

Abstract
Cancer is a heterogeneous disease harboring diverse subclonal populations that can be discriminated by their DNA mutations. Environmental pressure selects subclones that ultimately drive disease progression and tumor relapse. Circulating cell-free DNA (ccfDNA) can be used to approximate the mutational makeup of cancer lesions and can serve as a marker for monitoring disease progression at the molecular level without the need for invasively acquired samples from primary or metastatic lesions. This potential for molecular analysis makes ccfDNA attractive for the study of clonal evolution and for uncovering emerging therapeutic resistance or sensitivity. We assessed ccfDNA from colon and pancreatic adenocarcinoma patients using next generation sequencing of 56 cancer-associated genes at the time of primary resectable disease and metastatic progression and compared this to the mutational patterns of the primary tumor. 28%-47% of non-synonymous mutations in the primary tumors were also detected in the ccfDNA whilst 71%-78% mutations found in ccfDNA were not detected in the primary tumors. ccfDNA collected at the time of progression harbored 3-5 new mutations not detected in ccfDNA at the earlier collection time points. We conclude that incorporation of ccfDNA analysis provides crucial insights into the changing molecular makeup of progressive colon and pancreatic cancer.
Keywords: liquid biopsies, circulating tumor DNA, circulating cell-free DNA (ccfDNA), clonal evolution, pancreatic cancer, colon cancer
Introduction
Genomic mutations are one of the hallmarks of cancer [1]. The molecular characterization of a given cancer relies on the analysis of tissue specimen from a primary or metastatic lesion typically obtained at a single time point. However, due to intratumoral heterogeneity, the selection of cell subpopulations during cancer evolution and metastasis, the analysis of a single tissue specimen will provide only a limited characterization of the molecular makeup of the disease [2, 3]. Monitoring the molecular characteristics of cancer by serial analyses of circulating cell-free DNA (ccfDNA) enables capture of emerging heterogeneity of the disease and may support treatment decisions [4, 5]. ccfDNA analysis has evolved since its inception with improvements in the technologies and detection limits [6, 7] and represents a set of research tools that appear poised to enter routine clinical care [8, 9]. The recent FDA approval of a ccfDNA assay for the EGFR T790M mutation in lung cancer supports this notion [10].
Whether ccfDNA should complement tissue analyses in all cancer types remains to be studied, especially in early stage diseases [9, 11]. However, ccfDNA may be superior to tumor tissue DNA in the assessment of cancer heterogeneity and evolution during disease progression [12, 13]. Here we study the mutational landscape of ccfDNA at diagnosis and disease recurrence and compare it to that of DNA from the primary tumor tissues in ten patients with colon and pancreatic cancer.
Materials and Methods
Patient Samples
Patients with newly diagnosed colon adenocarcinoma (colon AC) or pancreatic ductal adenocarcinoma (PDAC) were recruited for blood and tissue collection under the IRB protocol 2007-345 “Establishment of the High Quality Tumor Biobank and Clinical Database” and the Non-Therapeutic Subject Registry (NTSR) Shared Resource protocol Pr000000007 at the Lombardi Comprehensive Cancer Center at Georgetown University after obtaining informed consent. Ten patients were retrospectively selected with the following inclusion criteria: initial diagnosis of treatment-naïve resectable primary adenocarcinoma (n=5 colon; n=5 pancreatic), surgical resection of the primary tumor, and development of progressive disease after surgery. None of the patients had a previous malignant disease.
Peripheral venous blood samples were collected in EDTA plasma tubes before the surgical removal of the primary tumors as well as at time of metastatic disease progression (1-70 months after surgery). The blood samples were centrifuged at ≤1300 RCF for 10 min within 2 hours of blood collection, after which plasma was separated and stored at -80 °C until further analysis.
Surgical specimen of the primary tumors were frozen in O.C.T and cryo-sectioned into 20 μm scrolls and examined by a pathologist for the presence of cancer cells.
DNA isolation
The plasma samples were thawed on ice and circulating cell-free circulating DNA (ccfDNA) was isolated from 2 × 100 μL plasma per patient, using the DNA extractor SP Kit (Wako cat. # 296-60501), following the manufacturer’s protocol. In brief, 200 μL Enzyme Reaction Solution and 5 μL Protein Digestion Solution was added to 100 μL plasma and mixed by vortexing. The samples were incubated at 56 °C for 10 min. Thereafter 300 μL of Sodium Iodide Solution and 600 μL Alcohol Solution were added and mixed by vortexing. After 10 min incubation at room temperature, the samples were centrifuged at 16,000 × g for 10 min at room temperature. The supernatant was discarded and ccfDNA pellets were washed with 1 mL Washing Solution A by vortexing. After 5 min centrifugation at 16,000 × g the supernatant was discarded. The ccfDNA pellets were washed with 1 mL Washing Solution B and centrifuged once more for 5 min. The supernatant was discarded again and DNA pellets were allowed to dry. The ccfDNA was diluted in 15 μL of ultra pure water and quantitated with the NanoDrop 2000c (Thermo Scientific) and the Promega Quantifluor ONE dsDNA Fluorescence Assay (Promega).
