

Abstract
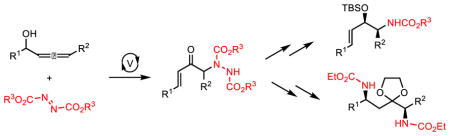
A vanadium catalyzed 1,3-rearrarngement of allenols to form transient vanadium enolates that selectively couple with electrophilic nitrogen sources is reported even in the presence of competing simple protonation and Alder-ene pathways. The hydrazine products can be cyclized in a 6-endo-trig fashion which, upon reductive cleavage of the N-N bond, yield 1,4-diamines. Additionally, cleavage of the N-N bond before cyclization can be achieved to form β-hydroxy amines, a common structural motif of biologically active compounds.
Graphical Abstract
Enolates are perhaps the most fundamental and broadly applied reactive intermediate for the formation of chemical bonds. While significant progress has been made over the last several decades addressing the requirement of stoichiometric enolate activation for aldehydes and certain classes of activated ketones, a significant need for the development of highly chemo- and regioselective methods for the generation of ketone enolates that also avoid cryogenic temperatures, strong pyrophoric bases, and stoichiometric activation still remains.1
Our group has pioneered such a mild, chemo- and regioselective method for the catalytic formation of ketone enolates from the vanadium catalyzed 1,3-rearrangement of allenols.2 This atom economic methodology relies upon the sigmatropic rearrangement of an allenol-metal oxo complex to form a metal enolate that undergoes subsequent trapping with a non-proton electrophile. Such a process, which has yet to be given a formal name, may be refered to as sigmatropic functionalization. The challenge with developing such methodology is that electrophilic trapping of the transient metal enolate must compete with simple protonation which results in formation of the Meyer-Schuster product.3
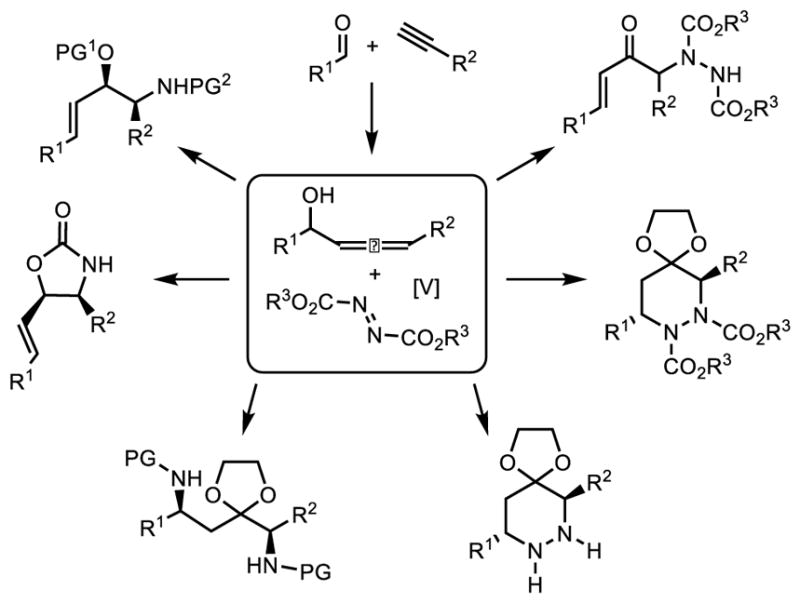
One important aspect of enolate chemistry is the selective introduction of heteroatoms, especially nitrogen, due to the importance and ubiquitous nature of the carbon-heteroatom bond in pharmaceutical and biologically active compounds.4 However, the ability of nitrogen to coordinate to vanadium raises questions about whether such basic entities would be compatible with sigmatropic functionalization, which relies upon the Lewis acidity of the vanadium catalyst for reactivity. Additionally, we were aware of the unique challenge that would be posed by the known, direct stoichiometric Alder-ene reaction of allenes with azo compounds that could be competitive with the catalytic 1,3-transposition (Figure 1).5 As such, we sought to answer these questions and determine whether the concept of sigmatropic functionalization can be expanded to include electrophilic amination. Such a process would generate synthetically useful α-amino-α′,β′-unsaturated ketones whose dense functionality would serve as important chemical building blocks.
