

Abstract
Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome (OMIM# 601338) is a rare autosomal dominant disorder characterized by episodic, fever-induced ataxic encephalopathy in childhood with residual symptoms. All identified patients have the same heterozygous missense variant c.2452G>A (p.Glu818Lys) in the ATP1A3 gene, encoding Na+/K+ ATPase α3. We describe a large CAPOS pedigree with three generations of affected members, the first ascertained in the United States. Deafness, optic atrophy, and pes cavus were present in all three members of the family evaluated. In addition, one of the affected individuals experienced markedly worsening features during her three pregnancies and in the immediate postpartum period, a potential element of the natural history of CAPOS previously unreported. We conclude that the triggering factors and clinical spectrum of pathogenic ATP1A3 variants may be broader than previously described. Targeted sequencing of ATP1A3 should be considered in any patient presenting with cerebellar ataxia triggered by febrile illness, or pregnancy and delivery, especially in the presence of sensorineural hearing loss, optic atrophy, pes cavus, or early childhood history of acute encephalopathic ataxia. Prophylactic administration of acetazolamide or flunarizine may prevent acute episodes of ataxia or mitigate neurologic symptoms, although their efficacies have not been well studied.
Keywords: ATP1A3, CAPOS syndrome, cerebellar ataxia, optic atrophy, pregnancy, sensorineural hearing loss
1 | INTRODUCTION
Pathogenic variants in ATP1A3 have been implicated in a phenotypic spectrum of autosomal dominant disorders including Alternating Hemiplegia of Childhood (AHC, OMIM 614820), Rapid-onset Dystonia-Parkinsonism (RDP, OMIM 128235) and Cerebellar ataxia, Areflexia, Pes cavus, Optic atrophy, and Sensorineural hearing loss syndrome (CAPOS syndrome, OMIM 601338) (de Carvalho Aguiar et al., 2004; Demos et al., 2014; Heinzen et al., 2014; Paciorkowski et al., 2015; Rosewich et al., 2012; Sweney, Newcomb, & Swoboda, 2015). Marked genetic heterogeneity exists for AHC and RDP, including possible genotype-phenotype correlation (Yang et al., 2014) and mosaicism (Hully et al., 2017). By way of contrast, thus far a single recurrent missense pathogenic variant c.2452G>A (p.Glu818Lys) in the ATP1A3 gene, encoding a P-type cation transporter Na+/K+ adenosine triphosphatase (ATPase), has been identified for all individuals with CAPOS syndrome (Demos et al., 2014; Maas, Schieving, Schouten, Kamsteeg, & van de Warrenburg, 2016; Nicolaides, Appleton, & Fryer, 1996). CAPOS syndrome is characterized by episodic, fever-induced ataxic encephalopathy in childhood with variable chronic progressive course without full recovery.
We describe a large CAPOS pedigree with three generations of affected members, the first such family ascertained in the United States. One of the affected individuals experienced markedly worsening features during her three pregnancies and in the immediate postpartum period, a feature of the natural history of CAPOS syndrome previously unreported.
2 | CLINICAL REPORT
Informed consent was obtained from the family. Participation in our report was exempt from IRB review given the small sample size.
2.1 | Patient 1
The proband initially presented at 9 months of age to an outside hospital with sudden-onset weakness and ataxia after 10 days of febrile illness and was transferred to our institution on day 5 of admission. He was born to a 26 year old G4P2012 woman at full term via spontaneous vaginal delivery. Past medical history was significant for a skull fracture at 8 months of age when his mother fell while carrying him. He achieved normal developmental milestones until presentation at 9 months of age. On physical examination at admission, he was non-dysmorphic and had normal growth parameters (i.e., length 69.0 cm [8.8th percentile], weight 9.30 kg [40.5th percentile], head circumference 48.8 cm [+2.25 SD]). He was noted to have head titubation, optic nerve pallor, horizontal nystagmus, diffuse hyporeflexia, mild truncal hypotonia, and pes cavus. Initial blood culture at the outside hospital grew gram-positive cocci in chains due to possible contamination, and he was empirically treated with ceftriaxone; repeat septic workup, stool, and respiratory PCR were negative. Lumbar puncture was performed and CSF culture and gram stain were negative. CSF red blood cells, nucleated cells, protein, and glucose were within normal limits. Blood glucose, ammonia, lactic acid, and complete blood count were within normal limits. Brain MRI and electroencephalogram were normal; hearing test showed bilateral moderate sensorineural hearing loss. Comprehensive urine toxicology was unremarkable. To date, the patient has had two acute episodes of ataxia, both associated with febrile illness. He has not been treated with acetazolamide.
