

Abstract
Hepatitis C Virus(HCV) represents a significant global disease burden with an estimated 130 – 150 million people worldwide living with chronic HCV infection. Within the 6 major clinical HCV genotypes, genotype 3 represents 22–30% of all infection, and is described as a unique entity with higher rates of steatosis, faster progression to cirrhosis, and higher rates of hepatocellular carcinoma. Hepatic steatosis in the setting of hepatitis C genotype 3 (HCV-3) is driven by viral influence on three major pathways: microsomal triglyceride transfer protein, sterol regulatory element-binding protein-1c, and peroxisome proliferator associated receptor-α. Historically with DAAs, the rates of cure for HCV-3 therapies lagged behind the other genotypes. As current therapies for HCV genotype 3 continue to close this gap, it is important to be cognizant of common drug interactions such as acid suppressing medication and amiodarone. In this review, we discuss the rates of steatosis in HCV-3, the mechanisms behind HCV-3 specific steatosis, and current and future therapies.
Introduction
Hepatitis C virus (HCV) represents a significant global burden of disease with an estimated 130 – 150 million people worldwide living with chronic HCV (CHC)infection[1, 2]. There are six major clinical HCV genotypes and over 50 subtypes; however, genotype 3 infection represents a unique entity, with higher rates of steatosis and more rapid fibrosis progression [3]. In the direct acting antiviral (DAA) era, cure rates for genotype 3 infection have lagged behind the other genotypes until the approval of daclatasvir and sofosbuvir in 2015 and more recently, the approval of the fixed dose combination sofosbuvir and velpatasvir[4, 5]. This review will discuss the pathogenesis of accelerated fibrosis and current treatment options for HCV genotype 3 infection.
1 Epidemiology
Globally, HCV genotype 3 (HCV-3) infection accounts for 22–30% of all HCV infection, second only to HCV genotype 1 (HCV-1) infection(Figure 1)[6, 7]. The highest global prevalence is in South and Central Asia, where it represents 71.6% of all HCV infection. In Western Europe the overall prevalence of HCV-3 is 24.8%, with Norway (50%), England (47%), Finland (46%), and Denmark (43%) among those countries with the highest prevalence[6]. South America is the next geographic region of highest HCV-3 prevalence at 26.9%, with rates of up to 30% in Brazil. There is a significantly lower prevalence in Africa[8, 7, 9], while North America is split with HCV-3 representing only 10–12% of all chronic HCV infection (CHC) infection in the United States, while accounting for 22% in Canada.
Figure 1.
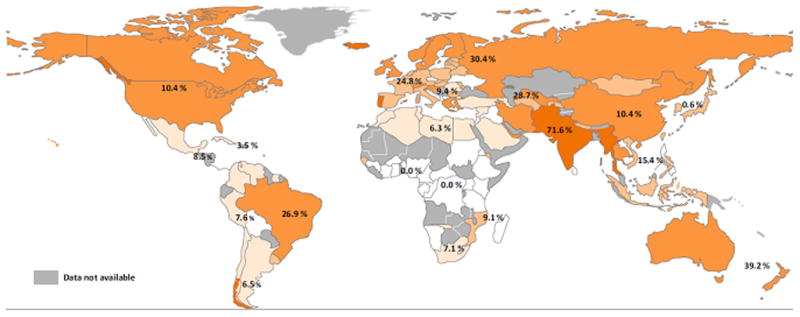
Global Prevalence of HCV Genotype 3
2 Fibrogenesis in Genotype 3 Infection
Fibrosis is a wound healing response that occurs in the setting of chronic liver injury. In chronic HCV infection there are multiple factors thought to play a role in the rate of fibrosis progression including age, sex, coinfection with HIV or HBV, alcohol intake, and potentially HCV genotype. There were early studies suggesting a potential association with the HCV-3 genotype and greater severity of fibrosis, but these retrospective studies were limited by small cohorts, variability in patient characteristics such as insulin resistance or Body Mass Index (BMI) or HCV genotype distribution, and inconsistencies in methodology, particularly with respect to the grading of steatosis[13,14]. One large retrospective analysis of the Swiss Hepatitis C study cohort of 1189 patients, indicated that HCV-3 was independently associated with fibrosis across multiple different estimates of progression rates [3]. A meta-analysis of 8 single biopsy studies representing 2349 patients with CHC confirmed an association between fibrosis and genotype 3, with an odds ratio of 1.52 for an accelerated fibrosis progression rate[10]. The same meta-analysis included 8 paired biopsy studies which were underpowered and did not reveal a similar association[10]. The primary limitation of these studies reporting an association between HCV-3 and fibrosis was they did not account for steatosis. Meanwhile, a large meta-analysis of 3068 CHC patients from N. America, Europe and Australia reported that HCV-3 was associated with steatosis, not fibrosis, and a multivariate analysis of fibrosis identified steatosis and level of inflammatory activity on histopathology as independent predictors of disease, not HCV-3. [11]. There are multiple studies supporting the association of higher grades of steatosis with higher rates of fibrosis progression[12]. There are also other studies supporting the concept that liver fibrosis is predominantly associated with steatosis in HCV-3 infection [13–15]. Thus, the burden of data does not support pathogenic evidence for enhanced direct viral mediated hepatic fibrogenesis for HCV-3 compared to other genotypes, and while the pathogenesis of disease progression in HCV-3 remains unclear, it is at least in part related to the higher rates of steatosis and hepatic inflammation reported in HCV-3 infection.
3 HCV Genotype 3 Infection and Steatosis
Hepatic steatosis is a common histological feature of patients with CHC infection and is multifactorial in etiology[16]. Observational data from several sources have indicated that steatosis is an independent variable that is associated with both severity and progression of fibrosis in CHC patients, and increases the risk of HCC[17, 18]. In non-alcoholic fatty liver disease (NAFLD), steatosis is considered to be the initial histologic manifestation of the metabolic syndrome and is associated with risk factors such as obesity, type 2 diabetes mellitus, and dyslipidemia[19]. In CHC infection, specifically in genotypes 1 and 4, hepatic steatosis is associated with insulin resistance and appears associated with more historical host factors[20].
Hepatic steatosis in the setting of HCV-3 is a unique entity in CHC, and while it is likely both viral and host mediated, viral factors appear more central in HCV-3 as compared to other genotypes. HCV-3 infection is typically associated with moderate-to-severe steatosis and a significant association between viral load and grade of steatosis has been observed[21, 15, 22–24]. Further support for direct viral mediated steatosis in HCV-3 was obtained from early clinical studies that demonstrated a significant improvement in biopsy proven steatosis in most HCV-3 infected patients following sustained virologic response, which was independent of changes in BMI[21, 22, 25]. Steatosis may also be seen in non-obese genotype 3a patients[14], further supporting the concept of direct viral-mediated steatosis in genotype-3 infected patients. In contrast, HCV RNA levels do not correlate with the degree of steatosis in non-genotype 3 infection[14, 12]. In these patients, steatosis appears to be associated with host metabolic factors such as BMI and visceral obesity and there is no significant improvement in steatosis when patients achieve viral clearance.
HCV-3 infection is associated with higher rates of steatosis, more rapid progression to liver disease and higher risk for hepatocellular carcinoma[26–29]. Even after SVR12, HCV-3 infection is associated with higher rates of HCC, likely as a result of more advanced liver disease[30]. With viral mediated steatosis a central driver of pathogenesis in HCV-3 infection, it is important to note that steatosis itself does not induce a pro-inflammatory state, but likely reflects the presence of other lipogenic pathways, such as lipid peroxidation and insulin resistance, that result in enhanced pro-fibrogenic stimuli. Lipids play an important role in several key aspects of the HCV lifecycle, including formation of the virion structure, cell receptor recognition, membrane fusion, viral replication, assembly, and export[31]. Although the precise pathogenic mechanisms of HCV-3 mediated steatosis are still unknown, HCV-3 modulates host lipid metabolism and appears to influence unique mechanisms of fat metabolism and transportation within the liver including microsomal triglyceride transfer protein (MTTP), sterol regulatory element binding protein 1c (SREBP-1c), and peroxisome proliferator associated receptor alpha (PPAR-α)[20].
The next section will explore the specific mechanisms by which HCV-3 is able to modulate host lipid metabolic pathways resulting in increased fatty acid accumulation and disease progression.