Two frozen tumor tissue scrolls of 20 μm thickness per patient with an average weight of 75 μg and surface of 1.15 cm2 were used for genomic DNA isolation. DNA was isolated using the PrepEase Genomic DNA Isolation Kit (USB), following the manufacturer’s protocol. In brief, the tissue was homogenized in 240 μL Homogenization Buffer in MagNA Lyser Green Beads (Roche) in the MagNA Lyser (Roche). A mixture of 200 μL Chloroform/Isoamyl Alcohol (24:1), as well as 800 μL Protein Precipitation Buffer were added to the lysates. Samples were mixed by vortexing and centrifuged at 13,000 × g for 4 min at room temperature. 880 μL of the upper aqueous phase of the sample was transferred to a new microcentrifuge tube containing 620 μL isopropanol. The samples were mixed by inverting the tubes and centrifuged at 13,000 × g for 4 min. The supernatant was discarded and DNA pellets were washed with 1 mL of 70% ethanol by vortexing. The samples were centrifuged for 2 min and DNA pellets were allowed to dry. The tumor DNA was diluted in 15 μL of ultra pure water and quantitated with the NanoDrop 2000c (Thermo Scientific) and the Promega Quantifluor ONE dsDNA Fluorescence Assay (Promega).
56G Oncology Panel Sequencing Library preparation
DNA mutation analysis was conducted using a Targeted Next Generation Sequencing Library Preparation Kit that is compatible with circulating cell-free DNA and the Illumina MiSeq Platform: the 56G Oncology Panel v2 from Swift Biosciences (Cat. # AL-56248). This panel contains 263 amplicons sized 92-184 bp that covers hotspots, exonic SNPs and contiguous regions of 56 human genes. The list of genes and number of amplicons is provided in Table 1. The kit contains a DNA standard with a set of 11 defined allelelic frequencies for major oncology targets to be used as a sequencing control and DNA from HCT116, RKO and SW48 colon cancer cell lines. The 56G Oncology library was prepared according to the manufacturer’s protocol. In brief, 10 ng DNA per sample was used for the Multiplex PCR Step using the Reaction Mix, and the following Thermocycler Program: 30 sec at 98 °C, 4 cycles of 10 sec at 98 °C, 5 min at 63 °C, 1 min at 65 °C, followed by 21 cycles of 10 sec at 98 °C, 1 min at 64 °C, followed by 1 min at 65 °C and hold at 4 °C. The resulting amplicons were purified using SPRIselect beads (Beckman Coulter, Cat. #B23318) and a DynaMag magnetic rack (Invitrogen). Next, a unique combination of Index D50X + Index D7XX was added to each sample bead pellet, together with the Indexing Reaction Mix (Swift Biosciences). The samples were incubated at 37 °C for 20 min with the lid heating turned off. The libraries were purified once more with SPRIselect beads and quantitated in triplicates by qRT-PCR in a 20 μL reaction using the iQ SYBR Green Supermix (BioRad), containing 10 μL of SYBR Green mix, 10 μL of diluted library (1:1000), and 500 nM of the following primers: 5′ AATGATACGGCGACCACCGAGAT 3′; and 5′ CAAGCAGAAGACGGCATACGA 3′. Serial dilutions of the PhiX Sequencing Control v3, (Illumina Cat. # FC-110-3001) was used as standard. After quantitation, the libraries were normalized to a concentration of 2 nM and pooled together.
Table 1.
Genes included in the analysis. The amplicon panel is from Swift Biosciences “Accel-Amplicon 56G Oncology Panel v2”. The number of amplicons (# ampl) for each gene is shown
| gene name | # amp l | gene name | # amp l |
|---|---|---|---|
|
| |||
| ABL1 | 5 | IDH2 | 2 |
| AKT1 | 2 | JAK2 | 2 |
| ALK | 2 | JAK3 | 3 |
| APC | 9 | KDR | 9 |
| ATM | 19 | KIT | 14 |
| BRAF | 2 | KRAS | 3 |
| CDH1 | 3 | MAP2K1 | 5 |
| CDKN2A | 2 | MET | 6 |
| CSF1R | 2 | MLH1 | 1 |
| CTNNB1 | 1 | MPL | 1 |
| DDR2 | 1 | MSH6 | 4 |
| DNMT3A | 1 | NOTCH1 | 3 |
| EGFR (HER1) | 9 | NPM1 | 1 |
| ERBB2 (HER2) | 4 | NRAS | 3 |
| ERBB4 (HER4) | 8 | PDGFRA | 4 |
| EZH2 | 1 | PIK3CA | 11 |
| FBXW7 | 6 | PTEN | 14 |
| FGFR1 | 2 | PTPN11 | 2 |
| FGFR2 | 4 | RB1 | 12 |
| FGFR3 | 6 | RET | 6 |
| FLT3 | 4 | STK11 | 5 |
| FOXL2 | 1 | SMAD4 | 10 |
| GNA11 | 2 | SMARCB1 | 4 |
| GNAQ | 2 | SMO | 5 |
| GNAS | 2 | SRC | 1 |
| HNF1A | 4 | TP53 (P53) | 21 |
| HRAS | 2 | TSC1 | 1 |
| IDH1 | 1 | VHL | 3 |
MiSeq Loading
The library pool was sequenced using the MiSeq v2 300 cycle Reagent Kit (Illumina). Five μL of the pooled amplicon library was denatured with 5 μL 0.2N NAOH for 5 min at room temperature. The library pool was then diluted to 8 pM with chilled HT1 buffer and 10% PhiX v3 control (Illumina) was spiked into the diluted library pool. Six hundred μL of the diluted pooled library with PhiX spike-in was loaded into the MiSeq reagent cartridge.
