

Abstract
Circulating tumor cells (CTCs) are extremely rare cells found in blood of metastatic cancer patients. There is a need for inexpensive technologies for fast enrichment of CTCs from large blood volumes. Previous data showed that antibody-conjugated lipid shell immuno-microbubbles (MBs) bind and isolate cells from biological fluids by flotation. Here, blood-stable MBs targeted to several surface markers for isolation of breast tumor cells were developed. MBs coated with anti-human EpCAM antibodies showed efficient binding of EpCAM+ breast cancer cell lines SKBR-3, MCF-7, and MDA-MB-453, whereas anti-human EGFR MBs showed binding of EpCAMLOW/NEGATIVE cell lines MDA-MB-231 and BT-549. Multitargeted anti-human EpCAM/EGFR MBs bound all cell lines with over 95% efficiency. Highly concentrated MB-bound tumor cells were collected in a microliter volume via an inverted vacuum-assisted harvesting setup. Using anti-EpCAM and/or anti-EpCAM/EGFR MBs, an efficient (70–90%) recovery and fast (30 min) isolation of the above-mentioned cells and cell clusters was achieved from 7.5 mL of spiked human blood. Using anti-EpCAM MBs and anti-EpCAM/EGFR MBs, cytokeratin-positive, CD45-negative CTCs were detected in 62.5% (10/16) of patients with metastatic breast cancer and CTC clusters were detected in 41.7% (5/12) of CTC-positive samples. Moreover, in some samples MBs isolated cytokeratin positive, CD45 negative tumor-derived microparticles. None of these structures were detected in blood from non-epithelial malignancies. The fast and inexpensive multitargeted platform for batch isolation of CTCs can promote research and clinical applications involving primary tumors and metastases.
Keywords: CTC, microbubbles, EpCAM, antibody, breast cancer, EGFR
Graphical Abstract
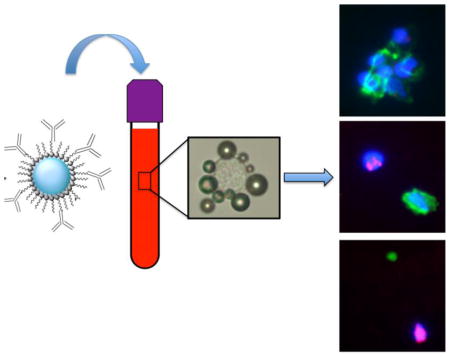
INTRODUCTION
Circulating tumor cells (CTCs) are cells of solid tumor origin found in the blood of metastatic cancer patients 1–3. In accord with the personalized therapy concept, CTC could provide invaluable information for monitoring of metastatic process and could be used to test novel therapies 1,4,5. CTCs could be used to study cancer biology, clinical prognosis, responsiveness to anticancer treatment, neoantigen discovery for immunotherapy, and in vitro culturing 6–10. For these purposes, technologies that can selectively and inexpensively isolate CTCs are extremely important. In the field of CTC enrichment and isolation, several approaches exist: a) positive selection based on surface markers or physical properties (mostly size and rigidity); b) negative selection through depletion of majority (but not all) leukocytes; 3) selection-free methods based on immunostaining with subsequent high throughput imaging or flow cytometry. 7,11 These approaches are implemented using various platform technologies, such as immunomagnetic beads, filter membranes or microfluidic chips. Each of these platforms has advantages and disadvantages, mostly related to the possibility of missing certain cell types, sample preparation time, final CTC quality and purity, or requirement for expensive high throughput processing equipment and technical skills.
Positive selection was the first clinically adopted approach to isolate CTCs. The main platforms used for positive enrichment are immunomagnetic beads (e.g., FDA-approved CTC isolation kit CellSearch®) and microfluidics (e.g., micropost CTC chip 12,13, most of them utilizing Epithelial Cell Adhesion Molecule (EpCAM) as the selection marker. The sensitivity of detection of EpCAM positive cells with these platforms is high, often as few as 1 CTC per 106–108 leukocytes 14. The main concern with the positive enrichment is that some tumor cells could be missed. Indeed, some CTCs appear to have low or no expression of EpCAM due to epithelial-to-mesenchymal transition (EMT 15,16). While the role of the EMT in the metastatic process is still being elucidated 17, there is massive clinical evidence that EpCAM positive CTCs are an independent prognostic factor 18–20. Moreover, several laboratories demonstrated the utility of multiple positive enrichment markers, such as epidermal growth factor receptor (EGFR), CD44 and human epidermal growth factor receptor (Her2/neu) 15,21–24. The apparent advantages of positive enrichment approaches are sample purity, sensitivity and ability to concentrate cells in a small volume. These advantages are important enough to pursue further development and clinical implementation of positive enrichment technologies.
