRAS and BRAF mutants, PI3K/AKT signaling and c-MYC overexpression, either directly or via upregulation of HIFs (Barthel et al., 1999; Kim et al., 2007; Yun et al., 2009). In fact, transformation of primary fibroblasts driven by oncogenic HRAS increases dependence on glucose (Ramanathan et al., 2005), and glucose starvation can select for acquisition of activating KRAS pathway mutations (Yun et al., 2009).
The reprogrammed glucose metabolism of cancer cells distinguishes them from most normal cells and has been exploited as an in vivo diagnostic and prognostic marker using [18F]fluoro-2-deoxyglucose (FDG) positron emission tomography (PET) imaging (Patel et al., 2010). However, therapeutic targeting of the Warburg effect in cancer cells has proven difficult, owing to their use of the same glycolytic enzymes as normal cells and the consequent risk of on-target adverse effects, as well as the expression of multiple isoforms of these enzymes (Hay, 2016; Vander Heiden and DeBerardinis, 2017). Although 2DG has been explored as a therapeutic agent (Landau et al., 1958; Raez et al., 2013), poor efficacy and undesired side effects such as hypoglycemia, fatigue, and tachycardia have limited its application as a single agent therapy (Bost et al., 2016). Interestingly, 2DG does seem to hold some promise in sensitizing cancer cells when used in combination with other pharmacological agents, such as Metformin (Cheong et al., 2011), Docetaxel (Raez et al., 2013), and BCL2 antagonists (Yamaguchi et al., 2011).
The discovery of CDK8 as a CRC oncogene has generated considerable interest in its therapeutic targeting in various cancer types, and numerous small molecule inhibitors have been reported recently, all of which inhibit both CDK8 and its highly similar paralog CDK19 (Clarke et al., 2016). Many of these compounds have demonstrated efficacy in vitro and in preclinical models, most notably Cortistatin A in AML cells (Pelish et al., 2015), Senexin A in fibrosarcoma cells (Porter et al., 2012), and CCT251921 and MSC2530818 in CRC cells (Clarke et al., 2016). However, the most comprehensive evaluation of the therapeutic potential of dual CDK8/19 inhibitors to date, using two different classes of compounds, demonstrated only modest activity in a β-catenin-driven mouse model of CRC, and various adverse effects in rat and dog tolerability studies (Clarke et al., 2016). Although the mouse model was not driven by CDK8 amplification, these results nonetheless suggest that on-target activity will lead to adverse effects that would limit the therapeutic application of these dual CDK8/19 inhibitors as single agents. Identification of CDK8-based combination strategies may allow for more effective tumor responses at lower doses and shorter times that would minimize adverse side-effects. In this context, our findings reported here suggest that combined inhibition of CDK8 and glycolysis as a therapeutic strategy warrants further investigation.
As discussed above, there are several reports demonstrating that dual CDK8/19 inhibitors are able to suppress in vitro or in vivo growth of different cancer types. However, it is not yet clear the extent to which these effects are due to inhibition of CDK8 versus CDK19. In addition to overcoming the challenges associated with adverse side-effects, predicting responses to CDK8/19 inhibitors will benefit from a detailed understanding of the functional specialization of these the two kinases. In CRC, only CDK8 is subject to recurrent amplification (Firestein et al., 2009), and we previously reported that CDK8, not CDK19, is a widespread coactivator of hypoxia-inducible genes (Galbraith et al., 2013). Here, we report a requirement for the kinase activity of CDK8 in expression of glycolytic genes. While we found measurable defects in glucose uptake and the rate of glycolysis in cells with impaired CDK8 activity, we cannot exclude effects on additional metabolic pathways that rely on glucose uptake, such as the pentose phosphate pathway and glutathione/redox homeostasis. It would be interesting to determine if CDK8 kinase activity has more global effects on metabolism beyond glycolysis. Microarray data for depletion of either kinase, and analysis of expression data for human cancers suggest that CDK8 plays the dominant role in expression of these genes, particularly in certain cancer types such as lung adenocarcinoma. Therefore, tumors that overexpress CDK8 may be more likely to respond to combination treatments with inhibitors of CDK8/19 and glycolysis than tumors overexpressing CDK19.