Sequencing data analysis
Adapter trimming was conducted per Swift Biosciences recommendation, using cutadapt [14]. Paired-end FASTQ samples were aligned to GRCh38 with BWA-MEM. Sorting and indexing was done using samtools. Base quality score recalibration followed by local re-alignment was done using the GATK Java package in conjunction with dbSNP annotation (b149) [15]. Variant calling was conducted using the LoFreqV2 (LoFreq*) mutation caller [16]. Visualizations were created in R using custom scripts (available on request) employing Bioconductor package VariantAnnotation [17] as well as the plotting framework ggplot2 [18] and further adapted in Excel. After mutations were called, percent representation was established as (number of variant reads)/(number of reference reads + number of variant reads). Variants leading to amino acid changes, with a minimum read count of 5 reads and a frequency above 1% in at least one of the DNA samples are shown. The variant frequencies of the expected mutations that were detected in the DNA standard are shown in the Supplementary Figure 1. Three variants detected in amplicons from patient samples and the DNA standard were discarded as false positives (FGFR1 D166del; MSH6 F1088frameshift and TP53 P72R).
Results
Patient characteristics and analysis approach
Patients with treatment-naïve adenocarcinomas of the colon (n=5) or the pancreas (n=5) that were considered resectable at the time of initial diagnosis were included in this study (Table 2). Plasma samples were collected before surgical removal of the primary tumors and at the time of disease progression. Resected tumor tissues were cryo-sectioned and evaluated by a pathologist to assess cancer cell and stroma abundance. After surgery, patients received different adjuvant therapies that are listed in Table 2. To assess the mutation patterns in early and late stage colon and pancreatic cancer we compared plasma DNA at the time of initial diagnosis to that of the primary tumor, as well to the plasma DNA at the time of metastatic disease (Figure 1). For DNA mutation analysis, we sequenced 30 DNA samples from 10 patients in a single, next generation deep-sequencing run of 263 mutation hotspots in a set of 56 cancer-related genes. The same platform was used for the analysis of tissue and circulating cell-free DNA (ccfDNA) to avoid discrepancies in mutation findings due to variability in sequencing methods or reagents.
Table 2.
Patient characteristics
| Disease | Colon adenocarcinoma | Pancreatic adenocarcinoma | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Patient # | 1 (1A0383) | 2 (1A0390) | 3 (1A0690) | 4 (1A0816) | 5 (1A1118) | 6 (1A0431) | 7 (1A0522) | 8 (1A0551) | 9 (1A0843) | 10 (1A1128) |
| Pathological stage | 3c T3N2M0 | 4a T3N1M1 | 3b T3N2(4/17)M0 | 2 pT4pN0M0 | 4a TxNxM1a | 2b pT3N1M0 | 2b pT3pN2 | 2b pT3N1M0 | 2a pT3N0M0 | 2b T3N1M0 |
| Age [years] | 67 | 62 | 50 | 66 | 51 | 54 | 69 | 66 | 45 | 57 |
| Gender | Male | Female | Female | Male | Male | Male | Female | Male | Female | Female |
| Surgery | Hemicolectomy | Colon tumor resection | Colon resection | Right colectomy | Hemicolectomy | Total pancreatectomy, splenectomy | Whipple | Whippl | Distal pancreatectomy, splenectomy | Distal pancreatectomy, splenectomy |
| Histology tumor scroll | 50% tumor, 50% fibrosis | 70% tumor, 30% fibrosis | 90% tumor, 10% fibrosis | 70% tumor, 30% necrosis | 90% tumor, 10% fibrosis | 30% tumor, 70% fibrosis | 40% tumor, 60% fibrosis | 50% tumor, 50% fibrosis | 50% tumor, 50% fibrosis | 40% tumor, 60% fibrosis |
| Time to metastasis after surgery [months] | 70 | 20 | 12 | 5 | 1 | 16 | 9 | 18 | 15 | 7 |
| Metastatic site | Mesentery, Anastomosis | Liver, Lung | Peritoneum, Liver | Liver | Peritoneum, Liver | Peritoneum, Live | Live | Lung | Ovary | Liver |
| Adjuvant therapy before 2nd plasma sample |
|
|
|
/ | / |
|
|
|
|
|
FOLFOX (Folinic acid + Fluorouracil + Oxaliplatin); FOLFIRI (Folinic acid, Fluorouracil and Irinotecan); GI4000 (vaccine against mutated Ras); Bevacizumab (anti-VEGF-A antibody); 5-FU (Fluorouracil); Veliparib (poly(ADP-ribose) polymerase (PARP) -1 and -2 inhibitor); MEDI-565 (anti-CEA/CD3 antibody; PF05082566 (anti-CD137 stimulating antibody).
Figure 1. Overview of the study.