Recently a novel approach was described for buoyancy-based isolation of tumor cells using lipid microbubbles tagged with antibodies 25–27. Perfluorocarbon-filled microbubbles (MBs) coated with lipids or with serum albumin are biocompatible and have been used as ultrasound contrast agents in the clinic 28,29. It was demonstrated 25,26 that anti-EpCAM antibody tagged lipid MBs could efficiently bind and isolate tumor cells from spiked mouse and human blood, and another group demonstrated sorting of tumor cells in PBS using buoyant albumin MBs30. The advantage of MB technology is that it does not involve sophisticated equipment and requires only a low-speed centrifugation step to quickly separate the MB-attached cells from blood. Moreover, due to MBs being composed of inert gas, they can be easily eliminated from the sample. At the same time, the intrinsic instability of lipid and albumin-based MBs required extensive washing of blood cells to remove plasma. This work reports on the development of blood-stable PEGylated MBs tethered with antibodies against several cell surface markers to capture tumor cells with variable EpCAM expression. We were able to conjugate 10 times more antibodies per MBs than in the previous work25–27. The improved MB formulation enabled fast enrichment of breast cancer cells and cell clusters from spiked normal blood. Moreover, we report that MBs isolate single CTC, CTC clusters and tumor microparticles (TMPs) from blood of metastatic breast cancer patients, which opens up exciting opportunities in liquid biopsy.
MATERIAL AND METHODS
Materials
1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-350] (DSPE-PEG-350), DSPE-PEG-750, DSPE-PEG-1000 and DSPE-PEG-2000 were purchased from Avanti Polar Lipids (Alabaster, AL, USA). 2-distearoyl-sn-glycero-3-phospho-ethanolamine-N-[maleimide(polyethylene glycol)-3400] (DSPE-PEG3400-Malemide) was purchased from Laysan Bio, Inc. (Arab, AL, USA). Polyoxyethylene (40) stearate (PEG40) was purchased from Sigma Aldrich (St. Louis, MO, USA). All lipids were stored in chloroform solution under argon at −20 °C. DiR (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindotricarbocyanine Iodide) was from Biotium (Hayward, CA, USA) and was stored as 2 mM sock in ethanol. Perflorohexane was purchased from Alfa Aesar, Ward Hill, CA. Traut’s reagent (2-Iminothiolane) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). The reagent was dissolved in 1X phosphate-buffered saline (PBS) at 5 mg/ml and stored in aliquots at −20 °C. ZebaTM Spin Desalting Columns were purchased from Thermo Fisher Scientific. Anti-human EpCAM (epithelial cell adhesion molecule) antibody ING-131 was provided by XOMA Corporation (San Francisco, USA). ERBITUX® (cetuximab), the anti-human epidermal growth factor receptor (EGFR) antibody, were obtained from Cancer Center pharmacy of the University of Colorado Denver. Alexa Fluor 488 mouse anti-pan Cytokeratin antibody (clone AE1/AE3) was purchased from eBioscience (San Diego, CA, USA). Alexa Fluor 594 mouse anti-CD45 was purchased from BioLegend (San Diego, CA, USA). All the antibodies that were used in our studies were listed in Table S1. Nuclear staining reagent Hoechst 33342 trihydrochloride tirihydrate was purchased from Life Technologies (Carlsbad, CA, USA). Live/Dead® cell viability assay was purchased from Thermo Fisher Scientific. BD Cytofix/CytopermTM kit was purchased from BD Biosciences (San Diego, CA, USA).