3.1 Microsomal Triglyceride Transfer Protein (MTTP) Inhibition
HCV-3 is associated with lower levels of low density lipoprotein (LDL), hypobetalipoproteinemia, and steatosis due to viral-mediated inhibition of MTTP, which plays an important role in triglyceride secretion from the liver[32]. MTTP is primarily responsible for assembly of lipid molecules with Apo lipoprotein B (apoB), which forms very low density lipoprotein (VLDL) that exports triglycerides into the bloodstream[32]. Mutations in apoB produce a disease state called apoB lipoproteinemia that is characterized by low levels of circulating apoB in addition to hepatic steatosis[33]. This pathway was initially implicated in HCV disease by Rubbia-Brandt et al who described decreased levels of circulating apoB in patients with HCV-3 infection and hepatic steatosis[23]. In a transgenic mouse model, Perlemuter et al noted that HCV core protein overexpression inhibited the ability of MTTP to transfer lipid molecules to apoB, thus resulting in increased apoB degradation and decreased production of VLDL[34]. Although all genotypes had some capability to inhibit the function of MTTP, Mirandola et al noted that this effect was greatest with HCV- 3a core proteins[35]. Cells transfected with HCV demonstrate co-localization of HCV core protein [36] and HCV non-structural protein 5A (NS5A) [37] to intra-cytoplasmic triglyceride-rich lipid droplets. Analysis of the primary sequence of the HCV core protein has revealed a unique domain necessary for association of the core protein with lipid droplets [38]. Our group has previously shown that specific HCV core protein polymorphisms are associated with intrahepatic lipid accumulation in HCV-3a, providing further evidence for viral and genotype specific steatosis [39]. Also, HCV-3 appears to selectively disrupt de novo lipogenesis in the distal cholesterol biosynthesis pathway [40], with restoration of distal lipid metabolites following successful DAA therapy [41]. This is in keeping with observed restoration in total cholesterol and apoB levels following viral clearance[42]. In summary, viral inhibition of MTTP, reduced apoB levels and selective disturbance in sterol synthesis may results in overall decreased hepatocyte lipid export, and may represent a pathway to hepatic steatosis in HCV-3 infection (Figure 2).
Figure 2.

HCV-3 Viral Mediated Mechanisms of Steatosis. A) HCV-3 core inhibits MTTP affecting assembly of ApoB and lipid to VLDL. B) HCV-3 core induces the PI3K-Akt pathway, increasing activity of SREBP-1c and increasing FAS. C) HCV core increases levels of PPAR-α leading to hepatic lipid accumulation
3.2 SREBP-1 Activation
Sterol Regulatory Element-binding Protein-1c (SREBP-1c) are transcription factors that regulate lipogenic pathways, including fatty acid synthesis, and have been investigated for associations with HCV infection [43]. SREBP-1c controls both cholesterol and fatty acid synthesis and serves as a transcription factor for multiple downstream enzymes such as fatty acid synthase(FAS)[44]. FAS plays an important role in lipid synthesis and triglyceride accumulation in hepatocytes by catalyzing the reaction of acetyl-CoA and malonyl-CoA in the synthesis pathway for triglycerides. With respect to HCV-3, Jackel-Cram et al demonstrated that SREBP-1c activity and subsequent FAS promoter up-regulation was increased in the presence of HCV-3a core protein (Figure 2) [45]. This finding would indicate that HCV-3a core protein was capable of increasing triglyceride production within the liver. Further studies noted that SREBP-1c activity was likely driven by upstream factors phosphoinositide-3-kinase (PI3K) and Protein Kinase B, also designated AKT. Directly downstream of the insulin receptor, the PI3K-AKT pathway is a highly conserved master regulatory pathway involved in cell proliferation, genetic stability, and apoptosis. Therefore, it is possible that there are pathways other than up-regulation of fatty acid synthase that are altered in the setting of HCV-3a core protein. Other literature suggests that viral proteins NS5A and NS4B also are capable of SREBP-1c activation, however it is unclear if this is unique to HCV-3 or also occurs in other genotypes[46]. The exact mechanism of HCV-3a core protein mediated SREBP-1c activation remains unknown, but several pathways have been implicated including insulin receptor signaling[45].
3.3 Peroxisome Proliferator Associated Receptor-α (PPAR-α) Inhibition
Another contributor to HCV-3 steatogenesis is inhibition of the PPAR-α pathway involved in metabolic regulation. PPAR-α is a transcription factor that induces hepatic fatty acid oxidation and ketogenesis while also up-regulating hepatic glucose production, the primary adaptive response to fasting[47]. PPAR-α is the pharmacologic target for the fibrate class of cholesterol lowering medications such as gemfibrozil and fenofibrate which function as PPAR-α agonists. Other PPAR agonists are also under current evaluation for cardiometabolic disease and NAFLD [48]. Conversely, inhibition of PPAR-α results in decreased triglyceride breakdown and accumulation of intrahepatic fatty acids. HCV-3a core protein has been demonstrated to function as an inhibitor of PPAR-α activity in vitro (Figure 2)[49]. De Gottardi et al compared the expression of PPAR-α in HCV-1b versus HCV-3a infection using liver biopsies of infected patients and in vitro models. This study noted that levels of PPAR-α mRNA were significantly decreased in HCV-3a compared to HCV-1b, independent of the extent of liver steatosis. However, inhibition of PPAR-α does not appear to be limited to HCV-3. Initial associations of PPAR-α inhibition were noted with HCV-1b core protein, although the effect appears less potent compared to that observed with HCV-3a[50, 51].
3.4 Interleukin-28B and Patatin-like Phospholiase Domain-Containing Protein 3 (PNPLA3) in HCV-3 related steatosis
IL28B and PNPLA3 genetic polymorphisms have been identified as important prognostic factors for progression of steatosis and treatment response in CHC[52, 53]. IL28B is a gene that codes for interferon-lambda, and the favorable polymorphism, rs12979860 CC, has been associated with improved responsiveness to interferon-containing therapies, and greater rates of natural clearance of HCV infection[54]. This polymorphism has also been associated with a decreased prevalence of steatosis in the setting of CHC as compared to the unfavorable wild type TT or heterozygote CT genotypes[55, 53, 56]. The PNPLA3 gene codes for a hydrolase against triglycerides and retinyl esters in hepatic stellate cells. The polymorphism rs738409 GG was initially identified as a risk factor for nonalcoholic fatty liver disease[57] and was associated with an increased risk of steatosis and fibrosis in the setting of CHC[58, 59]. Unfortunately, studies assessing genetic polymorphisms and CHC have included few HCV-3 patients, and the effect of IL28 CC or PNPLA3 GG on genotype-specific steatosis remains uncertain in HCV-3.
4 Treatment of HCV Genotype 3 Infection
The treatment of HCV-3 infection has generally paralleled the advances in HCV therapy as a whole, though with some key distinctions. In the era of peginterferon (PEG) and ribavirin (RBV), HCV-3 had higher rates of sustained virologic response (SVR) as compared to HCV-1 and 4, with rates of SVR ranging from 66–80%[60–62]. In addition, the treatment course for HCV-3 was shorter than HCV-1 and 4, usually 6 months. Unfortunately, the first generation DAA agents telaprevir and boceprevir, while demonstrating in vitro activity against HCV-3, failed to demonstrate any significant additional clinical benefit and PEG/RBV remained the standard of care until 2013[63, 64].
4.1 Sofosbuvir
Sofosbuvir (SOF), an NS5B polymerase inhibitor, represented the first major advance in HCV-3 therapy[65]. Four large trials assessed the efficacy of sofosbuvir with weight based ribavirin (SOF/RBV) for HCV-3: FISSION, FUSION, POSITRON, and VALENCE (Table 1). These studies included both HCV-2 and HCV-3 patients.
Table 1.