Figure 1.
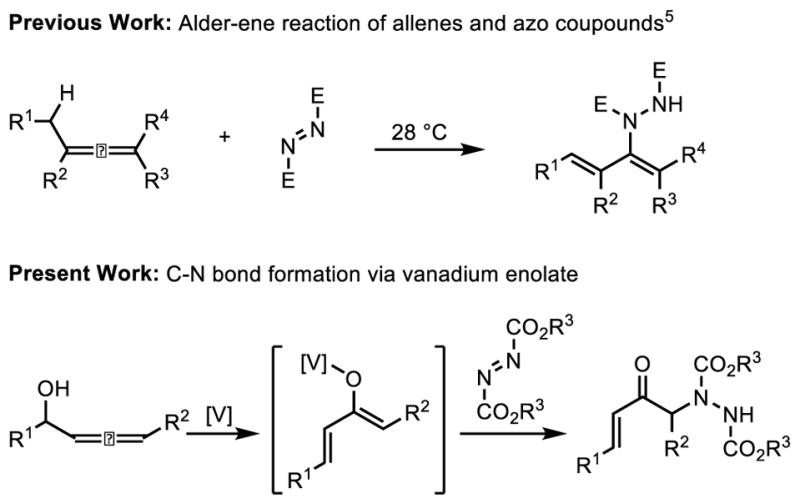
Sigmatropic Functionalization Extended to C-N Bond Formation.a
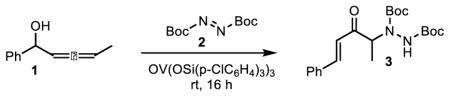
We initiated our studies on allenol 1. While nitroso electrophiles resulted in inhibition of the desired 1,3-transposition, dialkyl azodicarboxylates proved to be effective trapping agents.6 The reaction of allenic alcohol 1 with di-tert-butyl azodicarboxylate (1.4 equiv) in the presence of 7 mol % VO(OSi(p-ClC6H4)3)3 in 1,2-dichloroethane (DCE) (0.5 M) at room temperature was finished in 16 h and resulted in an isolated yield of 92% of desired product 3 with no trace of Alder-ene product (Table 1, entry 1). Reducing the electrophile loading to 1.2 or 1.0 equiv. resulted in a lower isolated yield as did switching catalysts to the more electron rich OV(OSiPh3)3 (Table 1, entries 2–4). The catalyst loading could be reduced to as little as 2.5 mol % while increasing concentration to 1.0 M without any impact on yield although further reductions in loading resulted in a modest drop in yield (Table 1, entries 5–8). Finally, a brief screen of solvents showed DCE to be the ideal solvent, although it is worth noting that toluene could be used with only a modest reduction in yield (Table 1, entries 9–10).
Table 1.
Optimization Studies for the Sigmatropic Amination of Allenols.a
| |||||
|---|---|---|---|---|---|
| entry | catalyst loading (mol %) | solvent | concn of 1 (M) | equiv of 2 | isolated yield of 3 (%) |
| 1 | 7 | DCE | 0.5 | 1.4 | 92 |
| 2 | 7 | DCE | 0.5 | 1.2 | 82 |
| 3 | 7 | DCE | 0.5 | 1.0 | 77 |
| 4b | 7 | DCE | 0.5 | 1.4 | 80 |
| 5 | 5 | DCE | 0.5 | 1.4 | 88 |
| 6 | 2.5 | DCE | 1.0 | 1.4 | 94 |
| 7 | 1 | DCE | 1.0 | 1.4 | 78 |
| 8 | 1 | DCE | 2.5 | 1.4 | 67 |
| 9 | 2.5 | PhMe | 1.0 | 1.4 | 77 |
| 10 | 2.5 | THF | 1.0 | 1.4 | 28 |
Unless otherwise noted, reactions were conducted with catalyst OV(OSi(p-ClC6H4)3)3 and 1 equiv of 1. Reaction times are 16 h.