Synthesis of lipid-formulated MBs
MBs were synthesized using DSPC, PEG40, DSPE-PEG3400-Malemide and DSPE-PEG with different PEG length ranging from 350 to 2000 Da. MBs formulations are shown in Table S2. The protocol for immuno-MB synthesis was described by us elsewhere. 25–27. MB size distribution was determined by measuring diameter of MBs in microscopical images taken at 400x magnification using ImageJ software. At least 100 MBs were measured for each formulation. The diameters were plotted as a histogram using Prism 6 (GraphPad). Human anti-human EpCAM antibody on MB surface was detected by fluorescent microscopy using anti-human AlexaFluor 594 IgG (Thermo Fisher), whereas the chimeric mouse/human anti-human EGFR (cetuximab) antibody was detected with anti-mouse AlexaFluor 488 IgG (Thermo Fisher). The number of antibodies per MB was quantified by a dot blot immunoassay as described by us previously.32 Briefly, MBs were destroyed by sonication, and amount equivalent to 2x105 MBs in a 2 μL volume was applied as a dot on a nitrocellulose membrane (Bio-Rad). Standard dilutions of human IgG (Jackson ImmunoResearch) or cetuximab were applied as 2 μL dots on the same membrane (between 12.5 ng and 100 ng protein per dot). The membrane was blocked in 5% milk in T-PBS (0.1% Tween in PBS), and probed with anti-human IRDye800 antibody (Li-COR Biosciences). The number of antibodies per MB was calculated from the standard curve using ImageJ software.
Recovery of MBs from human blood
Human blood samples were purchased from Bioreclamation IVT (Hicksville, NY, USA) with different anticoagulants, including Na heparin, Li heparin, Na2 EDTA, K2 EDTA, and Na citrate. MBs with or without conjugated antibody (0.5x108 MBs) were added to 1 ml of human blood in 2 ml borosilicate glass sample vial, incubated for 15 min with rotation, and then centrifuged at 100g for 30 sec to separate the MBs. Additional PBS was added to blood to bring the level of fluid above the neck of the vial. The layer of MBs was collected from the meniscus onto a piece of clean Parafilm M and then transferred to a 1.5 ml eppendorf tube. MBs were loaded into a hemocytometer for counting, and the percentage of recovered MBs was calculated.
Cell lines
Human breast cancer cell lines (SKBR-3, MCF-7, MDA-MB-231, MDA-MB-453, and BT-549) and human leukemia cell line (MOLM-13) were cultured in proper medium in a cell incubator with 5% CO2 at 37°C. SKBR-3 cells were maintained in McCoy’s 5A medium (Corning Life Sciences, Tewksbury, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Corning Life Sciences). MCF-7 cells were maintained in Minimum Essential Medium (MEM) (Corning Life Sciences) supplemented with 10% FBS. MDA-MB-231 cells were maintained in L-15 medium (ATCC, Manassas, VA, USA) supplemented with 10% FBS. MDA-MB-453 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Corning Life Sciences) supplemented with 10% FBS. BT-549 cells were maintained in Roswell Park Memorial Institute (RPMI) medium (Corning Life Sciences) supplemented with 10% FBS. MOLM-13 cells were maintained in RPMI-1640 medium (Corning Life Sciences) supplemented with 10% FBS and penicillin/streptomycin. .
Cell binding studies
Breast cancer cells at 2×106 cells/ml in PBS were mixed with MBs at MB/cell ratios of 50:1. After rotating at 30 rpm for 15 min, the mixture of cells with MBs was loaded into a hemocytometer for light microscope imaging and cell counting. After 5 min, the MB bound cells separated into a different focal plane from non-bound cells, which allowed bound and non-bound cells to be counted separately and calculate percentage of cells associated with MBs. At least 5 fields were counted for bound and non-bound cells.
Viability assay for cancer cells bound to MBs
MBs (approximately 7.5×105 in 20 μl) were added to 700 μl SKBR-3 cells at a concentration of 100 cells/μl and the mixture was gently rotated for 30 seconds for MB binding to cells. The cell viability assay was performed using live/dead viability/cytotoxicity kit (L3224, Thermo Fisher Scientific) following the manufacture’s protocol. After 1 h incubation of the Live/Dead reagent with cell/MB or cell only as a control at room temperature, the cell/MB mixture of 20 μl was added onto a glass slide and covered with a coverslip. All the groups were conducted in triplicate. Cells were imaged using a Nikon Fluorescence Microscope and live/dead cell numbers were counted.