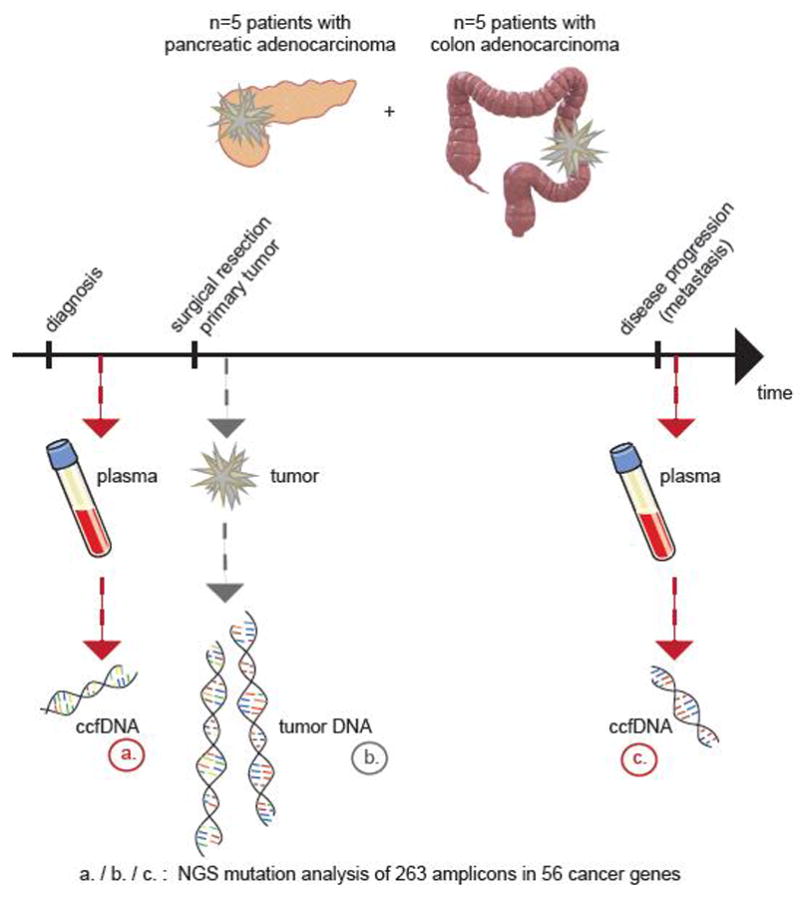
Ten patients were included in the analysis of circulating cell-free DNA (ccfDNA) at diagnosis of primary cancer and at the time of progressive disease. DNA from frozen primary tumors that were collected during surgery were used as a comparison. All 30 DNA samples were subjected to next generation sequencing (NGS) of 56 genes with cancer-associated mutations.
DNA mutations detected
Next generation deep-sequencing of amplicons showed a median depth of 754,000 reads per sample. In 9 of 10 pre-surgery plasma samples genomic alterations with a 1% frequency were detected, despite a relatively low read coverage in one of the ccfDNA samples (patient 9 pre-surgery). One out of 10 late stage plasma samples (ccfDNA patient 4 at metastasis) did not contain genomic alterations above 1% frequency due to hemolysis and wildtype cellular DNA contamination that diluted the ccfDNA. We focused on non-synonymous DNA alterations with a variant frequency of at least 1% in one of the samples per patient. In the 56 genes assessed we found an average of 10 mutations (range 4-15) in 17 genes in the ccfDNA of five colon cancer patients before surgery (Figure 2). The ccfDNA of the five patients with resectable PDAC contained fewer mutations, i.e. an average of 8 (range 5-12) mutations in 14 of the 56 genes assessed.
Figure 2. Circulating cell-free DNA mutation frequency before surgery.

Non-synonymous mutations at ≥1% variant frequency in at least one of the ccfDNA samples. * = premature stop codon; fs =frameshift; del=deletion; − = low coverage.
Surprisingly, the number of mutations detected in the primary tumors was much lower than that in the plasma: In the colon cancer tissues an average of 3 mutations (range 1-4) were found in 7 genes (APC, BRAF, CDKN2A, KIT, KRAS, PTEN, TP53; Figure 3). In the pancreatic cancer tissues 4 mutations (range 3-6) were found in 7 genes (CDNKN2A, JAK3, KDR, KIT, KRAS, SMAD4, TP53; Figure 3). Although our analysis detected mutations in only 7 genes, it is striking that each patient still had a unique combination of genomic alterations.
Figure 3. DNA mutation frequency in primary tumors.
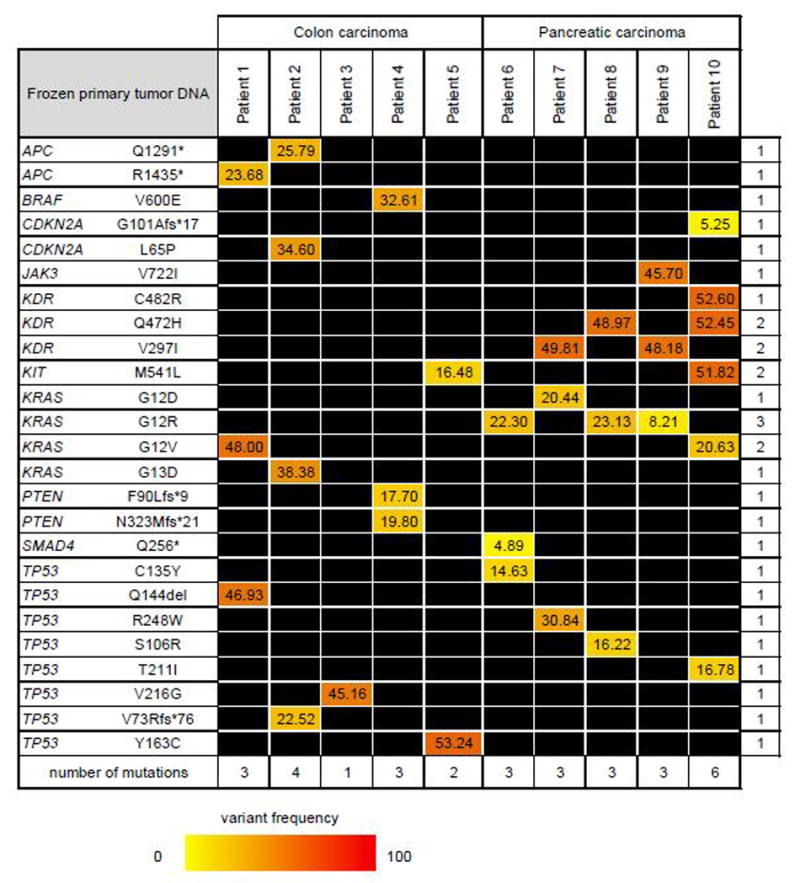
Non-synonymous mutations at ≥1% variant frequency in at least one of the primary tumors. * = premature stop codon; fs =frameshift; del=deletion.