EXPERIMENTAL PROCEDURES
Cell Culture and proliferation, growth, and viability assays
HCT116 human colon carcinoma cells were cultured in McCoy’s 5A (Gibco/Thermo Fisher Scientific) supplemented with 10% Fetal Bovine Serum (FBS, Peak Serum). SW480 human colon carcinoma and H460 human lung adenocarcinoma cells were cultured in RPMI 1640 medium (Gibco/Thermo Fisher Scientific) supplemented 10% FBS. A549 human lung adenocarcinoma cells were cultured in DMEM (4.5 g/l glucose, Gibco/Thermo Fisher Scientific) supplemented 10% FBS. All media were supplemented with 1% antibiotic/antimycotic solution (Gibco/Thermo Fisher Scientific). Accutase cell detachment solution (Thermo Fisher Scientific) or 0.25% Trypsin/EDTA solution (Thermo Fisher Scientific) was used to generate single cell suspensions for proliferation, clonogenic and soft-agar assays, and spheroid formation. For normoxic treatments, cells were maintained at 37°C in a humidified atmosphere with 5% CO2. For hypoxic treatments, cells were maintained in a humidified atmosphere at 37°C, with 1% O2 and 5% CO2, or were placed in humidified sealed incubation chambers (Billups-Rothenburg), flushed twice with 120 l of a mixture of 1% O2/5% CO2/94% N2 (Airgas), and maintained at 37°C. Assays for 2D proliferation rate, 3D tumor spheroid growth, soft agar colony formation, xenograft growth in nude mice, and Sulforhodamine B viability measurements are described in Supplemental Experimental Procedures.
AS CDK8 Genome Editing
Full details of TALEN design and assembly (Briggs et al., 2012), and rAAV homology donor construction (Kohli et al., 2004) are provided in Supplemental Experimental Procedures. To generate CDK8as/as cells, parental HCT116 cells were transfected with plasmids encoding the TALEN pair, followed by rAAV infection, selection, and single-cell cloning. Heterozygous clones were identified by PCR screening, pooled, and subjected to a second round of editing. A full description of clone validation by PCR, Southern hybridization, and sequencing can be found in Supplemental Experimental Procedures.
Western Blotting
Sample preparation, quantitation, and western blotting were carried out as described previously (Henry et al., 2012). Detection was by enhanced chemiluminescence, and images were captured using an ImageQuant LAS4000 digital camera system (GE Healthcare). Antibodies used are described in Supplemental Experimental Procedures.
CDK8 Immunoprecipitation Kinase Assay
Briefly, CDK8 was immunoprecipitated from whole cell lysates, and incubated with [γ-32P]-ATP in the presence of vehicle (DMSO) or 10 μM of the ATP analogs 3MB-PP1 or 1NM-PP1. Following SDS-PAGE, the dried gel was analyzed using a STORM 860 scanner (Molecular Dynamics). Complete details can be found in Supplemental Experimental Procedures.
RNA-seq and qRT-PCR
HCT116 cells were plated and treated as described under experimental procedures, followed by harvesting in cold PBS. Total RNA was extracted from cell pellets using TRI Reagent (Sigma Aldrich), according to the manufacturer’s instructions. RNA-seq library preparation and sequencing, and cDNA synthesis / qRT-PCR analysis are described in Supplemental Experimental Procedures.
RNA-seq data analysis
Data quality assessment, filtering, and mapping are described in Supplemental Experimental Procedures. Differential expression analysis was assessed using DESeq2 (Love et al., 2014). Full details are provided in Supplemental Experimental Procedures.
2NBDG Uptake Assay
Glucose uptake was measured using fluorescent glucose analog 2-NBDG (2-deoxy-2-[(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]-D-glucose, Cayman Chemical). Treatment conditions and flow cytometry are described in Supplemental Experimental Procedures.
Glycolysis Stress Test, ECAR and OCR Measurement
Extracellular Acidification Rate (ECAR) and Oxygen Consumption Rate (OCR) were measured on a Seahorse XFe24 Analyzer (Agilent). ECAR and OCR values were normalized to the total amount of protein per well. See Supplemental Experimental Procedures for additional details.
Supplementary Material
Document S1. Supplemental Experimental Procedures and Figures S1–S5.
Table S1. RNA-seq data, Related to Figures 4 and S4.
Table S2. Metascape analysis, Related to Figures 3 and 4.
HIGHLIGHTS.
A chemical genetics approach allows specific inhibition of CDK8
CDK8 kinase activity is required for expression of multiple glycolytic genes
Inhibition of CDK8 activity impairs glucose uptake and glycolysis
Inhibition of CDK8 activity sensitizes cancer cells to glycolysis inhibitors
Acknowledgments
We thank Dr. Adrian Briggs for TALEN construction plasmids and protocol advice, and Johan Hegg for helpful discussions. This work was supported by grants from the National Science Foundation (NSF MCB-1243522 and MCB-1627615), the National Institutes of Health (NIGMS R01GM120109), the Cancer League of Colorado (AWD-16323), and the Mary Miller & Charlotte Fonfara-LaRose Down Syndrome & Leukemia Research Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
The GEO accession number for the RNA-seq data reported in this paper is: GSE101526.