OV(OSiPh3)3 used as catalyst.
With a set of optimized conditions in hand (Table 1, entry 6), the scope of the reaction was explored. Steric bulk of the dialkyl group on the azodicarboxylate did not impact yield (Scheme 1, 3–6). Additionally, the reaction was amenable to scale up with no loss of yield observed when performed on a 2 mmol scale (6). Increasing the steric bulk of the R2 group on the allenol was well tolerated but surprisingly resulted in a slight decrease in the E:Z ratio (7 and 8), the opposite of what one may have expected. Replacement of the R2 group with a hydrogen resulted in excellent yield but required 5 mol % catalyst to ensure complete conversion (9). Allenols containing dialkyl substitution at R1 were activated enough to undergo the vanadium catalyzed 1,3-transposition under the originally developed reaction conditions (10–12). However, the 1,3-transposition proved sluggish when R1 was switched to a monoalkyl group and required slightly elevated temperatures as well as electrophile and catalyst loadings to obtain complete conversion and good yields (13–14). The sluggish nature of the monoalkyl R1 substituents can be explained by the development of a partial positive charge on the carbinol carbon during the transition state of the 1,3-transposition. This partial positive is less stabilized by a monoalkyl R1 than by an aromatic or dialkyl R1. More electron rich aromatic groups were tolerated, albeit with lower yields (15 and 16). In the case of R1 being indole, the substrate was too activated towards transposition and required a slow addition of the allenol to the reaction mixture in order to obtain an acceptable yield. Finally, both protected and free alcohols were tolerated (17 and 18).
Scheme 1. Reaction Scope of the Sigmatropic Amination of Allenols.a.

aUnless otherwise noted, reactions were conducted using 2.5 mol % OV(OSi(p-ClC6H4)3)3, 1 equiv of allenol and 1.4 equiv of dial-kyl azodicarboxylate in DCE (1.0 M) at rt. Product E/Z ratio >20/1 unless otherwise specified bReaction performed with 5 mol % catalyst. cReaction performed at the 2.0 mmol scale. dReaction performed at 45 °C. eReaction performed with 2.0 equiv of dialkyl azodicarboxylate. fSlow addition of the allenol over three hours. gReaction performed at 35 °C.
The unique juxtaposition of the hydrazine and enone moiety of our products led us to explore methods for their 6-endo-trig cyclization and subsequent reductive N-N bond cleavage to form 1,4-diamine products. While the corresponding 6-endo-trig cyclization was known for simple amines onto enones, the corresponding cyclization of a hydrazine was unknown. After failure to obtain any significant amount of cyclized product under Brønstead basic, Brønstead acidic, or Lewis acidic conditions, we were excited to see excellent conversion and diastereoselectivity under oxonium activation of the enone. This was best accomplished through the use of Noyori’s combination of 1,2-bis[(trimethylsilyl)oxy]ethane (BTSE) and catalytic trimethylsilyl trifluoromethanesulfonate (TMSOTf) at 45 °C in dichloromethane (DCM) for 16 h in a sealed tube (Scheme 2).7 While ethyl, isopropyl and benzyl carbamate protected hydrazines were well tolerated, use of a tert-butyl carbamate only resulted in decomposition of the starting material (19–21). In the case of the benzyl carbamate (Cbz), partial conversion as well as some mono-deprotected starting material were observed under the standard set of conditions. Switching to toluene at 65 °C resulted in complete conversion with a significant reduction in the amount of deprotected material recovered (21). Utilizing a more sterically demanding isopropyl group at R2 resulted in no change in the reactivity while changing R1 from an aromatic substituent to a dialkyl substituent was likewise tolerated (22 and 23). In the case of spirocycle product 24, little conversion was obtained under standard conditions, but increasing the temperature to 80 °C in toluene resulted in complete conversion and good yield. In all cases, the diastereoselectivity of this process was greater than 20:1 and was unambiguously assigned through the use of nOe experiments.