At the time of disease progression, the ccfDNA of five patients with metastasized colon cancer contains an average of 7 mutations (range 2-11) in 16 genes. In the plasma of the patients with metastatic PDAC an average of 9 mutations (range 3-12) in 20 genes (Figure 4). This number is close to the number of ccfDNA mutations before surgery. However, some of the ccfDNA mutations detected at the time of primary disease were lost at the time of metastatic disease, i.e. ABL1, ATM, DNMT3A, FLT3, HNF1A, NRAS and SMAD4. Possible explanations are that these mutations occured in cancer cell subpopulations in the primary cancers that were resected, or that the clones carrying the mutations were selected against during disease progression.
Figure 4. Circulating cell-free DNA mutation frequency at the time of metastasis.
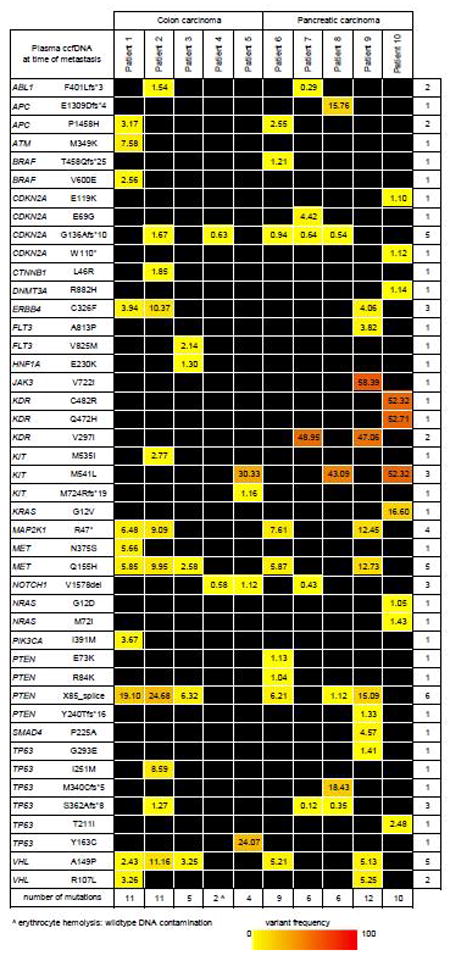
Non-synonymous mutations at ≥1% variant frequency in at least one of the ccfDNA samples. * = premature stop codon; fs =frameshift; del=deletion; ˆ = hemolysis.
Comparison of mutations in primary tumors and the circulation at early and late stage disease
We evaluated the concordance between DNA mutations in the primary tumors and the circulation. For each patient, the mutations above a 1% variant frequency threshold in at least one of the DNA samples are shown in Fig. 5. First we compared the similarity between primary tumor DNA and ccfDNA before surgery (Table 3). In the patients with colon cancer about half (47%) of tumor tissue mutations are also detected in the circulation though there is a wide variation between patients. In patients with primary PDAC this concordance is below one third (28%). On the other hand, it is notable that cancer heterogeneity within each patient was much more evident in the mutational landscape of the ccfDNA than in tumor tissue DNA. The majority of mutations in the ccfDNA were not detected in the primary tumor tissues: 71% in colon cancer and 78% of the mutations in PDAC (Table 3). This finding illustrates that a tissue section of a given tumor can fail to represent the molecular makeup of the entire cancer.
Figure 5. Mutation frequency in ccfDNA and tumors.

The results from individual patients are shown (n=10). Non-synonymous mutations with ≥1% variant frequency in at least one of the ccfDNA samples. * = premature stop codon; fs =frameshift; del=deletion; ˆ = hemolysis; - = low coverage.
Table 3.
Concordance and discordance between the number of mutations detected in plasma and primary tumor DNA at the time of initial diagnosis
| Primary tumor versus plasma at the time of diagnosis | ||||
|---|---|---|---|---|
| Patient | Disease | Tumor tissue DNA mutations detected in plasma samples | Plasma sample DNA mutations not detected in tumor tissues | Cancer cells in tumor |
| 1 | colon AC | 60% (3/5) | 79% (11/14) | 50% |
| 2 | colon AC | 0% (0/5) | 100% (5/5) | 70% |
| 3 | colon AC | 0% (0/1) | 100% (13/13) | 90% |
| 4 | colon AC | 75% (3/4) | 77% (10/13) | 70% |
| 5 | colon AC | 100% (3/3) | 0% (0/3) | 90% |
| average | 47% | 71.2% | 74% | |
| 6 | PDAC | 25% (1/4) | 92% (11/12) | 30% |
| 7 | PDAC | 33% (1/3) | 89% (8/9) | 40% |
| 8 | PDAC | 33% (1/3) | 86% (6/7) | 50% |
| 9 | PDAC | 0% (0/3) | 100% (8/8) | 50% |
| 10 | PDAC | 50% (3/6) | 25% (1/4) | 40% |
| average | 28.2% | 78.4% | 42% | |
AC= adenocarcinoma; PDAC= pancreatic ductal adenocarcinoma
For the current study we had selected a set of patients that developed metastatic disease after removal of the primary tumors. We evaluated the genomic evolution during this progression by comparing the ccfDNA at the time of surgery and metastasis. Table 4 summarizes the differences in the ccfDNA mutational landscape at the time of primary and metastatic cancer. In five patients with metastatic colon cancer, 34% of the ccfDNA mutations were not detected in the circulation at the time of primary disease. These emerging mutations indicate clonal evolution of the disease during malignant progression. In PDAC, the proportion of these metastasis-associated ccfDNA mutations is higher; 63% of the mutations were not detected during primary disease. Interestingly, the fraction of new mutations at the time of metastatic disease is not correlated with the length of time to progression, (Table 4) or the type of adjuvant therapy (Table 2). Although this is a small number of patients, it is noteworthy that approximately half of ccfDNA mutations detected at the time of primary disease were not detected after progression to metastatic disease (45% in colon AC, 59% in PDAC, Table 4). This would indicate that cancer cell subpopulations carrying these mutations were likely dominant in the primary tumor that was removed surgically. Also, these subpopulations did not play a significant role in the metastatic lesions.