Family history is significant for two older healthy siblings. The patient’s mother and maternal grandfather (patients 2 and 3, respectively) also have episodic and progressive ataxia, bilateral sensorineural hearing loss, optic atrophy, and areflexia. Family pedigree demonstrates an autosomal dominant inheritance pattern with likely complete penetrance across three generations (Figure 1). The remainder of the family history was unremarkable. The patient is of mixed Scandinavian heritage and there was no known consanguinity.
FIGURE 1.
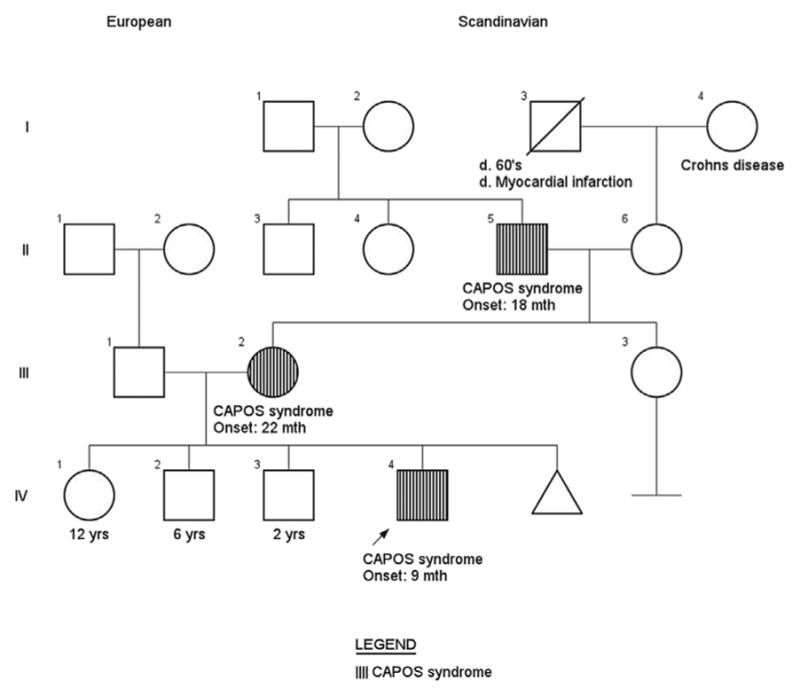
Family pedigree with three generations affected by CAPOS syndrome
Given this dominant pattern of inheritance and unique constellation of features, genomic DNA of all 23 coding exons and intronic/exonic boundaries of the ATP1A3 gene were sequenced by a CLIA-certified laboratory, revealing a heterozygous pathogenic missense variant c.2452G>A (p.Glu818Lys). This variant is consistently reported as pathogenic and disease-specific in multiple unrelated individuals with CAPOS syndrome (Demos et al., 2014; Heimer et al., 2015; Maas et al., 2016; Potic, Nmezi, & Padiath, 2015).
2.2 | Patient 2
The proband’s mother presented initially at 18 months of age with an acute episode of weakness and neurologic deterioration associated with a febrile illness. At the time of initial evaluation, she had mild gait and limb ataxia with areflexia, sensorineural hearing loss, and optic nerve pallor. These symptoms worsened acutely during subsequent febrile illnesses during childhood, but persisted and were also mildly progressive in between febrile episodes. She was evaluated in childhood by a neurogeneticist for persistent ataxia, optic nerve atrophy, and mild developmental delay. Genetic testing for triplet repeat expansions in the SCA1, 2, 3, 6, 7 genes associated with spinocerebellar ataxia was normal. Nerve conduction studies showed absent sural nerve response in the patient, and her head MRI was normal. The causative gene and diagnosis of CAPOS syndrome was unknown at the time of her evaluation, and further genetic testing was not pursued.