Scheme 2. Reaction Scope of the 6-endo-trig Cyclization of the Hydrazine Functionality onto the Enone.a.
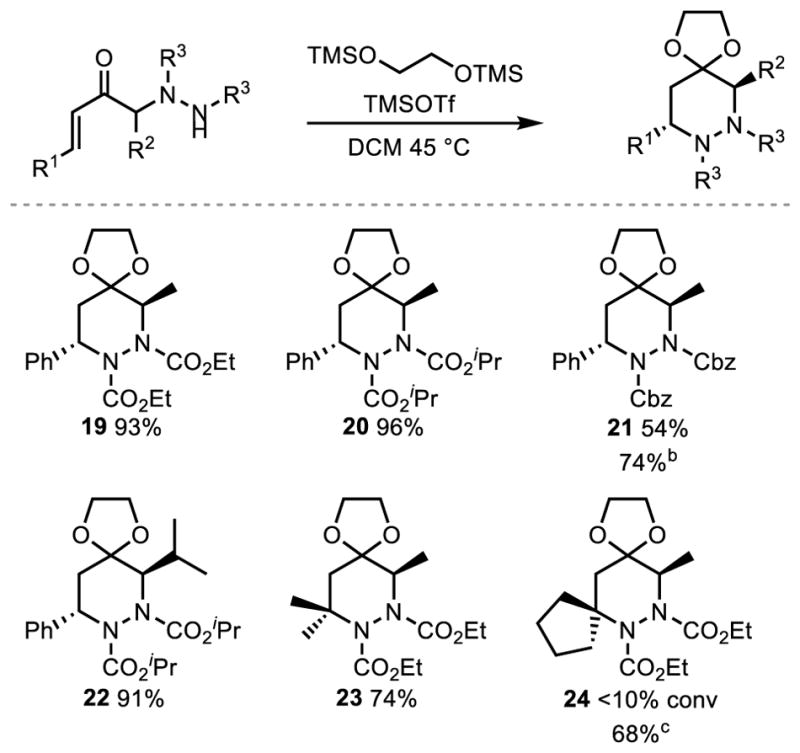
aUnless otherwise noted, reactions were conducted with 10 mol % TMSOTf, 2 equiv of BTSE and 1 equiv of ketone in DCM in a sealed tube 45 °C for 16 h. In all cases, the diastereoselectivity was >20:1. bReaction performed in toluene at 65 °C. cReacton performed in toluene at 80 °C.
The final step of this amination technique requires the cleavage of the hydrazine to reveal the desired amine products. Direct cleavage of the hydrazine N-N bond of the originally formed α′-hydrazino-α,β-unsaturated ketones proved challenging. Utilization of traditional single electron reduction conditions led to either decomposition, a mixture of 1,2- and 1,4- ketone reduction products, or a lack of reactivity. Similarly, utilization of Magnus’s alkylation followed by E1cB elimination resulted only in the formation of an α-iminoketone that underwent subsequent decomposition.8 To overcome these issues, enone 3 was reduced via the Luche reduction in 5.5:1 d.r. followed by protection of the alcohol to form either 25 or 26. From the protected alcohol, Magnus’s two step alkylation E1cB elimination yielded the desired β-hydroxy amines 27 and oxazolidone 28, respectively, in good yield (Scheme 3).
Scheme 3.