Table 4.
Concordance between the number of mutations detected in plasma at the time of initial diagnosis and detection of metastasis
| ccfDNA mutations at the time of primary versus metastatic disease | ||||
|---|---|---|---|---|
| Patient | Disease | Gain of mutations after metastasis | Loss of mutations after metastasis | Time to metastasis [months] |
| 1 | colon AC | 40% (4/10) | 38% (5/14) | 70 |
| 2 | colon AC | 64% (7/11) | 20% (1/5) | 20 |
| 3 | colon AC | 40% (2/5) | 77% (10/13) | 12 |
| 4 | colon AC | 0% (0/1) ˆ | 92% (12/13) | 5 |
| 5 | colon AC | 25% (1/4) | 0% (0/3) | 1 |
| average | 33.8% | 45.4% | 21.6 | |
| 6 | PDAC | 38% (3/8) | 58% (7/12) | 16 |
| 7 | PDAC | 67% (2/3) | 89% (8/9) | 9 |
| 8 | PDAC | 80% (4/5) | 86% (6/7) | 18 |
| 9 | PDAC | 58% (7/12) | 38% (3/8) | 15 |
| 10 | PDAC | 70% (7/10) | 25% (1/4) | 7 |
| average | 62.6% | 59.2% | 13 | |
AC, adenocarcinoma; PDAC, pancreatic ductal adenocarcinoma
hemolysis and wildtype cellular DNA contamination
In conclusion, we found that ccfDNA analysis complements the molecular insight into the genetic make-up of colon and pancreatic cancer and can be particularly helpful in monitoring molecular changes over time.
Discussion
The majority of molecular profiling of human tumors has relied on the analysis of aliquots of cancer tissues obtained from surgical resection specimen. Biopsies of cancerous lesions at disease progression are used rarely due to obvious disadvantages: They are potentially risky invasive procedures, time consuming and expensive. Also, cancer cells in tissue biopsies may be sparse due to the limited size of tissue recovered and against the background of wildtype stromal cells. Most importantly, subpopulations of a heterogeneous tumor may be poorly represented in biopsies. In the current study we sought to assess the changes in the mutational makeup of colon and pancreatic adenocarcinoma between the time of primary tumor surgery and detection of metastatic disease using ccfDNA. In this study we provide a direct comparison of mutation detection in primary tumor DNA and plasma ccfDNA at diagnosis and at disease recurrence. For this, amplicons covering 263 mutations in 56 cancer-associated genes were analyzed by deep-sequencing.
It is thought that ccfDNA can provide a better representation of the molecular makeup of a malignant disease than a single section from a surgical tumor specimen or a tissue biopsy. Also, blood samples can be drawn at deliberate intervals because they only require a minimally invasive procedure. In the present study the notion of a broader molecular representation is supported by the fact that the ccfDNA revealed approximately twice as many mutations as tumor tissue DNA. Very likely the tissue sections analyzed missed portions of the primary tumor that carried subpopulations with these additional mutations. It is also conceivable that patients had already developed occult metastatic disease at the time of the initial diagnosis and the additional ccfDNA mutations found represented the cancer cell subpopulations in the metastatic lesions. It appears that the ccfDNA provides a more complex picture of the disease.
Most studies focus on the presence of ccfDNA mutations in one or two genes to compare their presence to clinical outcome [19, 20]. In our study, we sought to assess the mutation patterns in a broad set of genes to highlight tumor heterogeneity and demonstrate clonal evolution over the course of disease progression. In colon cancer, for example, ccfDNA has been used to track clonal evolution during treatment with the epidermal growth factor receptor (EGFR)-specific antibodies. Alterations in KRAS, NRAS, MET, ERBB2, FLT3, EGFR and MAP2K1 were detected in ccfDNA of patients with primary or acquired resistance to EGFR blockade [21]. Using a broad panel of cancer-associated genes rather than frequently altered candidate oncogenes such as KRAS overcame one of the potential pitfalls encountered. None of the KRAS mutations from the seven patients with KRAS mutant primary tumors were detected in the ccfDNA at the time of diagnosis, while mutations in other genes were detected. Mutant KRAS ccfDNA was, however, found by others in 10 of 34 pancreatic patients (29%) [22] or in 136 of 188 (72.3%) of patients with metastatic PDAC [23]. Another study showed that mutant KRAS ccfDNA was detected in 14.8%, 45.5%, 30.8%, and 57.9% of age-matched controls, localized, locally advanced, and metastatic PDAC patients, respectively [20]. In circulating exosomal DNA the percentages of mutant KRAS in these groups were even higher, i.e. 7.4%, 66.7%, 80%, and 85% respectively [20].
At the time of surgical removal of the primary tumors between 28% and 47% of tumor mutations were also detected in ccfDNA. Others have found that in formalin-fixed paraffin-embedded cancer tissues and plasma from patients with different types of cancer an overall concordance of 60% in mutations of 19 genes analyzed [24]. Thus, the concordance found here is relatively low. In contrast, over 70% of mutations detected in ccfDNA were not found in the tumor samples. This could be due to metastatic disease at the time of diagnosis or poor representation of the primary tumor composition by the histological section obtained for the DNA analysis.