Patient 2’s symptoms remained relatively stable in early adulthood, and she was able to attain a high school degree with educational assistance. She then experienced severe, step-wise exacerbation of her neurologic symptoms during all three pregnancies. In her first pregnancy, her sensorineural hearing loss progressively worsened, culminating in severity during the intrapartum and postpartum period, and she had hearing aids placed. During her second and third pregnancies, she experienced both deteriorating vision and ataxia. After her second pregnancy, she met criteria for legal blindness. Her gait ataxia was progressively exacerbated during the third pregnancy and post-partum period, which contributed to her fall while carrying the proband. She also reports migraine headaches and anxiety. She has had six lifetime episodes of acute neurologic deterioration; the most severe of these were associated with her three pregnancies.
2.3 | Patient 3
The proband’s maternal grandfather presented at 22 months of age with ataxic gait and hearing loss after a febrile episode. He has had three lifetime episodes and is currently in his late 50s with almost complete bilateral hearing loss and vision loss. He otherwise is healthy without any cognitive deficits. None of the three patients have had seizures or been treated with acetazolamide.
3 | DISCUSSION
ATP1A3 encodes the α3 isomer of Na+/K+ ATPase in humans, which is predominantly expressed in neural tissues and heterogeneously expressed in other tissues such as inner ear membranes, optic nerves, and muscle spindle fibers (Bøttger et al., 2011; Dobretsov & Stimers, 2005). In the nervous system, ATP1A3 is highly expressed in the dorsal root ganglia, cerebellar cortex, and GABAergic neurons in the basal ganglia, which aligns with the diverse spectrum of motor and extrapyramidal symptoms seen in ATP1A3-associated disorders (Bøttger et al., 2011; Romanovsky, Moseley, Mrak, Taylor, & Dobretsov, 2007). Within the cochlea, it is found in membranes of the spiral ganglion somata and organ of Corti, affecting the innervation pathways of inner hair cell synapses (McLean, Smith, Glowatzki, & Pyott, 2009). In the eyes, ATP1A3 is predominantly expressed in the inner segments of photoreceptor inner segments and optic nerve fibers (McGrail & Sweadner, 1990). Taken together, the distribution and pattern of expression of the α3 isoform in different tissues unify the interesting constellation of features seen nearly ubiquitously in all patients with CAPOS syndrome. Clinical features of the 25 reported cases of CAPOS syndrome in the literature are summarized in Table 1.
TABLE 1.
Clinical features of patients with CAPOS syndrome
| Nicolaides et al. (1996) | Demos et al. (2014) | Rosewich et al. (2014) | Heimer et al. (2015) | Potic et al. (2015) | Maas et al. (2016) | Current cases 2017 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | 25 |
| Demographics | |||||||||||||||||||||||||
| Gender | M | F | F | F | M | M | M | F | F | M | M | F | M | F | M | M | F | M | F | F | M | F | M | F | M |
| Age of onset | 16 m | 18 m | 9 m | 5 y | 9 m | 18 m | 6 m | 3 y | 1 y | 3 y | 20 m | 2 y | 9 m | 4 y | 4 y | 3 y | 4 y | 4 y | 1 y | 4 y | 3 y | 3 y | 22 m | 18 m | 9 m |
| Number of episodes | 3 | 1 | 1 | 1 | 3 | 1 | 2 | 3 | 3 | 2 | 2 | 2 | 2 | 2 | 1 | 1 | 2 | 1 | 2 | 2 | 1 | 3 | 3 | 6 | 2 |
| Ethnicity | British | French Canadian | British | German | Israeli | Slavic | Dutch | Mixed European | |||||||||||||||||
| Symptoms | |||||||||||||||||||||||||
| Cerebellar ataxia | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Areflexia | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Hearing loss | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Optic atrophy | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Pes cavus | + | + | + | + | + | + | + | + | + | ||||||||||||||||
| Dysarthria | + | + | + | + | + | + | + | + | + | ||||||||||||||||
| Dysphagia | + | + | + | + | + | ||||||||||||||||||||
| Hypotonia | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||||
| Lethargy | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |||||||||||
| Strabismus | + | + | + | + | + | + | + | + | |||||||||||||||||
| Nystagmus | + | + | + | + | + | + | + | + | + | + | + | + | + | ||||||||||||
| Dystonia | + | + | + | + | |||||||||||||||||||||
| Autism | + | + | + | ||||||||||||||||||||||
| Anxiety | + | + | + | + | |||||||||||||||||||||