Isolation of cancer cells from spiked human blood
Anonymous human blood samples with K2 EDTA as the anticoagulant were obtained from Blood Donation Center at the Children’s Hospital Colorado. For DiI labeling of cells, trypsinized breast cancer cells (1×107) were incubated at 100 μM DiI in PBS for 30 min at room temperature. The labeled cells were washed with PBS and stored on ice before use. Before spiking, cells were diluted in 1% BSA/PBS to 1,500–5,000 cells/ml. The blood samples (7.5 ml) were spiked with exactly 20 μL of cell suspension. The loading control group in each experiment was the same volume of DiI labeled cells (20 μL), but directly added to the nitrocellulose membrane. All spiking and loading control experiments were conducted a minimum of three times. After spiking, blood was centrifuged at 1,500 g for 15 min to remove the plasma and resuspended in 5 mL PBS. The samples were then transferred into a 10 mL pipette tip (Mettler-Toledo-Rainin, LLC, Oakland, CA, USA) equipped with a cut 10 mL syringe plunger (BD, Franklin Lakes, NJ, USA), and additional PBS was added until the blood reached the top opening. Anti-human EpCAM MBs, anti-human EGFR MBs or anti-human EpCAM/EGFR MBs were then added to the samples. The complete isolation procedure is described in Fig. 4. The top opening was then sealed, device was rotated at 30 rpm for 15 min, and centrifuged for 1–2 min (100 g) using Thermo RC 6+ centrifuge equipped with FIBERLite® F21S-8×50y fixed angle rotor (with lid removed). After centrifugation, the plunger was pushed up to move the layer of MBs above the tip of the tube. The MB-containing meniscus was vacuum-collected using a customized nitrocellulose membrane disc (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were washed under vacuum with 1 ml ice-cold isopropanol to destroy MBs and then the cells on the membrane were fixed using 4% paraformaldehyde for 30 min at RT. After washing, cell nuclei were stained with Hoechst. The membrane was placed onto a glass slide and DiI labeled cells were counted using a Nikon Fluorescence Microscope with CFI Plan Apochromat Lambda 2x and 20x objective. The cell number for the loading control group was counted and used as the 100% spiked cells for that experiment, and the isolation efficiency was determined as the ratio of cells in the isolated samples to the cells in the loading control sample.
Figure 4. MB harvesting using inverted vacuum setup.

A) Scheme of MB separation setup with open-ended tip and rubber fitted plunger. Depending on the tube size, the setup can accommodate between 5 and 20 ml of blood; B) entire process of cell isolation. 1-MBs are added to blood sample; 2-sample is rotated; 3-separated MB layer after centrifugation; 4-MB layer on top of the meniscus after pushing the plunger up. Inset shows meniscus with separated MB layer (white); 5-harvesting of the layer on a nitrocellulose membrane attached to a vacuum (arrow); 6-harvested MB layer on the membrane. Inset shows that the same MB layer can be transferred to parafilm as a miniscule, highly concentrated droplet.
Isolation of CTCs from blood of breast cancer patients
De-identified metastatic breast cancer blood samples were obtained from consented patients at the University of Colorado Cancer Center under Colorado Multiple Institutional Review Board-approved (COMIRB) protocol #16-0610 and #16-1001. All samples were delivered for CTC isolation within 2 hours post-collection. De-identified blood samples from non-epithelial cancer patients were obtained from consented patients at UC San Diego Moores Cancer Center (IRB protocol #121046) and shipped within 1 day to the University of Colorado (Simberg lab). The blood samples were centrifuged at 1,500 g for 10 min to separate plasma and processed as described above for spiked blood. Anti-human EpCAM MBs or anti-human EpCAM/EGFR MBs were used. After immobilizing MBs and the associated cells on the membrane, the MBs were destroyed by addition of 1 mL of ice-cold isopropanol while keeping the membrane attached to the vacuum. This procedure destroyed MBs but kept all the cells on the membrane. Cells were fixed with 4% formalin, permeabilized using BD Cytofix/CytopermTM kit for 4 h at RT, and then stained using AlexaFluor 488 labeled anti-cytokeratin antibody and AlexaFluor 594 labeled anti-CD45 antibody overnight at 4 °C. After washing, cell nuclei were stained with Hoechst. The membrane was placed onto a glass slide and imaged using a Nikon Fluorescence Microscope with 10x and 20x Objective Lenses. The CTCs were identified based on previously published criteria,33 including CK+/CD45−/Hoechst+ staining, presence of a defined nucleus, and dimensions larger than 4x4 μm. Tumor microparticles were identified as CK+/CD45−/Hoechst- events sized smaller than 4 μm.