AUTHOR CONTRIBUTIONS
M.D.G. conceived and designed experiments; acquired, analyzed and interpreted data; and drafted and edited the manuscript. A.P. analyzed data. Z.A., M.H., E.A.B., and A.A.H. acquired and analyzed data. K.D.S. and J.M.E. conceived and designed experiments; interpreted data; and edited the manuscript.
References
- Bancerek J, Poss ZC, Steinparzer I, Sedlyarov V, Pfaffenwimmer T, Mikulic I, Dolken L, Strobl B, Muller M, Taatjes DJ, et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity. 2013;38:250–262. doi: 10.1016/j.immuni.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- Blethrow J, Zhang C, Shokat KM, Weiss EL. Design and use of analog-sensitive protein kinases. Curr Protoc Mol Biol Chapter. 2004;18 doi: 10.1002/0471142727.mb1811s66. Unit 18 11. [DOI] [PubMed] [Google Scholar]
- Bost F, Decoux-Poullot AG, Tanti JF, Clavel S. Energy disruptors: rising stars in anticancer therapy? Oncogenesis. 2016;5:e188. doi: 10.1038/oncsis.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs AW, Rios X, Chari R, Yang L, Zhang F, Mali P, Church GM. Iterative capped assembly: rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012;40:e117. doi: 10.1093/nar/gks624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cee VJ, Chen DY, Lee MR, Nicolaou KC. Cortistatin A is a high-affinity ligand of protein kinases ROCK, CDK8, and CDK11. Angew Chem Int Ed Engl. 2009;48:8952–8957. doi: 10.1002/anie.200904778. [DOI] [PubMed] [Google Scholar]
- Chen X, Lu P, Zhou S, Zhang L, Zhao JH, Tang JH. Predictive value of glucose transporter-1 and glucose transporter-3 for survival of cancer patients: A meta-analysis. Oncotarget. 2017;8:13206–13213. doi: 10.18632/oncotarget.14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, Wa Cheng K, Hall H, Zhang D, Lu Y, et al. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Mol Cancer Ther. 2011;10:2350–2362. doi: 10.1158/1535-7163.MCT-11-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AD, Oldenbroek M, Boyer TG. Mediator kinase module and human tumorigenesis. Critical Reviews in Biochemistry and Molecular Biology. 2015:1–34. doi: 10.3109/10409238.2015.1064854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PA, Ortiz-Ruiz MJ, TePoele R, Adeniji-Popoola O, Box G, Court W, Czasch S, El Bawab S, Esdar C, Ewan K, et al. Assessing the mechanism and therapeutic potential of modulators of the human Mediator complex-associated protein kinases. Elife. 2016;5 doi: 10.7554/eLife.20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czodrowski P, Mallinger A, Wienke D, Esdar C, Poschke O, Busch M, Rohdich F, Eccles SA, Ortiz-Ruiz MJ, Schneider R, et al. Structure-Based Optimization of Potent, Selective, and Orally Bioavailable CDK8 Inhibitors Discovered by High-Throughput Screening. J Med Chem. 2016;59:9337–9349. doi: 10.1021/acs.jmedchem.6b00597. [DOI] [PubMed] [Google Scholar]
- Dale T, Clarke PA, Esdar C, Waalboer D, Adeniji-Popoola O, Ortiz-Ruiz MJ, Mallinger A, Samant RS, Czodrowski P, Musil D, et al. A selective chemical probe for exploring the role of CDK8 and CDK19 in human disease. Nat Chem Biol. 2015;11:973–980. doi: 10.1038/nchembio.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DL, Ford M, Schwinn MK, Benink H, Galbraith MD, Amunugama R, Jones R, Allen D, Okazaki N, Yamakawa H, et al. Mutual Exclusivity of MED12/MED12L, MED13/13L, and CDK8/19 Paralogs Revealed within the CDK-Mediator Kinase Module. Journal of Proteomics and Bioinformatics. 2013;S2 [Google Scholar]
- Donner AJ, Ebmeier CC, Taatjes DJ, Espinosa JM. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat Struct Mol Biol. 2010;17:194–201. doi: 10.1038/nsmb.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, Freed E, Ligon AH, Vena N, Ogino S, et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R, Shima K, Nosho K, Irahara N, Baba Y, Bojarski E, Giovannucci E, Fuchs C, Ogino S. CDK8 expression in 470 colorectal cancers in relation to beta-catenin activation, other molecular alterations and patient survival. International journal of cancer Journal international du cancer. 