| LV enlargement | + | + | |||||||||||||||||||||||
| Full CAPOS | + | + | + | + | + | + | + | + | + | ||||||||||||||||
Although the specific pathogenic mechanism of the α3 isomer remains to be fully elucidated, it is likely required for neurotransmitter reuptake and rapid restoration of the resting membrane potential after depolarization (Kristensen et al., 2011). Functional disruption of the Na+/K+ ATPase in murine models has led to varying motor deficits, learning disability, gait imbalance, and seizures (DeAndrade, Yokoi, van Groen, Lingrel, & Li, 2011; Holm & Lykke-Hartmann, 2016; Ikeda et al., 2013; Sugimoto, Ikeda, & Kawakami, 2014). Heat increases transmembrane ion permeability in P-type ATPases and causes improper coupling of the ATPase cycle likely through a gain-of-function mechanism (Kaneko, Desai, & Cook, 2014; Lingrel, Williams, Vorhees, & Moseley, 2007; Moseley et al., 2007; Shull et al., 2003). In drosophila studies, subsequently replicated in mammalian cells, the motor dyscoordination phenotype and ion leakage were exacerbated by exposure to higher temperatures (i.e., between 37°C and 42°C) (Kaneko et al., 2014). The p.Glu818Lys pathogenic variant in ATP1A3 is located within a mutation cluster associated with milder phenotypes, close to the transmembrane domain M6 encoded by exon 17 (Panagiotakaki et al., 2015). The substitution of a negatively charged glutamic acid with a positively charged lysine is also thought to destabilize the structure and alter functioning of ATPase (Demos et al., 2014; Heimer et al., 2015).
During pregnancy, maternal core and skin temperature may increase by 0.5°C due to increased metabolic demands and basal metabolic rate, thermogenic effects of progesterone, and fetal heat generation (Blackburn, 2014), potentially explaining why Patient 2 had significantly more episodes and more severe neurologic manifestations compared to other patients previously reported in literature. During the intrapartum period, maternal body temperature also rises due to increased physical exertion from uterine contractions and hypothalamic thermoregulatory center stimulation from fetal-placental factors (Schouten et al., 2008). In a study of 147 women during the intrapartum period, 95% had body temperatures between 36.2°C and 37.8°C (Bartholomew, Ashkin, Schiffman, & Larsen, 2002). Epidural analgesia and prolonged labor have also been associated with increased maternal body temperature (Lieberman et al., 1997; Mercier & Benhamou, 1997).
Acetazolamide, a carbonic anhydrase inhibitor, and flunarizine, a non-selective calcium receptor inhibitor, have been demonstrated to reduce the severity and frequency of hemiplegic attacks in patients with AHC (Camfield & Andermann, 2006; Chi et al., 2012). Acetazolamide has also been previously reported to prevent ataxic episodes in two patients with CAPOS syndrome, although these treatment efficacies have not been systematically studied (Maas et al., 2016).
We conclude that the triggering factors and clinical spectrum of pathogenic ATP1A3 variants may be broader than previously described. Recently, sudden unexpected death in epilepsy, cardiac structural abnormalities, episodic prolonged apnea, and postnatal microcephaly have been reported in patients with ATP1A3-related disorders (Dard et al., 2015; Paciorkowski et al., 2015; Rosewich et al., 2012; Rosewich, Weise, Ohlenbusch, Gärtner, and Brockmann, 2014). Atypical and overlapping features of AHC, RDP, and CAPOS syndrome with intermediate phenotypes have been reported in patients with novel variants p.Arg756His and p.Arg756Cys in ATP1A3 (Jaffer et al., 2017; Kanemasa et al., 2016). While clinical diagnostic criteria for AHC and RDP have been established, supportive features remain the only diagnostic clues for CAPOS syndrome (Rosewich et al., 2017). We propose widening of these criteria to recommend targeted sequencing of ATP1A3 in any patient presenting with cerebellar ataxia triggered by a febrile illness, especially in the presence of any or all of the following: sensorineural hearing loss, optic atrophy, and pes cavus. If these features are exacerbated during pregnancy, careful ascertainment of medical and family history should be obtained, with low threshold for ATP1A3 molecular analysis. Further research into the cognitive behavioral phenotypes and pathophysiologic impact of pregnancy on progression of disease may further elucidate the exact mechanism of neurologic exacerbations.