Summary of HCV-3 Trials and Rates of SVR12
| Trial Name | Regimen | Tx Duration (weeks) | SVR12 for Treatment Naïve, Non-cirrhotic (%) | SVR12 for Treatment Naïve, Cirrhotic (%) | SVR12 for Treatment Experienced, Non-cirrhotic (%) | SVR12 for Treatment Experienced, Cirrhotic (%) |
|---|---|---|---|---|---|---|
| FUSION | SOF+RBV | 12 | 37 | 19 | -- | -- |
| SOF+RBV | 16 | 63 | 61 | -- | -- | |
| POSITRON | SOF+RBV | 12 | 68 | 21 | -- | -- |
| VALENCE | SOF+RBV | 24 | 95 | 92 | 87 | 62 |
| BOSON | SOF+RBV | 16 | 83 | 57 | 76 | 47 |
| 24 | 90 | 82 | 81 | 76 | ||
| ALLY-3 | DAC+SOF | 12 | 90 | 58 | 86 | 69 |
| ALLY-3+ | DAC+SOF+RBV | 12 | 100 | 88 | -- | 93 |
| 16 | 100 | 89 | -- | 86 | ||
| ASTRAL-3 | SOF+VEL | 12 | 98 | 91 | 93 | 89 |
FISSION enrolled 527 total patients, including 359 treatment naïve patients with HCV- 3 infection who were all randomized to PEG/RBV for 24 weeks or SOF/RBV for 12 weeks. Sustained virologic response rates for the SOF/RBV arm was 56% as compared to 63% for PEG/RBV [66]. Predictors of treatment failure included HCV-3 and presence of cirrhosis.
POSITRON and FUSION trials further characterized the efficacy of SOF/RBV as a salvage regimen for patients who either had a contraindication or intolerance to interferon or who were treatment experienced with PEG/RBV, respectively[67]. The POSITRON was a placebo controlled evaluation of SOF/RBV for 12 weeks and FUSION was a randomized controlled trial of SOF/RBV for 12 weeks versus 16 weeks. POSITRON reported an SVR12 of 68% (n = 57/84) after 12 weeks of SOF/RBV for HCV-3 subjects without cirrhosis and 21% (n = 3/14) SVR12 for those with cirrhosis, confirming overall low rate of SVR12 in HCV-3 with 12 weeks of therapy, and an unacceptable relapse rate in patients with cirrhosis in particular. FUSION was the first study to assess an extension of therapy in more difficult to treat patients and noted that 16 weeks of SOF/RBV had higher overall rates of SVR12 (62% vs. 30%) as compared to 12 weeks in HCV-3 infection.[67]. Again, HCV-3 infection and cirrhosis were predictors of treatment failure.
VALENCE was initially designed as a placebo controlled multi-center Phase 3 trial of SOF/RBV for 12 weeks versus placebo in HCV-2 and 3 infection[68]. The study included both treatment experienced patients and patients with cirrhosis. Results of the FUSION trial suggesting HCV-3 response rates were higher with extension of therapy to 16 weeks were published while the VALENCE trial was ongoing. Based on these results, the study was unblinded, the placebo group was terminated, and all HCV-3 infected patients were extended to 24 weeks of SOF/RBV. Patients with HCV-3 who received 24 weeks of SOF/RBV, achieved an overall SVR12 rate of 85% (n = 213/250), the highest reported with this regimen in HCV-3 infection. VALENCE also provided insight into the impact of prior treatment failure and cirrhosis on SVR. Treatment naïve patients achieved SVR12 of 92% with cirrhosis and 95% without cirrhosis, while treatment experienced patients achieved SVR12 of 62% with cirrhosis and 87% without cirrhosis [68].
Recognizing the limitations of the SOF/RBV regimen in particularly hard to treat HCV-3 infected patients, the BOSON study set out to find an optimized regimen. BOSON was a randomized phase 3, open label trial that included treatment experienced and naive patients, randomizing them to SOF/RBV for 16 or 24 weeks, and SOF/PEG/RBV for 12 weeks[69]. The overall SVR12 rate in HCV-3 infected patients was 71% in the 16 week arm, 84% in the 24 week arm, and 93% for those who received SOF/PEG/RBV for 12 weeks. The differences between all groups were statistically significant, suggesting the most efficacious regimen for HCV-3 infection was SOF/PEG/RBV[69]. The higher SVR of the SOF/PEG/RBV regimen held true regardless of treatment experience and presence of cirrhosis with all subgroups achieving SVR12 > 90% with one exception: treatment experienced patients with cirrhosis achieved an SVR12 of 86% (30/35), though still superior to the other regimen. Both sofosbuvir containing regimens remain recommended per the current European Association for the Study of Liver Disease (EASL) HCV Treatment Guidelines, but were recently removed from the American Association for the Study of Liver Disease/Infectious Diseases Society of America (AASLD/IDSA) HCV Treatment Guidance due to the availability of several DAA combination therapies [70, 71].
4.2 Daclatasvir
In 2013, the approval of daclatasvir (DCV), a pangenotypic NS5A inhibitor, in combination with sofosbuvir for the treatment of HCV-3 infection represented the beginning of a new era for HCV-3 therapy[5]. Although interferon and ribavirin-free DAA combination therapies had already been approved for genotype 1 infection, these regimens did not extend to the HCV-3 infected population.
ALLY-3 study was an open-label single arm study of DCV (60 mg daily) in combination with SOF (400 mg daily) for 12 weeks (DCV+SOF) in all patients, including those with cirrhosis and who had failed prior treatment with interferon based therapies[72]. The overall SVR12 rate was 90% in treatment naïve patients and 86% in treatment experienced patients with HCV-3 infection. However, as in the prior SOF/RBV studies, rates of SVR12 in patients with cirrhosis lagged behind with SVR12 of 58% (11/19) in treatment naïve patients with cirrhosis and 69% (9/13) for treatment experienced patients with cirrhosis (Table 1). This was the first study that suggested a role for baseline NS5A resistance mutations in predicting treatment failure. Although the numbers were small, two resistance associated polymorphisms (RAPs) were associated with lower SVR12: (1) fourteen patients had evidence of the A30 polymorphism at baseline with SVR12 of 100% (9/9) in patients without cirrhosis and 20% (1/5) in patients with cirrhosis; (2) thirteen patients had evidence of the Y93H polymorphism at baseline with SVR12 of 67% (6/9) in patients without cirrhosis and 25% (1/4) in patients with cirrhosis. These 10 failures accounted for more than half of all treatment failures although the 2 RAPs were only detected in 17% of patients.
ALLY-3+ was a small randomized controlled trial of DCV+SOF with the primary objective to investigate the impact of weight-based ribavirin and treatment extension (12 versus 16 weeks) on response rates in HCV-3 infection[73]. The study included treatment naïve and experienced patients with severe fibrosis (N=14) and compensated cirrhosis (N=36). The overall SVR12 was 88% (21/24) in the 12-week arm and 92% (24/26) in the 16-week arm (Table 1). Specifically, in patients with cirrhosis, the SVR12 was 83% (15/18) and 89% (16/18), respectively. There were only 4 relapses in the study, 2 in each arm. The numbers were small but overall, ribavirin seemed to decrease relapse compared to ALLY-3 and extension of therapy did not significantly improve outcome. The study was too small to sufficiently investigate the impact of NS5A RAPs on treatment outcome.
Thus, while DCV+SOF provided a highly effective treatment option for HCV-3 patients without cirrhosis, the management of those with cirrhosis remained a challenge. The addition of ribavirin appeared to play a role in decreasing relapse, but the lack of a randomized study made it difficult to know how great that impact was. Furthermore, the potential for benefit by extending to 24 weeks as was seen in the SOF/RBV studies resulted in a knowledge gap created by piecemeal studies. This regimen is recommended for the treatment of HCV-3 infection in both the EASL and AASLD/IDSA HCV treatment guidelines in which both recommend extending to 24 weeks of therapy with the addition of ribavirin when possible in patients with cirrhosis, and a recommendation for NS5A resistance testing in specific subgroups (Table 2).
Table 2.
Indications for RAP Testing in HCV-3
| Treatment Regimen | Presence of Cirrhosis | Prior Treatment | NS5a RAP Testing Indication |
|---|---|---|---|
| DAC+SOF | − | − | − |
| + | − | + | |
| − | + | + | |
| + | + | −a | |
| SOF+VEL | − | − | − |
| + | − | + | |
| − | + | + | |
| + | + | −b |
Add weight based ribavirin and treat for 24 weeks regardless of presence of NS5a RAP
Add weight based ribavirin regardless of presence of NS5a RAP
4.3 Velpatasvir
In June of 2016, the approval of velpatasvir (VEL, 100 mg daily), a pangenotypic NS5A inhibitor, in combination with sofosbuvir, ushered in the first fixed-dose pan-genotypic regimen (SOF/VEL) for the treatment of hepatitis C infection[74]. This regimen was the first combination DAA therapy approved for the treatment of all HCV clinical genotypes 1–6.