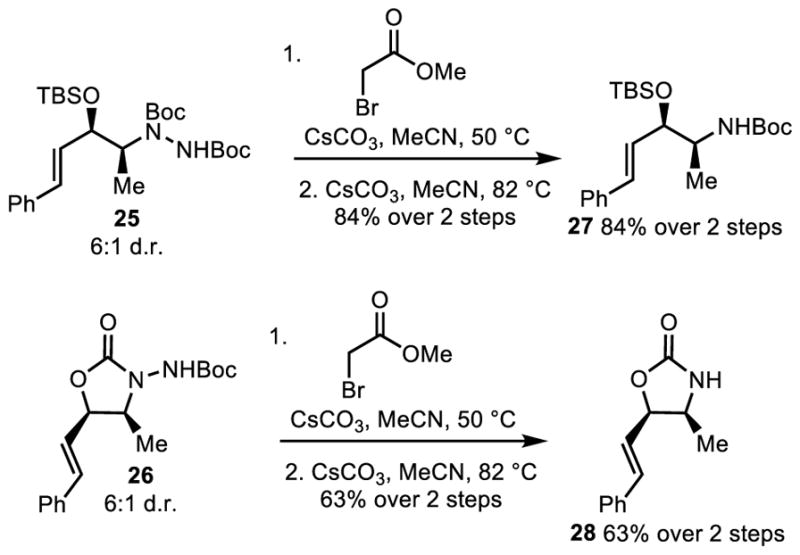
Acyclic Hydrazine N-N Bond Cleavage.
The cyclic hydrazine was also found to be resistant to most single electron reductions as well as hydrogenations. However, when the Cbz protected cyclic hydrazine 21 was subjected to Raney-nickel hydrogenolysis, the free hydrazine could be isolated in good yield. (Scheme 4). These types of cyclic hydrazines are important building blocks for biologically active compounds and are seen heavily in the pinoxaden class of herbicidal compounds reported by Syngenta.9
Scheme 4.
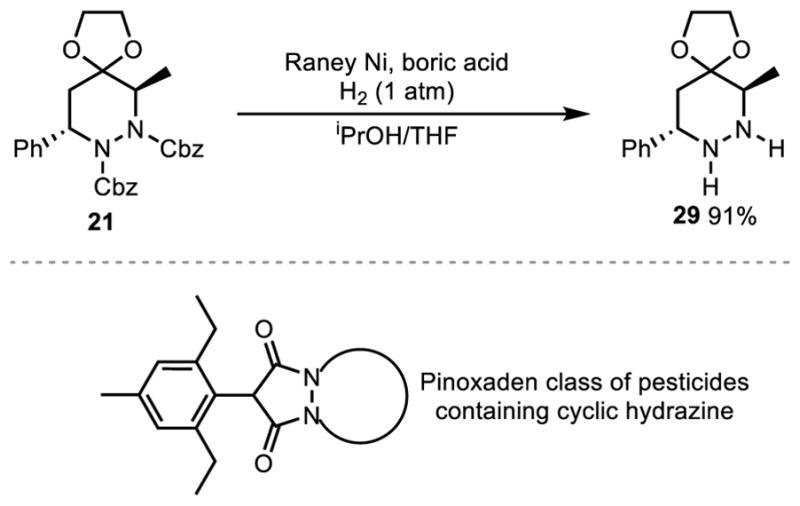
Raney Nickel Hydrogenolysis.
Further treatment of free hydrazine 29 with powdered zinc in acetic acid with 2 equivalents of trifloroacetic acid (TFA) followed by protection of the free amines resulted in isolation of desired 1,4-diamine 30 in 37% yield over three steps starting from protected hydrazine 21 (Table 2, entry 1). Hypothesizing that the reductive cleavage may initiate through a single electron transfer to the doubly protonated hydrazine dication, we proceeded to screen the amount of added TFA in order to modulate the reaction pH. Consistent with this hypothesis, use of 15 volume percent TFA relative to DCM resulted in an improved yield of 50% over three steps (Table 2, entry 2). Further decreasing reaction pH through the addition of 100 volume % of TFA relative to DCM resulted in a diminished yield (Table 3, entry 3).
Table 2.