Acknowledgments
Funding information
NIH Clinical Center, Grant number: T32GM007454
We thank the family for cooperating in our study. We also thank our neurology colleagues Dr. John Carter and Jason Lockrow for their clinical expertise. IC is supported by the National Institutes of Health T32GM007454.
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no relevant financial conflicts of interests.
References
- Bartholomew ML, Ashkin E, Schiffman A, Larsen JW. Maternal temperature variation during parturition. Obstetrics and Gynecology. 2002;100:642–647. doi: 10.1016/s0029-7844(02)02163-4. [DOI] [PubMed] [Google Scholar]
- Blackburn ST. Maternal, Fetal, and Neonatal Physiology: A Clinical Perspective. 4. Maryland Heights: MO: Elsevier Saunders; 2014. pp. 657–660. [Google Scholar]
- Bøttger P, Tracz Z, Heuck A, Nissen P, Romero-Ramos M, Lykke-Hartmann K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. Journal of Comparative Neurology. 2011;519:376–404. doi: 10.1002/cne.22524. [DOI] [PubMed] [Google Scholar]
- Camfield P, Andermann F. Successful treatment of two cases of alternating hemiplegia with acetazolamide. Neuropediatrics. 2006;37:PS2_5_ 3. [Google Scholar]
- Chi LY, Zhao XH, Liu XW, Jiang WJ, Chi ZF, Wang SJ. Alternating hemiplegia of childhood in chinese following long-term treatment with flunarizine or topiramate. International Journal of Neuroscience. 2012;122:506–510. doi: 10.3109/00207454.2012.683216. [DOI] [PubMed] [Google Scholar]
- Dard R, Mignot C, Durr A, Lesca G, Sanlaville D, Roze E, Mochel F. Relapsing encephalopathy with cerebellar ataxia related to an ATP1A3 mutation. Developmental Medicine & Child Neurology. 2015;57:1183–1186. doi: 10.1111/dmcn.12927. [DOI] [PubMed] [Google Scholar]
- de Carvalho Aguiar P, Sweadner KJ, Penniston JT, Zaremba J, Liu L, Caton M, … Ozelius LJ. Mutations in the Na+/K+ –ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia parkinsonism. Neuron. 2004;43:169–175. doi: 10.1016/j.neuron.2004.06.028. [DOI] [PubMed] [Google Scholar]
- DeAndrade MP, Yokoi F, van Groen T, Lingrel JB, Li Y. Characterization of Atp1a3 mutant mice as a model of rapid-onset dystonia with parkinsonism. Behavioural Brain Research. 2011;216:659–665. doi: 10.1016/j.bbr.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demos MK, van Karnebeek CD, Ross CJ, Adam S, Shen Y, … Zhan SH. A novel recurrent mutation in ATP1A3 causes CAPOS syndrome. Orphanet Journal of Rare Diseases. 2014;9:15. doi: 10.1186/1750-1172-9-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobretsov M, Stimers JR. Neuronal function and alpha3 isoform of the Na/K-ATPase. Frontiers in Bioscience: A Journal and Virtual Library. 2005;10:2373–2396. doi: 10.2741/1704. [DOI] [PubMed] [Google Scholar]
- Heimer G, Sadaka Y, Israelian L, Feiglin A, Ruggieri A, Marshall CR, … Ben Zeev B. Caos—episodic cerebellar ataxia, areflexia, optic atrophy, and sensorineural hearing loss. Journal of Child Neurology. 2015;30:1749–1756. doi: 10.1177/0883073815579708. [DOI] [PubMed] [Google Scholar]