ASTRAL-3 was an open-label, randomized trial comparing SOF/VEL for 12 weeks to SOF/RBV for 24 weeks in treatment naïve and experienced HCV-3 infected patients[75]. Overall SVR12 was 95% versus 80%, respectively, confirming superiority of the combination DAA therapy (Table 1). Yet a similar trend emerged: SVR12 was 91% in patients with cirrhosis versus 97% in those without cirrhosis and 89% in the most difficult treatment experienced patients with cirrhosis. Furthermore, of the 25 patients with the Y93H NS5A RAP at baseline, 84% (21/25) achieved SVR12, compared to 97% (225/231) of the patients without NS5A RAPs.
Thus, while SOF/VEL brings great hope for the majority of HCV-3 infected patients, the higher relapse rate in HCV-3 infection (N=11/277) vs all other genotypes (N=3/758) in the HCV mono-infected registration program of patients without cirrhosis or with compensated cirrhosis suggests there is room for improvement, in particular for patients with cirrhosis. It is likely that multiple baseline predictors add up to increase the risk of treatment failure including prior treatment failure, presence of cirrhosis, and presence of high fold NS5A RAPs like the Y93H variant. For this reason, the AALSD/IDSA HCV Treatment Guidance Panel recommends NS5A testing in treatment experienced patients without cirrhosis and treatment naïve patients with cirrhosis and adding weight-based ribavirin when the Y93H RAP is detected (Table 2). Due to the presence of 2 of these 3 negative predictors, patients that are both prior treatment failures and have evidence of cirrhosis are recommended to receive weight based ribavirin regardless of NS5A testing results. An active study (NCT02781558) is expected to provide more data on the impact of ribavirin on treatment response to SOF/VEL in HCV-3 infected patients with cirrhosis.
4.4 Elbasvir/grazoprevir
Although not approved in the United States, the fixed dose combination of elbasvir (EBR), an NS5A inhibitor, and grazoprevir (GZR), a next generation NS3/4 protease inhibitor, in combination with sofosbuvir for 12 weeks has demonstrated efficacy for the treatment of HCV-3 infection in treatment naïve patients[76].
C-SWIFT was a randomized trial of combined DAA regimens EBR/GZR+SOF for 8 (N=15) and 12 (N=26) weeks in treatment naïve HCV-3 infected patients with and without cirrhosis[77]. Overall SVR12 was achieved in 93% (14/15) of patients treated for 8 weeks, 100% (14/14) of patients without cirrhosis treated for 12 weeks, and 83% (10/12) of patients with cirrhosis treated for 12 weeks. It is difficult to draw conclusions as to overall cure rates for this regimen because of the small numbers, but this should be considered as a possible salvage regimen.
4.5 Future DAA Therapies
Now that interferon and ribavirin free treatment regimens for all genotypes have been established, future HCV regimens have pivoted towards the potential to shorten treatment duration and optimize treatment outcomes for the most difficult to treat populations including combination DAA failure with multidrug resistance and HCV-3 infected patients with cirrhosis. There are several new regimens in human study that share these objectives.
SURVEYOR- 2 was a phase 2 trial investigating the safety and efficacy of the pangenotypic dual combination of ABT-493 + ABT-530 ± RBV. ABT-493 is an NS3/4A protease inhibitor and ABT-530 is an NS5a inhibitor, both representing next generation DAA agents in their respective classes[78]. Overall SVR12 was 97%(n = 28/29) for treatment naïve HCV-3 infected patients without cirrhosis who received 8 weeks of therapy without ribavirin and 100% (48/48) in treatment naive HCV-3 infected patients with cirrhosis who received 12 weeks of therapy with (N=24) and without (N=24) ribavirin[79].
C-CREST 2 was a phase 2 trial investigating the safety and efficacy of the triple combination therapy of MK-3682/GZR/MK-8408. MK-3682 is an NS5B inhibitor and MK-8408 is a second generation NS5A inhibitor[80]. Patients were randomized to 4 therapy arms investigating dosing of MK-3682 and elbasvir versus the new MK-8408. The treatment duration for all regimens was 8 weeks. Overall rate of SVR12 in HCV-3 infected treatment naive patients without cirrhosis was 86–95%, suggesting 8 weeks will not be the optimal therapy with this regimen for this patient population.
LEPTON was a phase 2 trial investigating the safety and efficacy of a pangenotype triple DAA therapy including SOF/VEL and GS-9857, a next generation NS3/4A protease inhibitor[81]. Three HCV-3 infected groups of patients were included: treatment naïve with compensated cirrhosis, PEG+RBV failures with cirrhosis, and DAA-failures with and without cirrhosis. The treatment naïve group received 6 weeks of triple therapy and the two treatment experienced groups received 8 weeks of triple therapy. SVR12 was 83% (15/18) in the treatment naïve group and 100% (23/23) in the treatment experienced groups including 4 who failed prior DAA therapy.
4.6 Resistance Associated Polymorphisms (RAPs)
Resistance associated polymorphisms, also referred to as resistance associated variants or resistance associated substitutions, are point mutations that are associated with drug resistance in vitro. However, the genotypic presence of a RAP does not necessarily translate to a phenotypic treatment failure. Like advanced cirrhosis or prior treatment experience, the presence of RAPs represent an important factor in overall treatment outcomes, and when combined with other negative predictors may result in treatment failure. In many cases RAPs can be overcome by potent combination DAA therapies, extension of therapy, and/or the addition of ribavirin. The two primary techniques for genotype sequencing include next generation (clonal) sequencing which can detect down to a frequency of 0.5–1% of the viral variants[82] and population sequencing which can detect a frequency of approximately 20% of the viral variants[83]. Based on the current literature, population level sequencing is the most clinically relevant[84].
With only one exception, the clinical relevance of resistance testing has been limited to RAPs in the NS5A gene. Two RAPS in particular, Y93H and A30K have emerged as the most clinically relevant polymorphisms in HCV-3, and are present at baseline in up to 8.3% and 6.3% of all HCV-3 infected patients, respectively[85–87]. The ALLY-3 trial of DCV+SOF in HCV-3 infection patients reported that both Y93H and A30K polymorphisms were associated with higher rates of treatment failure, especially in those patients with cirrhosis[72]. Similarly, the ASTRAL-3 trial of SOF/VEL reported a lower SVR12 in those patients with the Y93H polymorphism at baseline (84%) as compared to those without the polymorphism (97%)[75]. As a result, the current recommendation from the AASLD/IDSA guideline panel when a provider is planning to treat HCV-3 infection with SOF/VEL or DCV+SOF is to perform population level genotyping in patients who are either treatment-naïve with cirrhosis or who are treatment-experienced without cirrhosis (Table 2). For those patients who are treatment-experienced with cirrhosis, ribavirin should be added regardless of the results of resistance testing[70].
5 Clinically Relevant Drug Interactions
Although the overall safety profile of the DAA agents is excellent, it is important to recognize that there are drug interactions that can either impact the antiviral potency of the DAA regimen or potentiate an adverse effect of either the DAA or the concomitant medication. For example, daclatasvir is a substrate of CYP3A4 and therefore must be dose adjusted when administered with either inhibitors or inducers of the enzyme. The University of Liverpool hosts a comprehensive, easy-to-use, and up to date website that contains all relevant drug-drug interactions for the different DAA regimens (www.hep-druginteractions.org) [88]. It is important to review all potential drug interactions prior to starting any DAA therapy. In this section, we will review the most common and severe drug interactions that one may encounter in the treatment of HCV-3.
5.1 Sofosbuvir Associated Bradycardia
Both of the currently recommended DAA regimens, SOF/VEL and DCV+SOF, for treatment of HCV-3 infection are well tolerated with minimal side effects and an overall favorable drug interaction profile. However, there are some notable interactions and toxicities that have recently emerged since approval by both the United States Food and Drug Administration (FDA) and European Medical Agency (EMA). In the spring of 2015, the FDA and EMA warned that bradycardia could occur when amiodarone was co-administered with sofosbuvir as part of a DAA combination regimen[89, 90]. Sofosbuvir now has a package insert warning, strongly cautioning against use in combination with amiodarone[91]. A case series published by Renet et al describes two patients who developed symptomatic bradycardia following administration of SOF and amiodarone. The first patient was a 61 year-old female with compensated cirrhosis(CP-A6) who was receiving DCV+SOF for HCV-1b disease. The second patient was a 50 year-old male with decompensated cirrhosis (CP-B9) who was receiving DCV+SOF for HCV-1b. This case series and FDA guidance was followed by a third case series by Fontaine et al which describes 3 patients who developed symptomatic bradycardia on SOF[92]. Of note, one of those patients was not on amiodarone and only receiving propranolol while another was not receiving any AV nodal agents of any kind. In search of a potential mechanism of action for this toxicity, Liu et al used an in vitro model to describe decreased AV nodal conduction in the setting of multiple different DAA agents, with the most profound AV nodal blockade occurring with sofosbuvir[93]. In addition, they noted that the effect of nodal blockade was more than additive for infusions of sofosbuvir and amiodarone[93]. These findings were further expanded upon by Regan et al who was able to recreate the SOF+amiodarone induced bradycardia in animal models using guinea pigs and rhesus monkeys[94]. These data strongly suggest that sofosbuvir has an independent mechanism of AV nodal blockade and that co-administration with amiodarone can potentiate a life threatening bradycardia.