Optimization Studies for the Reductive Cleavage of Cyclic Hydrazines.a
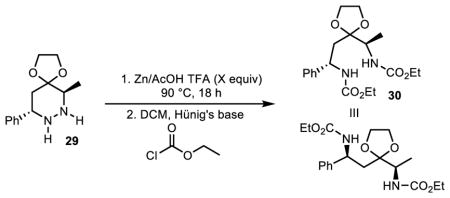
| ||
|---|---|---|
| entry | amount of TFA | yield of 30 over three steps from 21 (%) |
| 1 | 2 equiv | 37 |
| 2 | 15 volume % | 50 |
| 3 | 100 volume % | 22 |
The proposed catalytic cycle for this process is depicted in Scheme 5. Ligand exchange of the vanadium oxo catalyst 31 with allenol 32 generates vanadium-allenol complex 33. Subsequent 1,3-transposition results in vanadium enolate 34 that can undergo simple protonation to give the undesired Meyer-Schuster product 35 or undergo coupling with the dialkyl azodicarboxylate to form vanadium complex 36. Protonation of complex 36 results in generation of the desired coupling product 37 as well as regeneration of the catalyst.
Scheme 5.
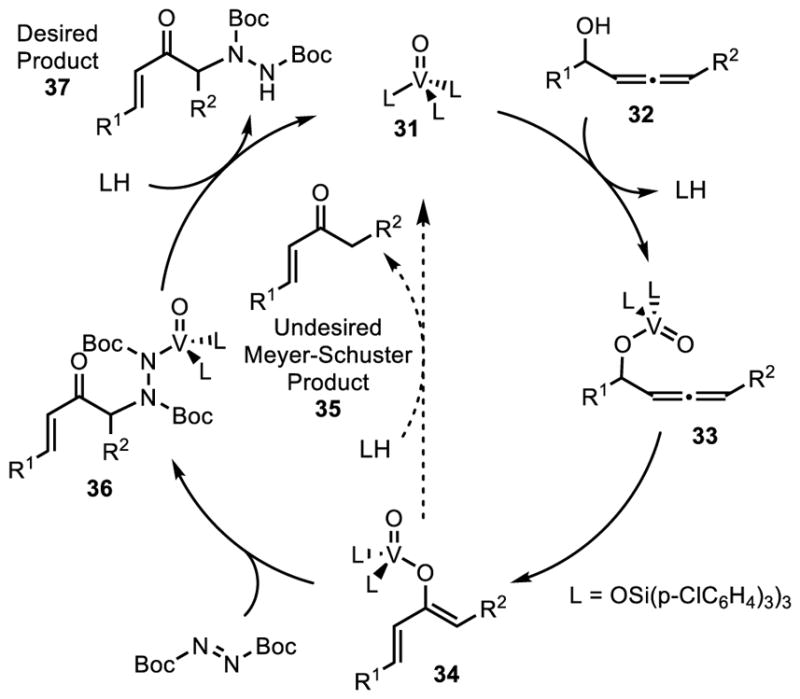
Proposed Catalytic Cycle.
In conclusion, we have developed a mild, chemo- and regioselective method for the catalytic in-situ generation of vanadium enolates starting from allenols for the formation of C-N bonds. Particularly noteworthy is the ability to introduce an acylic 1,4-diamino functionality with near perfect diastereoselectivity via a new 6-endo-trig cyclization (Scheme 6). This process represents the first time that sigmatropic functionalization has been used to form carbon nitrogen bonds. Additionally, the allenol starting materials can be rapidly accessed in as little as one step starting from commercially available alkynes and aldehydes.10 Thus, sigmatropic functionalization methodology provides a practical and convenient alternative approach to elaborated α-substituted-α′,β′-unsaturated ketones.
Scheme 6.
Useful Products Generated From the Sigma-tropic Amination of Allenols.