RESULTS
a) Development of blood-stable immunomicrobubbles targeted to multiple tumor markers
High MB stability and recovery is a prerequisite for rare CTCs isolation. MB are intrinsically unstable due to: a) Laplace pressure gradient 34; b) perfluorocarbon mixing and exchange with the blood gases 35. Lipid monolayer permeability and perfluorocarbon solubility play a major role in preventing gas exchange and MB collapse 36–38. For preparation of MBs, we used perflorohexane (boiling point 56°C) gas and DSPC (C18) as the main phospholipid due to a solid (gel) phase at ambient temperature (phase transition 55°C 39). In order to improve blood stability and decrease the non-specific binding by blood cells and proteins, phospholipid derivatives tethered with PEGs of different molecular weights (DSPE-PEG350, DSPE-PEG750, DSPE-PEG1000 and DSPE-PEG2000) were included. PEG40-stearate was included in all formulations as an emulsifying agent 36. Each MB formulation contained also DSPE-PEG3400-maleimide at for conjugation of human thiolated IgG via Michael addition (Fig. 1A and 25). Plain DSPC MBs and MBs made with DSPE-PEG2000 (formulations F0 and F1) and the corresponding antibody-conjugated formulations (F0A and F1A) resulted in stable MBs (count over 108/ml at 24h post-preparation), while MBs made with shorter PEG chains did not result in a high MB count (Fig. 1B and Table S2). After storage at 4°C for 7 days, formulations with antibody were more stable than without antibody, and F1A was visibly more stable than F0A (Fig. 1C). Formulations F1 and F1A were tested for stability in human blood anticoagulated with sodium EDTA, sodium heparin, lithium heparin, potassium EDTA and sodium citrate. According to Figure S1, whereas non-antibody MBs showed relatively low recovery (less than 5%) from all anticoagulated blood, the Ab-conjugated formulation F1A showed good recovery (35%) in sodium and potassium EDTA anticoagulated blood. Based on the stability studies the F1A formulation was selected to prepare MBs targeted to multiple tumor markers. Previous studies demonstrated that EGFR could be used as the enrichment marker for CTCs that lost the epithelial marker EpCAM expression due to EMT.23 MBs were conjugated with either single antibody species (EpCAM or EGFR) or with both (EpCAM/EGFR mixed in 1:1 ratio). According to size distribution (Fig. 1D) all MB formulations showed right skewed distribution, with EpCAM, EGFR and EpCAM/EGFR MBs showing 5.1, 5.4 and 3.7 μm median diameters, respectively. Fluorescent immunostaining confirmed efficient conjugation of singe or dual antibodies to MBs (Fig. 1E). Immuno-dot blot assay of antibody content showed that EpCAM, EGFR and EpCAM/EGFR MBs had 1.2x106, 2.0x106 and 1.6x106 IgG molecules per MB, respectively (Extended Supplemental Data). This number agrees well with theoretical calculations: assuming phospholipid crossection of 0.7 nm2, each 5-μm diameter MB should contain approximately 2x106 of DSPE-PEG3400-MAL molecules (for F1A formulation, Fig. 1B). Such high number of IgG molecules suggests that MB surface is entirely covered with IgG molecules (assuming 50nm2 average cross section of an IgG molecule).
Figure 1. Preparation of antibody-coated MBs.

A) Conjugation of Traut’s reagent-modified antibody (red residue) to preformed, perflorohexane gas-filled (blue), PEG-maleimide-decorated MBs; B) Formulations F0 and F1 that showed the best stability upon preparation were selected for Ab conjugation (full list of tested formulations is in Supplemental Table S2); C) Storage stability of MBs at 4°C for 7 days after preparation. Antibody decorated MBs (F0A and F1A) show better stability than the original formulations as evidenced by less coalescence and foaming of MBs; D) size (diameter) distribution of MBs; E) anti-EpCAM/EGFR MBs, anti-EpCAM MBs and anti-EGFR MBs were stained with secondary antibodies (anti-human IgG for anti-EpCAM, anti-mouse IgG for anti-EGFR and both antibodies for anti-EGFR/EpCAM and control MBs). Images confirm that MBs were decorated with one or both antibody types. Note that anti-EGFR MBs have much weaker fluorescence because only a small part of cetuximab is of murine origin. Size bar is 15 μm for all samples.