5.2 Velpatasvir and Acid Suppressing Medications
Velpatasvir relies on an acid environment for absorption. Initial pharmacokinetics studies described decreases in both maximum concentration (Cmax) and area under the curve (AUC) for velpatasvir serum drug levels when co-administered with famotidine and omeprazole[95, 96]. This drop in serum drug concentration is particularly pronounced for co-administration with omeprazole regardless of whether they are dosed separately or together. If H2-blocking agents are to be administered with SOF/VEL, the package insert recommends dosing simultaneously or 12 hours apart, with doses not to exceed an equivalent of 40 mg of famotidine[74]. For proton pump inhibitors (PPI) and SOF/VEL, the package insert recommends against co-administration, but notes that if it is necessary, it should be taken with food, four hours prior to a max dose of 20 mg of omeprazole[74]. There is currently no data on co-administration of SOF/VEL and acid suppressing medications in patients with CHC and any inferences on the subject must be drawn from data on ledipasvir in genotype 1. Like velpatasvir, ledipasvir (LDV) is a first generation NS5A inhibitor that relies on stomach acid for absorption. Similar to the registration program for velpatasvir, the program for ledipasvir excluded the concomitant use of acid suppressing medications. Initial data from the real world Target-C Cohort suggested that patients who were on any dose of PPI at the start of LDV+SOF therapy had a significantly lower SVR12 (93% vs 98%)[97]. These findings were expanded upon by Tapper et al who noted that in another real world cohort patients did not have any difference in SVR12 rates if they were on any PPI or were taking a PPI once a day at higher than the recommended dose[98]. However, they did report decreased rates of SVR12 in patients who were taking PPIs twice a day. The impact of twice daily dosed PPIs was notable regardless of the presence of cirrhosis, but did have the greatest impact in patients with cirrhosis taking PPIs twice a day with a reported 20% decrease in SVR12 (76.9% vs 96.3%) [98]. Given that velpatasvir had greater pharmacokinetic variability with acid suppressing medication, co-administration with PPIs should be avoided until there is further data in patients with CHC[74].
6 Conclusion
Genotype 3 represents a unique entity within HCV treatment. It is associated with genotype specific mechanisms of steatosis in addition to accelerated development of fibrosis and higher rates of hepatocellular carcinoma. These findings underscore the need for effective therapy for this group of patients. Although DCV+SOF and SOF/VEL has finally brought HCV-3 into the modern DAA era with cure rates comparable to the other genotypes, there remains room for improvement in particular for patients with cirrhosis and NS5A RAPs. These are issues that need to be addressed by the next generation of dual and triple pangenotypic regimens. Furthermore, how HCV eradication by current and future DAA regimens impacts the natural history of liver disease with this infection remains unclear and follow-up studies of steatosis resolution and fibrosis regression are needed. The DAA era has truly revolutionized HCV therapy, but we must still work to ensure that no subgroup, regardless of genotype, cirrhosis, or treatment experience, is left by the wayside.
Contributor Information
Austin Chan, Division of Infectious Diseases, Department of Medicine, Duke University School of Medicine.
Keyur Patel, Toronto Center for Liver Disease, Associate Professor of Medicine, University of Toronto, Toronto, ON.
Susanna Naggie, Division of Infectious Diseases, Associate Professor of Medicine, Department of Medicine, Duke University School of Medicine, Director, Infectious Diseases Research, Duke Clinical Research Institute.
References
- 1.WHO. Fact Sheet. WHO; 2016. [Accessed 8/29/2016]. Hepatitis C Fact Sheet. http://www.who.int/mediacentre/factsheets/fs164/en/ [Google Scholar]
- 2.Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology (Baltimore, Md) 2013;57(4):1333–42. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 3.Bochud PY, Cai T, Overbeck K, Bochud M, Dufour JF, Mullhaupt B, et al. Genotype 3 is associated with accelerated fibrosis progression in chronic hepatitis C. Journal of hepatology. 2009;51(4):655–66. doi: 10.1016/j.jhep.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 4.Kattakuzhy S, Levy R, Rosenthal E, Tang L, Wilson E, Kottilil S. Hepatitis C genotype 3 disease. Hepatology international. 2016 doi: 10.1007/s12072-016-9748-z. [DOI] [PubMed] [Google Scholar]
- 5.Daclatasvir Package Insert. Bristol Meyers Squib; 2015. [Google Scholar]
- 6.Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. Journal of hepatology. 2014;61(1 Suppl):S45–57. doi: 10.1016/j.jhep.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 7.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology (Baltimore, Md) 2015;61(1):77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruggmann P, Berg T, Ovrehus AL, Moreno C, Brandao Mello CE, Roudot-Thoraval F, et al. Historical epidemiology of hepatitis C virus (HCV) in selected countries. Journal of viral hepatitis. 2014;21(Suppl 1):5–33. doi: 10.1111/jvh.12247. [DOI] [PubMed] [Google Scholar]
- 9.Saraswat V, Norris S, de Knegt RJ, Sanchez Avila JF, Sonderup M, Zuckerman E, et al. Historical epidemiology of hepatitis C virus (HCV) in select countries - volume 2. Journal of viral hepatitis. 2015;22(Suppl 1):6–25. doi: 10.1111/jvh.12350. [DOI] [PubMed] [Google Scholar]
- 10.Probst A, Dang T, Bochud M, Egger M, Negro F, Bochud PY. Role of hepatitis C virus genotype 3 in liver fibrosis progression--a systematic review and meta-analysis. Journal of viral hepatitis. 2011;18(11):745–59. doi: 10.1111/j.1365-2893.2011.01481.x. [DOI] [PubMed] [Google Scholar]
- 11.Leandro G, Mangia A, Hui J, Fabris P, Rubbia-Brandt L, Colloredo G, et al. Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: a meta-analysis of individual patient data. Gastroenterology. 2006;130(6):1636–42. doi: 10.1053/j.gastro.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Adinolfi LE, Utili R, Andreana A, Tripodi MF, Marracino M, Gambardella M, et al. Serum HCV RNA levels correlate with histological liver damage and concur with steatosis in progression of chronic hepatitis C. Digestive diseases and sciences. 2001;46(8):1677–83. doi: 10.1023/a:1010697319589. [DOI] [PubMed] [Google Scholar]
- 13.Westin J, Nordlinder H, Lagging M, Norkrans G, Wejstal R. Steatosis accelerates fibrosis development over time in hepatitis C virus genotype 3 infected patients. Journal of hepatology. 2002;37(6):837–42. doi: 10.1016/s0168-8278(02)00299-4. [DOI] [PubMed] [Google Scholar]
- 14.Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology (Baltimore, Md) 2001;33(6):1358–64. doi: 10.1053/jhep.2001.24432. [DOI] [PubMed] [Google Scholar]
- 15.Rubbia-Brandt L, Fabris P, Paganin S, Leandro G, Male PJ, Giostra E, et al. Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut. 2004;53(3):406–12. doi: 10.1136/gut.2003.018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bedossa P, Moucari R, Chelbi E, Asselah T, Paradis V, Vidaud M, et al. Evidence for a role of nonalcoholic steatohepatitis in hepatitis C: a prospective study. Hepatology (Baltimore, Md) 2007;46(2):380–7. doi: 10.1002/hep.21711. [DOI] [PubMed] [Google Scholar]
- 17.Hwang SJ, Lee SD. Hepatic steatosis and hepatitis C: Still unhappy bedfellows? Journal of gastroenterology and hepatology. 2011;26(Suppl 1):96–101. doi: 10.1111/j.1440-1746.2010.06542.x. [DOI] [PubMed] [Google Scholar]
- 18.Roingeard P. Hepatitis C virus diversity and hepatic steatosis. Journal of viral hepatitis. 2013;20(2):77–84. doi: 10.1111/jvh.12035. [DOI] [PubMed] [Google Scholar]