- Heinzen EL, Arzimanoglou A, Brashear A, Clapcote SJ, Gurrieri F, Goldstein DB, … Vilsen B. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurology. 2014;13:503–514. doi: 10.1016/S1474-4422(14)70011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm TH, Lykke-Hartmann K. Insights into the pathology of the α3 Na(+)/K(+)-ATPase ion pump in neurological disorders; lessons from animal models. Frontiers in Physiology. 2016;7:209. doi: 10.3389/fphys.2016.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hully M, Ropars J, Hubert L, Boddaert N, Rio M, Bernardelli M, … Bahi-Buisson N. Mosaicism in ATP1A3-related disorders: Not just a theoretical risk. Neurogenetics. 2017;18:23–28. doi: 10.1007/s10048-016-0498-9. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Satake S, Onaka T, Sugimoto H, Takeda N, Imoto K, Kawakami K. Enhanced inhibitory neurotransmission in the cerebellar cortex of Atp1a3-deficient heterozygous mice. Journal of Physiology. 2013;591:3433–3449. doi: 10.1113/jphysiol.2012.247817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffer F, Fawcett K, Sims D, Heger A, Houlden H, Hanna MG, … Sisodiya SM. Familial childhood-onset progressive cerebellar syndrome associated with the ATP1A3 mutation. Neurology Genetics. 2017;3:e145. doi: 10.1212/NXG.0000000000000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanemasa H, Fukai R, Sakai Y, Torio M, Miyake N, Lee S, … Hara T. De novo p.Arg756Cys mutation of ATP1A3 causes an atypical form of alternating hemiplegia of childhood with prolonged paralysis and choreoathetosis. BMC Neurology. 2016;16:174. doi: 10.1186/s12883-016-0680-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Desai BS, Cook B. Ionic leakage underlies a gain-of-function effect of dominant disease mutations affecting diverse P-type ATPases. Nature Genetics. 2014;46:144–151. doi: 10.1038/ng.2850. [DOI] [PubMed] [Google Scholar]
- Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, … Gether U. SLC6 neurotransmitter transporters: Structure, function, and regulation. Pharmacological Reviews. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- Lieberman E, Lang JM, Frigoletto F, Richardson DK, Ringer SA, Cohen A. Epidural analgesia, intrapartum fever, and neonatal sepsis evaluation. Pediatrics. 1997;99:415–419. doi: 10.1542/peds.99.3.415. [DOI] [PubMed] [Google Scholar]
- Lingrel JB, Williams MT, Vorhees CV, Moseley AE. Na,K-ATPase and the role of α isoforms in behavior. Journal of Bioenergetics and Biomembranes. 2007;39:385–389. doi: 10.1007/s10863-007-9107-9. [DOI] [PubMed] [Google Scholar]
- Maas RPPWM, Schieving JH, Schouten M, Kamsteeg EJ, van de Warrenburg BPC. The genetic homogeneity of CAPOS syndrome: Four new cases with the c.2452G>A (p.Glu818Lys) mutation in the ATP1A3 gene. Pediatric Neurology. 2016;59:71–75. e1. doi: 10.1016/j.pediatrneurol.2016.02.010. [DOI] [PubMed] [Google Scholar]
- McGrail KM, Sweadner KJ. Complex expression patterns for Na+,K+–ATPase isoforms in retina and optic nerve. The European Journal of Neuroscience. 1990;2:170–176. doi: 10.1111/j.1460-9568.1990.tb00409.x. [DOI] [PubMed] [Google Scholar]
- McLean WJ, Smith KA, Glowatzki E, Pyott SJ. Distribution of the Na,K-ATPase alpha subunit in the rat spiral ganglion and organ of corti. Journal of the Association for Research in Otolaryngology. 2009;10:37–49. doi: 10.1007/s10162-008-0152-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier FJ, Benhamou D. Hyperthermia related to epidural analgesia during labor. International Journal of Obstetric Anesthesia. 1997;6:19–24. doi: 10.1016/s0959-289x(97)80047-7. [DOI] [PubMed] [Google Scholar]