Supplementary Material
Acknowledgments
We thank the NIH GM-033049and NSF CHE-1360634 for their generous support of our programs. J.S.T. acknowledges support from the John Stauffer Memorial Fellowship.
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details and spectroscopic data (PDF)
References
- 1.For selected reviews, see Mahrwald R. Modern Methods in Stereoselective Aldol Reactions. Wiley-VCH; Weinheim: 2013. Notz W, Tanaka F, Barbas CF., III Acc Chem Res. 2004;37:580–591. doi: 10.1021/ar0300468.Mukherjee S, Yang JW, Hoffman S, List B. Chem Rev. 2007;107:5471–5569. doi: 10.1021/cr0684016.MacMillan DWC. Nature. 2008;455:304–308. doi: 10.1038/nature07367.Shibasaki M, Kanai M, Matsunaga S, Kumagai N. Acc Chem Res. 2009;42:1117–1127. doi: 10.1021/ar9000108.Trost BM, Brindle CS. Chem Soc Rev. 2010;39:1600–1632. doi: 10.1039/b923537j.
- 2.(a) Trost BM, Jonasson C, Wuchrer M. J Am Chem Soc. 2001;123:12736–12737. doi: 10.1021/ja011351w. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Tracy JS. Chem Eur J. 2015;21:15108–15112. doi: 10.1002/chem.201502973. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Jonasson C. Angew Chem Int Ed. 2003;42:2063–2066. doi: 10.1002/anie.200350890. [DOI] [PubMed] [Google Scholar]
- 3.For a recent review on the metal-catalyzed Meyer-Schuster rearrangement see: Cadierno V, Crochet P, García-Garrido SE, Gimeno J. Dalton Trans. 2010;39:4015–4031. doi: 10.1039/b923602c.
- 4.Hili R, Yudin AK. Nat Chem Biol. 2006;2:284–287. doi: 10.1038/nchembio0606-284. [DOI] [PubMed] [Google Scholar]
- 5.(a) Lee CB, Newman JJ, Taylor DR. J Chem Soc, Perkin Trans I. 1978;10:1161–1168. [Google Scholar]; (b) Sabbasani VR, Huang G, Xia Y, Lee D. Chem Eur J. 2015;21:17210–17214. doi: 10.1002/chem.201503243. [DOI] [PubMed] [Google Scholar]
- 6.For a recent review of dialkyl azodicarboxylates as electrophiles see: Nair V, Biju AT, Mathew SC, Badu BP. Chem Asian J. 2008;3:810–820. doi: 10.1002/asia.200700341.
- 7.Tsunoda T, Suzuki M, Noyori R. Tetrahedron Lett. 1980;21:1357. [Google Scholar]
- 8.Magnus P, Garizi N, Seibert KA, Ornholt A. Org Lett. 2009;11:5646–5648. doi: 10.1021/ol902313v. [DOI] [PubMed] [Google Scholar]
- 9.Muehlebach M, Boeger M, Cederbaum F, Cornes D, Friedmann AA, Glock J, Niderman T, Stoller A, Wagner T. Bioorg Med Chem. 2009;4241–4256 doi: 10.1016/j.bmc.2008.12.062. [DOI] [PubMed] [Google Scholar]
- 10.While there are a number of one-step syntheses available, we settled on a three step synthesis by Landor due to its robustness and ability to rapidly generate scope. Select one-step syntheses Mundal DA, Lutz KE, Thomson RJ. J Am Chem Soc. 2012;134:5782–5785. doi: 10.1021/ja301489n.Kuang J, Luo H, Ma S. Adv Synth Catal. 2012;354:933–944.Three step Landor procedure: Cowie JS, Landor PD, Landor SR. J Chem Soc Perkin Trans 1. 1973:720–724.. For recent reviews on allene synthesis see: Yu S, Ma S. Chem Commun. 2011;47:5384–5418. doi: 10.1039/c0cc05640e.Neff RK, Frantz DE. ACS Catal. 2014;4:519–528.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.