- 19.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142(7):1592–609. doi: 10.1053/j.gastro.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Abenavoli L, Masarone M, Peta V, Milic N, Kobyliak N, Rouabhia S, et al. Insulin resistance and liver steatosis in chronic hepatitis C infection genotype 3. World journal of gastroenterology. 2014;20(41):15233–40. doi: 10.3748/wjg.v20.i41.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patton HM, Patel K, Behling C, Bylund D, Blatt LM, Vallee M, et al. The impact of steatosis on disease progression and early and sustained treatment response in chronic hepatitis C patients. Journal of hepatology. 2004;40(3):484–90. doi: 10.1016/j.jhep.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 22.Poynard T, Ratziu V, Charlotte F, Goodman Z, McHutchison J, Albrecht J. Rates and risk factors of liver fibrosis progression in patients with chronic hepatitis c. Journal of hepatology. 2001;34(5):730–9. doi: 10.1016/s0168-8278(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 23.Rubbia-Brandt L, Quadri R, Abid K, Giostra E, Male PJ, Mentha G, et al. Hepatocyte steatosis is a cytopathic effect of hepatitis C virus genotype 3. Journal of hepatology. 2000;33(1):106–15. doi: 10.1016/s0168-8278(00)80166-x. [DOI] [PubMed] [Google Scholar]
- 24.Hezode C, Roudot-Thoraval F, Zafrani ES, Dhumeaux D, Pawlotsky JM. Different mechanisms of steatosis in hepatitis C virus genotypes 1 and 3 infections. Journal of viral hepatitis. 2004;11(5):455–8. doi: 10.1111/j.1365-2893.2004.00528.x. [DOI] [PubMed] [Google Scholar]
- 25.Kumar D, Farrell GC, Fung C, George J. Hepatitis C virus genotype 3 is cytopathic to hepatocytes: Reversal of hepatic steatosis after sustained therapeutic response. Hepatology (Baltimore, Md) 2002;36(5):1266–72. doi: 10.1053/jhep.2002.36370. [DOI] [PubMed] [Google Scholar]
- 26.Nkontchou G, Ziol M, Aout M, Lhabadie M, Baazia Y, Mahmoudi A, et al. HCV genotype 3 is associated with a higher hepatocellular carcinoma incidence in patients with ongoing viral C cirrhosis. Journal of viral hepatitis. 2011;18(10):e516–22. doi: 10.1111/j.1365-2893.2011.01441.x. [DOI] [PubMed] [Google Scholar]
- 27.Shrivastava S, Meissner EG, Funk E, Poonia S, Shokeen V, Thakur A, et al. Elevated hepatic lipid and interferon stimulated gene expression in HCV GT3 patients relative to non-alcoholic steatohepatitis. Hepatology international. 2016 doi: 10.1007/s12072-016-9733-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanwal F, Kramer JR, Ilyas J, Duan Z, El-Serag HB. HCV genotype 3 is associated with an increased risk of cirrhosis and hepatocellular cancer in a national sample of U.S. Veterans with HCV. Hepatology (Baltimore, Md) 2014;60(1):98–105. doi: 10.1002/hep.27095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McMahon BJ, Bruden D, Townshend-Bulson L, Simons B, Spradling P, Livingston S, et al. Infection With Hepatitis C Virus Genotype 3 is an Independent Risk Factor for End-stage Liver Disease, Hepatocellular Carcinoma, and Liver-related Death. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2016 doi: 10.1016/j.cgh.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.El-Serag HB, Kanwal F, Richardson P, Kramer J. Risk of hepatocellular carcinoma after sustained virological response in Veterans with hepatitis C virus infection. Hepatology (Baltimore, Md) 2016;64(1):130–7. doi: 10.1002/hep.28535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends in endocrinology and metabolism: TEM. 2010;21(1):33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon DA, Jamil H. Progress towards understanding the role of microsomal triglyceride transfer protein in apolipoprotein-B lipoprotein assembly. Biochimica et biophysica acta. 2000;1486(1):72–83. doi: 10.1016/s1388-1981(00)00049-4. [DOI] [PubMed] [Google Scholar]
- 33.Di Leo E, Lancellotti S, Penacchioni JY, Cefalu AB, Averna M, Pisciotta L, et al. Mutations in MTP gene in abeta- and hypobeta-lipoproteinemia. Atherosclerosis. 2005;180(2):311–8. doi: 10.1016/j.atherosclerosis.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Perlemuter G, Sabile A, Letteron P, Vona G, Topilco A, Chretien Y, et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: a model of viral-related steatosis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2002;16(2):185–94. doi: 10.1096/fj.01-0396com. [DOI] [PubMed] [Google Scholar]
- 35.Mirandola S, Realdon S, Iqbal J, Gerotto M, Dal Pero F, Bortoletto G, et al. Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology. 2006;130(6):1661–9. doi: 10.1053/j.gastro.2006.02.035. [DOI] [PubMed] [Google Scholar]
- 36.Barba G, Harper F, Harada T, Kohara M, Goulinet S, Matsuura Y, et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(4):1200–5. doi: 10.1073/pnas.94.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi ST, Polyak SJ, Tu H, Taylor DR, Gretch DR, Lai MM. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology. 2002;292(2):198–210. doi: 10.1006/viro.2001.1225. [DOI] [PubMed] [Google Scholar]
- 38.Hope RG, McLauchlan J. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. The Journal of general virology. 2000;81(Pt 8):1913–25. doi: 10.1099/0022-1317-81-8-1913. [DOI] [PubMed] [Google Scholar]
- 39.Jhaveri R, McHutchison J, Patel K, Qiang G, Diehl AM. Specific polymorphisms in hepatitis C virus genotype 3 core protein associated with intracellular lipid accumulation. The Journal of infectious diseases. 2008;197(2):283–91. doi: 10.1086/524846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark PJ, Thompson AJ, Vock DM, Kratz LE, Tolun AA, Muir AJ, et al. Hepatitis C virus selectively perturbs the distal cholesterol synthesis pathway in a genotype-specific manner. Hepatology (Baltimore, Md) 2012;56(1):49–56. doi: 10.1002/hep.25631. [DOI] [PubMed] [Google Scholar]
- 41.Younossi ZM, Stepanova M, Estep M, Negro F, Clark PJ, Hunt S, et al. Dysregulation of distal cholesterol biosynthesis in association with relapse and advanced disease in CHC genotype 2 and 3 treated with sofosbuvir and ribavirin. Journal of hepatology. 2016;64(1):29–36. doi: 10.1016/j.jhep.2015.08.027. [DOI] [PubMed] [Google Scholar]
- 42.Poynard T, Ratziu V, McHutchison J, Manns M, Goodman Z, Zeuzem S, et al. Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology (Baltimore, Md) 2003;38(1):75–85. doi: 10.1053/jhep.2003.50267. [DOI] [PubMed] [Google Scholar]
- 43.Jackel-Cram C, Babiuk LA, Liu Q. Up-regulation of fatty acid synthase promoter by hepatitis C virus core protein: genotype-3a core has a stronger effect than genotype-1b core. Journal of hepatology. 2007;46(6):999–1008. doi: 10.1016/j.jhep.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 44.Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. The Journal of biological chemistry. 1999;274(50):35832–9. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- 45.Jackel-Cram C, Qiao L, Xiang Z, Brownlie R, Zhou Y, Babiuk L, et al. Hepatitis C virus genotype-3a core protein enhances sterol regulatory element-binding protein-1 activity through the phosphoinositide 3-kinase-Akt-2 pathway. The Journal of general virology. 2010;91(Pt 6):1388–95. doi: 10.1099/vir.0.017418-0. [DOI] [PubMed] [Google Scholar]
- 46.Xiang Z, Qiao L, Zhou Y, Babiuk LA, Liu Q. Hepatitis C virus nonstructural protein-5A activates sterol regulatory element-binding protein-1c through transcription factor Sp1. Biochemical and biophysical research communications. 2010;402(3):549–53. doi: 10.1016/j.bbrc.2010.10.081. [DOI] [PubMed] [Google Scholar]
- 47.Kersten S. Integrated physiology and systems biology of PPARalpha. Molecular metabolism. 2014;3(4):354–71. doi: 10.1016/j.molmet.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahebkar A, Chew GT, Watts GF. New peroxisome proliferator-activated receptor agonists: potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert opinion on pharmacotherapy. 2014;15(4):493–503. doi: 10.1517/14656566.2014.876992. [DOI] [PubMed] [Google Scholar]