Shed DNA in the circulation is fragmented into relatively short size fragments with tumor-derived DNA exhibiting even higher fragmentation than normal cellular DNA [25]. This was also found for specific examples such as the BRAF V600E mutant allele with a fragment size <145 base pairs. Indeed detection of EGFR T790M mutant DNA in the circulation of cancer patients was improved by selecting for shorter DNA fragment lengths [26]. Comparisons of tumor tissue somatic DNA and ccfDNA mutation rate can be impacted by the application of different sequencing technologies and amplicon sizes [27, 28] as well as read depths and these technical issues may bias the data interpretation [22]. To avoid this pitfall we used a platform that is adapted to the detection of short DNA fragments found as circulating cell-free DNA. Thus, we sought to avoid differences in amplicon generation, library preparation or sequencing depth and all samples were subjected to the same amplicon and library generation protocol and analyzed in parallel.
We also compared the clonal evolution of cancer in the ccfDNA mutations over the course of disease progression. After metastasis, new ccfDNA mutations are gained both in colon (33.8%) and pancreatic cancer (62.6%) and were not detected at the time of diagnosis of the primary cancer. This indicates clonal selection due to treatment, malignant progression or metastatic spread to different tissues with different microenvironmental selection pressure. Complementary to the gain of mutations after metastasis, we also observed a loss of approximately half of the ccfDNA mutations. This loss of cancer subpopulations will be due to surgical removal of the primary tumor as well as patient treatment.
One final caveat in ccfDNA mutation analysis is the assignment of mutant DNA to cancer lesions under study rather than spontaneous mutations that occured in other tissues and have no disease relevance. Recently reported analysis of mutation accumulation in human adult stem cells in different tissues showed that on average 40 new mutations arise per year during the life time of an individual [29]. In a study focused on the analysis of human skin, biopsies of physiologically normal skin showed 2 to 6 mutations per million bases per cell and matched to a large extent with cancer-associated mutations [30]. Notably, the frequency of mutations seen in normal skin is within the median range of 1 - 10 somatic mutations observed for human cancers [1]. We did not find detailled reports on ccfDNA mutation frequencies in healthy individuals but the occurence of spontaneous mutations in healthy tissues provide a caveat on interpreting mutant ccfDNA as evidence of the presence of a cancerous lesion.
In conclusion, we found that ccfDNA appears to represent the heterogeneity of colon and pancreatic cancer more extensively than tumor tissue DNA. The analysis of a relatively broad panel of cancer-related genes is feasible for ccfDNA and would allow monitoring of changes in the molecular makeup over time and under therapy. A challenge will be to what extent altered mutation patterns in ccfDNA could also prompt a change in treatment.
Supplementary Material
Highlights.
Cancer is a heterogeneous disease harboring diverse subclonal populations with different DNA mutations. We used circulating cell-free DNA (ccfDNA) to assess the mutational makeup and monitor changes during disease progression of pancreatic and colorectal cancers after surgery. A 56 cancer-associated gene panel showed that less than half of the mutations in the primary tumors were also detected in the ccfDNA. Also, additional mutations not detected in the primary tumors were found in the ccfDNA due to disease heterogeneity or metastatic spread at the time of diagnosis. At the time of disease progression 3-5 new mutations were detected in ccfDNA. We conclude that ccfDNA can represent the heterogeneity of colon and pancreatic cancer more extensively than tumor tissue DNA and allow for monitoring of changes in the molecular makeup during disease progression and therapy.
Acknowledgments
We thank the patients who donated blood and tissue samples for molecular analysis. We also thank pathologist Brent Harris, MD PhD (Georgetown University) for evaluating the tumors and Sarah Martinez Roth, MS (Georgetown University) for help with the experiments. EEV, AW, GG and JLM designed the research study, EEV, AJ and JNM performed the research, GTG and EEV analyzed the data, EEV and AW wrote the manuscript. This research was funded by the Ruesch Center for the Cure of GI Cancers (EEV, AW, JLM) and P30 CA51008 (AW)
Footnotes
Conflicts of interest
None of the authors have conflicts to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jager N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, Lopez-Otin C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdes-Mas R, van Buuren MM, van ’t Veer L, Vincent-Salomon A, Waddell N, Yates LR, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, Stratton MR. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, Mcdonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nature Reviews Cancer. 2012;12:323–34. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 4.Schiavon G, Hrebien S, Garcia-Murillas I, Cutts RJ, Pearson A, Tarazona N, Fenwick K, Kozarewa I, Lopez-Knowles E, Ribas R, Nerurkar A, Osin P, Chandarlapaty S, Martin L-A, Dowsett M, Smith IE, Turner NC. Analysis of ESR1 mutation in circulating tumor DNA demonstrates evolution during therapy for metastatic breast cancer. Sci Transl Med. 2015;7:313ra182. doi: 10.1126/scitranslmed.aac7551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, Cheang M, Osin P, Nerurkar A, Kozarewa I, Garrido JA, Dowsett M, Reis-Filho JS, Smith IE, Turner NC. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med. 2015;7:302ra133. doi: 10.1126/scitranslmed.aab0021. [DOI] [PubMed] [Google Scholar]