2009 doi: 10.1002/ijc.24908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith MD, Allen MA, Bensard CL, Wang X, Schwinn MK, Qin B, Long HW, Daniels DL, Hahn WC, Dowell RD, et al. HIF1A Employs CDK8-Mediator to Stimulate RNAPII Elongation in Response to Hypoxia. Cell. 2013;153:1327–1339. doi: 10.1016/j.cell.2013.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith MD, Donner AJ, Espinosa JM. CDK8: A positive regulator of transcription. Transcription. 2010;1:4–12. doi: 10.4161/trns.1.1.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Gu W, Wang C, Li W, Hsu FN, Tian L, Zhou J, Yuan C, Xie XJ, Jiang T, Addya S, et al. Tumor-suppressive effects of CDK8 in endometrial cancer cells. Cell Cycle. 2013;12:987–999. doi: 10.4161/cc.24003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16:635–649. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry RE, Andrysik Z, Paris R, Galbraith MD, Espinosa JM. A DR4:tBID axis drives the p53 apoptotic response by promoting oligomerization of poised BAX. EMBO J. 2012;31:1266–1278. doi: 10.1038/emboj.2011.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannou M, Paraskeva E, Baxevanidou K, Simos G, Papamichali R, Papacharalambous C, Samara M, Koukoulis G. HIF-1alpha in colorectal carcinoma: review of the literature. J BUON. 2015;20:680–689. [PubMed] [Google Scholar]
- Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105–1109. doi: 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381–7393. doi: 10.1128/MCB.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Donner AJ, Espinosa JM, Taatjes DJ. The human CDK8 subcomplex is a histone kinase that requires Med12 for activity and can function independently of mediator. Mol Cell Biol. 2009;29:650–661. doi: 10.1128/MCB.00993-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler MF, Bergeron P, Blackwood EM, Bowman K, Clark KR, Firestein R, Kiefer JR, Maskos K, McCleland ML, Orren L, et al. Development of a Potent, Specific CDK8 Kinase Inhibitor Which Phenocopies CDK8/19 Knockout Cells. ACS Med Chem Lett. 2016;7:223–228. doi: 10.1021/acsmedchemlett.5b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli M, Rago C, Lengauer C, Kinzler KW, Vogelstein B. Facile methods for generating human somatic cell gene knockouts using recombinant adeno-associated viruses. Nucleic Acids Res. 2004;32:e3. doi: 10.1093/nar/gnh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Kunkel M, Reichert TE, Benz P, Lehr HA, Jeong JH, Wieand S, Bartenstein P, Wagner W, Whiteside TL. Overexpression of Glut-1 and increased glucose metabolism in tumors are associated with a poor prognosis in patients with oral squamous cell carcinoma. Cancer. 2003;97:1015–1024. doi: 10.1002/cncr.11159. [DOI] [PubMed] [Google Scholar]
- Landau BR, Laszlo J, Stengle J, Burk D. Certain metabolic and pharmacologic effects in cancer patients given infusions of 2-deoxy-D-glucose. J Natl Cancer Inst. 1958;21:485–494. [PubMed] [Google Scholar]
- Larochelle S, Batliner J, Gamble MJ, Barboza NM, Kraybill BC, Blethrow JD, Shokat KM, Fisher RP. Dichotomous but stringent substrate selection by the dual-function Cdk7 complex revealed by chemical genetics. Nat Struct Mol Biol. 2006;13:55–62. doi: 10.1038/nsmb1028. [DOI] [PubMed] [Google Scholar]
- Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, Shokat KM, Jallepalli PV, Fisher RP. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25:839–850. doi: 10.1016/j.molcel.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Fassl A, Chick J, Inuzuka H, Li X, Mansour MR, Liu L, Wang H, King B, Shaik S, et al. Cyclin C is a haploinsufficient tumour suppressor. Nat Cell Biol. 2014;16:1080–1091. doi: 10.1038/ncb3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinger A, Crumpler S, Pichowicz M, Waalboer D, Stubbs M, Adeniji-Popoola O, Wood B, Smith E, Thai C, Henley AT, et al. Discovery of potent, orally bioavailable, small-molecule inhibitors of WNT signaling from a cell-based pathway screen. J Med Chem. 2015;58:1717–1735. doi: 10.1021/jm501436m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinger A, Schiemann K, Rink C, Sejberg J, Honey MA, Czodrowski P, Stubbs M, Poeschke O, Busch M, Schneider R, et