- 49.de Gottardi A, Pazienza V, Pugnale P, Bruttin F, Rubbia-Brandt L, Juge-Aubry CE, et al. Peroxisome proliferator-activated receptor-alpha and -gamma mRNA levels are reduced in chronic hepatitis C with steatosis and genotype 3 infection. Alimentary pharmacology & therapeutics. 2006;23(1):107–14. doi: 10.1111/j.1365-2036.2006.02729.x. [DOI] [PubMed] [Google Scholar]
- 50.Dharancy S, Malapel M, Perlemuter G, Roskams T, Cheng Y, Dubuquoy L, et al. Impaired expression of the peroxisome proliferator-activated receptor alpha during hepatitis C virus infection. Gastroenterology. 2005;128(2):334–42. doi: 10.1053/j.gastro.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 51.Cheng Y, Dharancy S, Malapel M, Desreumaux P. Hepatitis C virus infection down-regulates the expression of peroxisome proliferator-activated receptor alpha and carnitine palmitoyl acyl-CoA transferase 1A. World journal of gastroenterology. 2005;11(48):7591–6. doi: 10.3748/wjg.v11.i48.7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Singal AG, Manjunath H, Yopp AC, Beg MS, Marrero JA, Gopal P, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. The American journal of gastroenterology. 2014;109(3):325–34. doi: 10.1038/ajg.2013.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tillmann HL, Patel K, Muir AJ, Guy CD, Li JH, Lao XQ, et al. Beneficial IL28B genotype associated with lower frequency of hepatic steatosis in patients with chronic hepatitis C. Journal of hepatology. 2011;55(6):1195–200. doi: 10.1016/j.jhep.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li JH, Lao XQ, Tillmann HL, Rowell J, Patel K, Thompson A, et al. Interferon-lambda genotype and low serum low-density lipoprotein cholesterol levels in patients with chronic hepatitis C infection. Hepatology (Baltimore, Md) 2010;51(6):1904–11. doi: 10.1002/hep.23592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarrazin C, Susser S, Doehring A, Lange CM, Muller T, Schlecker C, et al. Importance of IL28B gene polymorphisms in hepatitis C virus genotype 2 and 3 infected patients. Journal of hepatology. 2011;54(3):415–21. doi: 10.1016/j.jhep.2010.07.041. [DOI] [PubMed] [Google Scholar]
- 56.Chen Y, Xu HX, Wang LJ, Liu XX, Mahato RI, Zhao YR. Meta-analysis: IL28B polymorphisms predict sustained viral response in HCV patients treated with pegylated interferon-alpha and ribavirin. Alimentary pharmacology & therapeutics. 2012;36(2):91–103. doi: 10.1111/j.1365-2036.2012.05131.x. [DOI] [PubMed] [Google Scholar]
- 57.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nature genetics. 2008;40(12):1461–5. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ali M, Yopp A, Gopal P, Beg MS, Zhu H, Lee W, et al. A Variant in PNPLA3 Associated With Fibrosis Progression but not Hepatocellular Carcinoma in Patients With Hepatitis C Virus Infection. Clinical gastroenterology and hepatology: the official clinical practice journal of the American Gastroenterological Association. 2016;14(2):295–300. doi: 10.1016/j.cgh.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 59.Huang CF, Chen JJ, Yeh ML, Huang CI, Hsieh MY, Yang HL, et al. PNPLA3 genetic variants determine hepatic steatosis in non-obese chronic hepatitis C patients. Scientific reports. 2015;5:11901. doi: 10.1038/srep11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manns M, Zeuzem S, Sood A, Lurie Y, Cornberg M, Klinker H, et al. Reduced dose and duration of peginterferon alfa-2b and weight-based ribavirin in patients with genotype 2 and 3 chronic hepatitis C. Journal of hepatology. 2011;55(3):554–63. doi: 10.1016/j.jhep.2010.12.024. [DOI] [PubMed] [Google Scholar]
- 61.Andriulli A, Mangia A, Iacobellis A, Ippolito A, Leandro G, Zeuzem S. Meta-analysis: the outcome of anti-viral therapy in HCV genotype 2 and genotype 3 infected patients with chronic hepatitis. Alimentary pharmacology & therapeutics. 2008;28(4):397–404. doi: 10.1111/j.1365-2036.2008.03763.x. [DOI] [PubMed] [Google Scholar]
- 62.Mangia A, Santoro R, Minerva N, Ricci GL, Carretta V, Persico M, et al. Peginterferon alfa-2b and ribavirin for 12 vs. 24 weeks in HCV genotype 2 or 3. The New England journal of medicine. 2005;352(25):2609–17. doi: 10.1056/NEJMoa042608. [DOI] [PubMed] [Google Scholar]
- 63.De Meyer S, Ghys A, Foster GR, Beumont M, Van Baelen B, Lin TI, et al. Analysis of genotype 2 and 3 hepatitis C virus variants in patients treated with telaprevir demonstrates a consistent resistance profile across genotypes. Journal of viral hepatitis. 2013;20(6):395–403. doi: 10.1111/jvh.12046. [DOI] [PubMed] [Google Scholar]
- 64.Silva MO, Treitel M, Graham DJ, Curry S, Frontera MJ, McMonagle P, et al. Antiviral activity of boceprevir monotherapy in treatment-naive subjects with chronic hepatitis C genotype 2/3. Journal of hepatology. 2013;59(1):31–7. doi: 10.1016/j.jhep.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 65.Sciences G; Administration FaD, editor. Sofosbuvir Package Insert. Foster City: Gilead Sciences; 2013. [Google Scholar]
- 66.Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. The New England journal of medicine. 2013;368(20):1878–87. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 67.Jacobson IM, Gordon SC, Kowdley KV, Yoshida EM, Rodriguez-Torres M, Sulkowski MS, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. The New England journal of medicine. 2013;368(20):1867–77. doi: 10.1056/NEJMoa1214854. [DOI] [PubMed] [Google Scholar]
- 68.Zeuzem S, Dusheiko GM, Salupere R, Mangia A, Flisiak R, Hyland RH, et al. Sofosbuvir and ribavirin in HCV genotypes 2 and 3. The New England journal of medicine. 2014;370(21):1993–2001. doi: 10.1056/NEJMoa1316145. [DOI] [PubMed] [Google Scholar]
- 69.Foster GR, Pianko S, Brown A, Forton D, Nahass RG, George J, et al. Efficacy of sofosbuvir plus ribavirin with or without peginterferon-alfa in patients with hepatitis C virus genotype 3 infection and treatment-experienced patients with cirrhosis and hepatitis C virus genotype 2 infection. Gastroenterology. 2015;149(6):1462–70. doi: 10.1053/j.gastro.2015.07.043. [DOI] [PubMed] [Google Scholar]
- 70. [Accessed August 5, 2016];HCV Treatment Guidelines. http://www.hcvguidelines.org.
- 71.EASL Recommendations on Treatment of Hepatitis C 2015. Journal of hepatology. 2015;63(1):199–236. doi: 10.1016/j.jhep.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 72.Nelson DR, Cooper JN, Lalezari JP, Lawitz E, Pockros PJ, Gitlin N, et al. All-oral 12-week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY-3 phase III study. Hepatology (Baltimore, Md) 2015;61(4):1127–35. doi: 10.1002/hep.27726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leroy V, Angus P, Bronowicki JP, Dore GJ, Hezode C, Pianko S, et al. Daclatasvir, sofosbuvir, and ribavirin for hepatitis C virus genotype 3 and advanced liver disease: A randomized phase III study (ALLY-3+) Hepatology (Baltimore, Md) 2016;63(5):1430–41. doi: 10.1002/hep.28473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sciences G, inventor. Epclusa Package Insert. United States: 2016. [Google Scholar]
- 75.Foster GR, Afdhal N, Roberts SK, Brau N, Gane EJ, Pianko S, et al. Sofosbuvir and Velpatasvir for HCV Genotype 2 and 3 Infection. The New England journal of medicine. 2015;373(27):2608–17. doi: 10.1056/NEJMoa1512612. [DOI] [PubMed] [Google Scholar]
- 76.Elbasvir Grazoprevir Package Insert. USA: 2016. [Google Scholar]
- 77.Poordad F, Lawitz Eric1, Gutierrez Julio A1, Evans Barbara2, Howe Anita2, Feng Hwa-Ping2, Li Jerry Jing2, Hwang Peggy2, Robertson Michael2, Wahl Janice2, Barr Eliav2, Haber Barbara2. [Accessed August 5 2016];C-SWIFT: GRAZOPREVIR/ELBASVIR + SOFOSBUVIR IN CIRRHOTIC AND NONCIRRHOTIC, TREATMENT-NAIVE PATIENTS WITH HEPATITIS C VIRUS GENOTYPE 1 INFECTION, FOR DURATIONS OF 4, 6 OR 8 WEEKS AND GENOTYPE 3 INFECTION FOR DURATIONS OF 8 OR 12 WEEKS. 2015 http://www.natap.org/2015/EASL/EASL_11.htm.