- 6.Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA, Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. PNAS. 2005;102:16368–73. doi: 10.1073/pnas.0507904102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–90. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10:472–84. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 9.Haber DA, Velculescu VE. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discovery. 2014;4:650–61. doi: 10.1158/2159-8290.CD-13-1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosell R, Karachaliou N. Lung cancer: Using ctDNA to track EGFR and KRAS mutations in advanced-stage disease. Nature Reviews Clinical Oncology. 2016;13:401–2. doi: 10.1038/nrclinonc.2016.83. [DOI] [PubMed] [Google Scholar]
- 11.Alix-Panabières C, Pantel K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discovery. 2016;6:479–91. doi: 10.1158/2159-8290.CD-15-1483. [DOI] [PubMed] [Google Scholar]
- 12.Rapisuwon S, Vietsch EE, Wellstein A. Circulating Biomarkers to Monitor Cancer Progression and Treatment. Computational and Structural Biology Journal. 2016;14:211–22. doi: 10.1016/j.csbj.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vietsch EE, van Eijck CH, Wellstein A. Circulating DNA and micro-RNA in patients with pancreatic cancer. Pancreatic Disorders & Therapy. 2015;5:1–11. doi: 10.4172/2165-7092.1000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011;17 [Google Scholar]
- 15.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilm A, Aw PP, Bertrand D, Yeo GH, Ong SH, Wong CH, Khor CC, Petric R, Hibberd ML, Nagarajan N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012;40:11189–201. doi: 10.1093/nar/gks918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, Morgan M. VariantAnnotation : a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics. 2014;30:2076–8. doi: 10.1093/bioinformatics/btu168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietrasz D, Pecuchet N, Garlan F, Didelot A, Dubreuil O, Doat S, Imbert-Bismut F, Karoui M, Vaillant JC, Taly V, Laurent-Puig P, Bachet JB. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clin Cancer Res. 2017;23:116–23. doi: 10.1158/1078-0432.CCR-16-0806. [DOI] [PubMed] [Google Scholar]
- 19.Scholer LV, Reinert T, Orntoft MW, Kassentoft CG, Arnadottir SS, Vang S, Nordentoft I, Knudsen M, Lamy P, Andreasen D, Mortensen FV, Knudsen AR, Stribolt K, Sivesgaard K, Mouritzen P, Nielsen HJ, Laurberg S, Orntoft TF, Andersen CL. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-17-0510. [DOI] [PubMed] [Google Scholar]
- 20.Allenson K, Castillo J, San Lucas FA, Scelo G, Kim DU, Bernard V, Davis G, Kumar T, Katz M, Overman MJ, Foretova L, Fabianova E, Holcatova I, Janout V, Meric-Bernstam F, Gascoyne P, Wistuba I, Varadhachary G, Brennan P, Hanash S, Li D, Maitra A, Alvarez H. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol. 2017;28:741–7. doi: 10.1093/annonc/mdx004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, Lamba S, Hobor S, Avallone A, Valtorta E, Rospo G, Medico E, Motta V, Antoniotti C, Tatangelo F, Bellosillo B, Veronese S, Budillon A, Montagut C, Racca P, Marsoni S, Falcone A, Corcoran RB, Di Nicolantonio F, Loupakis F, Siena S, Sartore-Bianchi A, Bardelli A. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pishvaian MJ, Bender RJ, Matrisian LM, Rahib L, Hendifar A, Hoos WA, Mikhail S, Chung V, Picozzi V, Heartwell C, Mason K, Varieur K, Aberra M, Madhavan S, Petricoin E, 3rd, Brody JR. A pilot study evaluating concordance between blood-based and patient-matched tumor molecular testing within pancreatic cancer patients participating in the Know Your Tumor (KYT) initiative. Oncotarget. 2016 doi: 10.18632/oncotarget.13225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng H, Liu C, Jiang J, Luo G, Lu Y, Jin K, Guo M, Zhang Z, Xu J, Liu L, Ni Q, Yu X. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int J Cancer. 2017;140:2344–50. doi: 10.1002/ijc.30650. [DOI] [PubMed] [Google Scholar]
- 24.Perkins G, Yap TA, Pope L, Cassidy AM, Dukes JP, Riisnaes R, Massard C, Cassier PA, Miranda S, Clark J, Denholm KA, Thway K, Gonzalez De Castro D, Attard G, Molife LR, Kaye SB, Banerji U, de Bono JS. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PLoS One. 2012;7:e47020. doi: 10.1371/journal.pone.0047020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, Gongora C, Thierry AR. High fragmentation characterizes tumour-derived circulating DNA. PloS one. 2011;6:e23418. doi: 10.1371/journal.pone.0023418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Underhill HR, Kitzman JO, Hellwig S, Welker NC, Daza R, Baker DN, Gligorich KM, Rostomily RC, Bronner MP, Shendure J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016;12:e1006162. doi: 10.1371/journal.pgen.1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang P, Chan CW-M, Chan KCA, Cheng SH, Wong J, Wong VW-S, Wong GLH, Chan SL, Mok TSK, Chan HLY, Lai PB-S, Chiu RWK, Lo YMD. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. PNAS. 2015;112:E1317–25. doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mouliere F, Rosenfeld N. Circulating tumor-derived DNA is shorter than somatic DNA in plasma. PNAS. 2015;112:3178–9. doi: 10.1073/pnas.1501321112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EES, Verstegen MMA, van der Laan LJW, de Jonge J, Ijzermans JNM, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, Van Boxtel R. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–4. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, Wedge DC, Fullam A, Alexandrov LB, Tubio JM, Stebbings L, Menzies A, Widaa S, Stratton MR, Jones PH, Campbell PJ. Tumor evolution High burden and pervasive positive selection of somatic mutations in normal human skin. Science (New York, NY) 2015;348:880–6. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.