- Moseley AE, Williams MT, Schaefer TL, Bohanan CS, Neumann JC, Behbehani MM, … Lingrel JB. Deficiency in Na,K-ATPase α isoform genes alters spatial learning, motor activity, and anxiety in mice. The Journal of Neuroscience. 2007;27:616–626. doi: 10.1523/JNEUROSCI.4464-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolaides P, Appleton RE, Fryer A. Cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS): A new syndrome. Journal of Medical Genetics. 1996;33:419–421. doi: 10.1136/jmg.33.5.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paciorkowski AR, McDaniel SS, Jansen LA, Tully H, Tuttle E, Ghoneim DH, … Hahn S. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia. 2015;56:422–430. doi: 10.1111/epi.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagiotakaki E, De Grandis E, Stagnaro M, Heinzen EL, Fons C, … Sisodiya S. Clinical profile of patients with ATP1A3 mutations in Alternating Hemiplegia of Childhood-a study of 155 patients. Orphanet Journal of Rare Diseases. 2015;10:123. doi: 10.1186/s13023-015-0335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potic A, Nmezi B, Padiath QS. CAPOS syndrome and hemiplegic migraine in a novel pedigree with the specific ATP1A3 mutation. Journal of the Neurological Sciences. 2015;358:453–456. doi: 10.1016/j.jns.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Romanovsky D, Moseley AE, Mrak RE, Taylor MD, Dobretsov M. Phylogenetic preservation of α3 Na+,K+-ATPase distribution in vertebrate peripheral nervous systems. Journal of Comparative Neurology. 2007;500:1106–1116. doi: 10.1002/cne.21218. [DOI] [PubMed] [Google Scholar]
- Rosewich H, Thiele H, Ohlenbusch A, Maschke U, Altmüller J, Frommolt P, … Gärtner J. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: A whole-exome sequencing gene-identification study. Lancet Neurology. 2012;11:764–773. doi: 10.1016/S1474-4422(12)70182-5. [DOI] [PubMed] [Google Scholar]
- Rosewich H, Weise D, Ohlenbusch A, Gärtner J, Brockmann K. Phenotypic overlap of alternating hemiplegia of childhood and CAPOS syndrome. Neurology. 2014;83:861–863. doi: 10.1212/WNL.0000000000000735. [DOI] [PubMed] [Google Scholar]
- Rosewich H, Sweney MT, DeBrosse S, Ess K, Ozelius L, Andermann E, … Swoboda K. Research conference summary from the 2014 international task force on ATP1A3-related disorders. Neurology Genetics. 2017;3:e139. doi: 10.1212/NXG.0000000000000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten FD, Wolf H, Smit BJ, Bekedam DJ, De Vos R, Wahlen I. Maternal temperature during labour. BJOG: An International Journal of Obstetrics & Gynaecology. 2008;115:1131–1137. doi: 10.1111/j.1471-0528.2008.01781.x. [DOI] [PubMed] [Google Scholar]
- Shull GE, Okunade G, Liu LH, Kozel P, Periasamy M, Lorenz JN, Prasad V. Physiological functions of plasma membrane and intracellular Ca2+ pumps revealed by analysis of null mutants. Annals of the New York Academy of Sciences. 2003;986:453–460. doi: 10.1111/j.1749-6632.2003.tb07229.x. [DOI] [PubMed] [Google Scholar]
- Sugimoto H, Ikeda K, Kawakami K. Heterozygous mice deficient in Atp1a3 exhibit motor deficits by chronic restraint stress. Behavioural Brain Research. 2014;272:100–110. doi: 10.1016/j.bbr.2014.06.048. [DOI] [PubMed] [Google Scholar]
- Sweney MT, Newcomb TM, Swoboda KJ. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, CAPOS and beyond. Pediatric eurology. 2015;52:56–64. doi: 10.1016/j.pediatrneurol.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Gao H, Zhang J, Xu X, Liu X, Wu X, … Zhang Y. ATP1A3 mutations and genotype-phenotype correlation of alternating hemiplegia of childhood in chinese patients. PLoS ONE. 2014;9:e97274. doi: 10.1371/journal.pone.0097274. [DOI] [PMC free article] [PubMed] [Google Scholar]