- 78.Poordad F, Felizarta Franco2, Asatryan Armen3, Hassanein Tarek4, Aguilar Humberto5, Lalezari Jacob6, Scott Overcas J7, Ng Teresa I3, Liu Ran3, Lin Chih-Wei3, Mensa Federico J3, Kort Jens3. SURVEYOR-I: 98% – 100% SVR4 in HCV Genotype 1 Non-Cirrhotic Treatment-Naïve or Pegylated Interferon/Ribavirin Null-Responders with the Combination of the Next Generation NS3/4A Protease Inhibitor ABT-493 and NS5A Inhibitor ABT-530. [Accessed August 8, 2016 2016];NATAP. ASLD. 2015 http://www.natap.org/2015/AASLD/AASLD_06.htm.
- 79.Muir ASS, Wang Stanley3, Shafran Stephen4, Bonacini Maurizio5, Kwo Paul Y6, Wyles David L7, Gane Edward8, Lovell Sandra S3, Lin Chih-Wei3, Ng Teresa I3, Kort Jens3, Mensa Federico J3. HIGH SVR RATES WITH ABT-493 + ABT-530 CO-ADMINISTERED FOR 8 WEEKS IN NON-CIRRHOTIC PATIENTS WITH HCV GENOTYPE 3 INFECTION. [Accessed August 5, 2016 2016];NATAP. 2016 http://www.natap.org/2016/EASL/EASL_30.htm.
- 80.Gane E, et al., editors. Phase 2, Randomized, Open-Label Clinical Trials of the Efficacy and Safety of Grazoprevir and MK-3682 (NS5B Polymerase Inhibitor) with Either Elbasvir or MK-8408 (NS5A Inhibitor) in patients with Chronic HCV GT1, 2, or 3 Infection (Part A of C-Crest 1 & 2). AASLD; November 13 – 17 2015; San Francisco. [Google Scholar]
- 81.Gane EJ, Schwabe C, Hyland RH, Yang Y, Svarovskaia E, Stamm LM, et al. Efficacy of the Combination of Sofosbuvir, Velpatasvir, and the NS3/4A Protease Inhibitor GS-9857 in Treatment-Naive or Previously Treated Patients With Hepatitis C Virus Genotype 1 or 3 Infections. Gastroenterology. 2016;151(3):448–56e1. doi: 10.1053/j.gastro.2016.05.021. [DOI] [PubMed] [Google Scholar]
- 82.Dietz J, Schelhorn SE, Fitting D, Mihm U, Susser S, Welker MW, et al. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. Journal of virology. 2013;87(11):6172–81. doi: 10.1128/jvi.02778-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barzon L, Lavezzo E, Militello V, Toppo S, Palu G. Applications of next-generation sequencing technologies to diagnostic virology. International journal of molecular sciences. 2011;12(11):7861–84. doi: 10.3390/ijms12117861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sarrazin C. The importance of resistance to direct antiviral drugs in HCV infection in clinical practice. Journal of hepatology. 2016;64(2):486–504. doi: 10.1016/j.jhep.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 85.Hernandez D, Zhou N, Ueland J, Monikowski A, McPhee F. Natural prevalence of NS5A polymorphisms in subjects infected with hepatitis C virus genotype 3 and their effects on the antiviral activity of NS5A inhibitors. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology. 2013;57(1):13–8. doi: 10.1016/j.jcv.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 86.Plaza Z, Soriano V, Vispo E, del Mar Gonzalez M, Barreiro P, Seclen E, et al. Prevalence of natural polymorphisms at the HCV NS5A gene associated with resistance to daclatasvir, an NS5A inhibitor. Antiviral therapy. 2012;17(5):921–6. doi: 10.3851/imp2091. [DOI] [PubMed] [Google Scholar]
- 87.Nakamoto S, Kanda T, Wu S, Shirasawa H, Yokosuka O. Hepatitis C virus NS5A inhibitors and drug resistance mutations. World journal of gastroenterology. 2014;20(11):2902–12. doi: 10.3748/wjg.v20.i11.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liverpool Uo. [Accessed 9/8/2016];Hep Drug Interaction Checker. 2016 http://www.hep-druginteractions.org/
- 89.Renet S, Chaumais MC, Antonini T, Zhao A, Thomas L, Savoure A, et al. Extreme bradycardia after first doses of sofosbuvir and daclatasvir in patients receiving amiodarone: 2 cases including a rechallenge. Gastroenterology. 2015;149(6):1378–80e1. doi: 10.1053/j.gastro.2015.07.051. [DOI] [PubMed] [Google Scholar]
- 90.FDA U. Hepatitis C Treatments Containing Sofosbuvir in Combination With Another Direct Acting Antiviral Drug: Drug Safety Communication - Serious Slowing of Heart Rate When Used With Antiarrhythmic Drug Amiodarone. US FDA; 2015. [Accessed August 6, 2016 2016]. http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm439662.htm. [Google Scholar]
- 91.Back DJ, Burger DM. Interaction between amiodarone and sofosbuvir-based treatment for hepatitis C virus infection: potential mechanisms and lessons to be learned. Gastroenterology. 2015;149(6):1315–7. doi: 10.1053/j.gastro.2015.09.031. [DOI] [PubMed] [Google Scholar]
- 92.Fontaine H, Lazarus A, Pol S, Pecriaux C, Bagate F, Sultanik P, et al. Bradyarrhythmias Associated with Sofosbuvir Treatment. The New England journal of medicine. 2015;373(19):1886–8. doi: 10.1056/NEJMc1505967#SA1. [DOI] [PubMed] [Google Scholar]
- 93.Liu GTC, Rajamani S, Ray A, Stamm L, Vick Effect of amiodarone and HCV direct-acting antiviral agents on cardiac conduction in nonclinical studies. In the 2016 International Liver Congress; 2016. [Google Scholar]
- 94.Regan CP, Morissette P, Regan HK, Travis JJ, Gerenser P, Wen J, et al. Assessment of the clinical cardiac drug-drug interaction associated with the combination of hepatitis C virus nucleotide inhibitors and amiodarone in guinea pigs and rhesus monkeys. Hepatology (Baltimore, Md) 2016 doi: 10.1002/hep.28752. [DOI] [PubMed] [Google Scholar]
- 95.Sciences G. Package Insert. 2014. Harvoni: Highlights of Prescribing Information. [Google Scholar]
- 96.SCiences G. EPCLUSA Highlights of Prescribing Information. 2016 https://www.gilead.com/~/media/files/pdfs/medicines/liver-disease/epclusa/epclusa_pi.pdf?la=en.
- 97.Terrault N, ZSDBA, Lim JK, Pockros PJ, Frazier LM, Kuo A, Lok AS, Shiffman ML, Ben, Ari ZST, Sulkowski MS, Fried MW, Nelson DR. Treatment outcomes with 8, 12 and 24 week regimens of ledipasvir/sofosbuvir for the treatment of hepatitis C infection: analysis of a multicenter prospective, observational study. Hepatology (Baltimore, Md) 62:256A. [Google Scholar]
- 98.Tapper EB, Bacon BR, Curry MP, Dieterich DT, Flamm SL, Guest LE, et al. Evaluation of Proton Pump Inhibitor Use on Treatment Outcomes with Ledipasvir and Sofosbuvir in a Real-World Cohort Study. Hepatology (Baltimore, Md) 2016 doi: 10.1002/hep.28782. [DOI] [PubMed] [Google Scholar]