al. 2,8-Disubstituted-1,6-Naphthyridines and 4,6-Disubstituted-Isoquinolines with Potent, Selective Affinity for CDK8/19. ACS Med Chem Lett. 2016a;7:573–578. doi: 10.1021/acsmedchemlett.6b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallinger A, Schiemann K, Rink C, Stieber F, Calderini M, Crumpler S, Stubbs M, Adeniji-Popoola O, Poeschke O, Busch M, et al. Discovery of Potent, Selective, and Orally Bioavailable Small-Molecule Modulators of the Mediator Complex-Associated Kinases CDK8 and CDK19. J Med Chem. 2016b;59:1078–1101. doi: 10.1021/acs.jmedchem.5b01685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CN, Goldstone AR, Chowdhury FU, Scarsbrook AF. FDG PET/CT in oncology: “raising the bar”. Clin Radiol. 2010;65:522–535. doi: 10.1016/j.crad.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- Pelish HE, Liau BB, Nitulescu II, Tangpeerachaikul A, Poss ZC, Da Silva DH, Caruso BT, Arefolov A, Fadeyi O, Christie AL, et al. Mediator kinase inhibition further activates super-enhancer-associated genes in AML. Nature. 2015;526:273–276. doi: 10.1038/nature14904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Perri JI, Dengler VL, Audetat KA, Pandey A, Bonner EA, Urh M, Mendez J, Daniels DL, Wappner P, Galbraith MD, et al. The TIP60 Complex Is a Conserved Coactivator of HIF1A. Cell Rep. 2016;16:37–47. doi: 10.1016/j.celrep.2016.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DC, Farmaki E, Altilia S, Schools GP, West DK, Chen M, Chang BD, Puzyrev AT, Lim CU, Rokow-Kittell R, et al. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc Natl Acad Sci U S A. 2012;109:13799–13804. doi: 10.1073/pnas.1206906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Odell AT, Tangpeerachaikul A, Lee T, Pelish HE, Shair MD, Dowell RD, Old WM, Taatjes DJ. Identification of Mediator Kinase Substrates in Human Cells using Cortistatin A and Quantitative Phosphoproteomics. Cell Rep. 2016;15:436–450. doi: 10.1016/j.celrep.2016.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss ZC, Ebmeier CC, Taatjes DJ. The Mediator complex and transcription regulation. Crit Rev Biochem Mol Biol. 2013;48:575–608. doi: 10.3109/10409238.2013.840259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ, Tolba K, Langmuir VK, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:523–530. doi: 10.1007/s00280-012-2045-1. [DOI] [PubMed] [Google Scholar]
- Ramanathan A, Wang C, Schreiber SL. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci U S A. 2005;102:5992–5997. doi: 10.1073/pnas.0502267102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanso M, Levin RS, Lipp JJ, Wang VY, Greifenberg AK, Quezada EM, Ali A, Ghosh A, Larochelle S, Rana TM, et al. P-TEFb regulation of transcription termination factor Xrn2 revealed by a chemical genetic screen for Cdk9 substrates. Genes Dev. 2016;30:117–131. doi: 10.1101/gad.269589.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann K, Mallinger A, Wienke D, Esdar C, Poeschke O, Busch M, Rohdich F, Eccles SA, Schneider R, Raynaud FI, et al. Discovery of potent and selective CDK8 inhibitors from an HSP90 pharmacophore. Bioorg Med Chem Lett. 2016;26:1443–1451. doi: 10.1016/j.bmcl.2016.01.062. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe. 2015;18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerling T, Kuuluvainen E, Makela TP. Cdk8 is essential for preimplantation mouse development. Mol Cell Biol. 2007;27:6177–6182. doi: 10.1128/MCB.01302-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R, Janssen E, Perkins G, Ellisman M, Kitada S, Reed JC. Efficient elimination of cancer cells by deoxyglucose-ABT-263/737 combination therapy. PLoS ONE. 2011;6:e24102. doi: 10.1371/journal.pone.0024102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JW, Wang G. The Mediator complex: a master coordinator of transcription and cell lineage development. Development. 2014;141:977–987. doi: 10.1242/dev.098392. [DOI] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Supplemental Experimental Procedures and Figures S1–S5.
Table S1. RNA-seq data, Related to Figures 4 and S4.
Table S2. Metascape analysis, Related to Figures 3 and 4.