To test the binding efficiency of single-targeted and multitargeted MBs, human breast adenocarcinoma cell lines SKBR-3, MCF-7, MDA-MB-231, BT-549, and MDA-MB-453 with different levels of EpCAM and EGFR expression were used (Fig. 2A). The expression of these markers on these cells has been thoroughly validated in the literature. 40–42 In particular, EpCAMLOW/NEGATIVE status of MDA-MB-231 and BT-549 has been conformed in numerous studies. 23,40,41,43,44 In addition, the human leukemia cell line MOLM-13 that does not express EpCAM or EGFR was used as a negative control. As summarized in Fig. 2B, the binding efficiency of anti-EpCAM MBs to SKBR-3, MCF-7 and MDA-MB-453 cells was over 95%, but less than 20% to MDA-MB-231 and less then 5% to BT-549 cells. The binding efficiency of anti-EGFR MBs to SKBR-3, BT-549 and MDA-MB-231 cells was over 95%, but less then 35% to EGFRLOW MCF-7 and less then 3% to EGFR- MDA-MB-453 cells. Dual targeted anti-EpCAM/EGFR MBs bound to all breast cancer cell lines with over 95% efficiency. The binding efficiency to MOLM-13 cells that do not express these markers was negligible for all MB types (Fig. 2B). As shown in the representative images (Fig. 2C), anti-EpCAM MBs formed rosettes with SKBR-3 and MCF-7 cells but only few MBs bound to MDA-MB-231 cells. Anti-EGFR MBs entirely covered SKBR-3 and MDA MB-231 cells but showed much less efficient binding to MCF-7 cells. Interestingly, cell clusters were densely coated with MBs (Fig. 2C), suggesting that clusters also could be captured by MBs. These data demonstrate that combination of anti-EpCAM and anti-EGFR can be used to capture cells with variable EpCAM expression.
Figure 2. Binding of antibody-decorated MBs to breast cancer cells with different receptor expression.

A) Marker expression on the breast cancer cell lines (summarized based on published literature 23,40,41,43,44) and B) binding efficiency (graph). Binding efficiency was calculated as described in Methods. Dual EpCAM/EGFR MBs have over 95% binding efficiency to the breast cancer cells with or without EpCAM expression. N=3–5; C) representative cropped microscopical images show MBs and cells on a slide. Whenever binding occurred, MBs and cells formed characteristic rosettes (cells surrounded by multiple dark spheres). Due to buoyancy, MB-bound cells are in the same focal plane with MBs, whereas non-bound cells are in a different focal plane, which facilitated the counting of bound and unbound cells. Note that cell clusters show efficient binding to MBs. Some binding of anti-EpCAM MBs is visible for MDA-MB-231 cells, but much less than for SKBR-3 and MCF-7 cells with high EpCAM expression. No binding was observed for MOLM-13 cells. For MOLM-13 cells, 10x objective was used; for other cells, 40x objective was used.
Perfluorocarbon-based MBs have been used safely in clinic as ultrasound contrast media,28,29 however the effect of targeted MBs on viability of tumor cells is not known. Live/Dead® assay showed that over 98% of SKBR-3 cells bound to anti-EpCAM MBs were viable after 1h of incubation (Fig. 3A and Fig. S1). In order to tested whether MB binding inhibits cell growth, DiI labeled SKBR3 cells were mixed with anti-EpCAM MBs at a ratio 1:20, and seeded in a 96-well plate in the inverted configuration (Fig. 3B). The same number of cells was seeded in the upright configuration as a control (Fig. 3B). After 24h, control cells homogenously grow in the entire well, whereas MB coated cells distributed in the periphery of the wells (Fig. 3B), possibly due to the surface tension and formation of inverted meniscus. The cells were able to attach to the well and proliferate with MBs still bound to the membrane (Fig. 3C–D).
Fig. 3. Effect of MB binding on cell viability and growth.

A) Live/Dead assay showed that MB-bound cells are viable; B) MBs were mixed with DiI labeled SKBR-3 cells and plated in a 96-well plate in the inverted position (10,000 cells/well). Control non-coated cells were grown in the upright position. Low magnification images show the entire well bottom. Cells with and without MBs show different growth patterns in the well after 24 h of culture, likely due to the preferential accumulation of MBs at the edges of the well; C) Cells attached and proliferated in the wells (arrow). Size bar=15 μm
b) Isolation of single breast cancer cells and cell clusters from spiked human blood using targeted MBs
Following separation from blood, the buoyant MB layer needs to be effectively skimmed for subsequent analysis. An inverted isolation device consisting of a 10 mL open-ended narrow tip equipped with a rubber fitted plunger (Fig. 4A) have been used for MB harvesting. Following mixing of MBs with blood (15 min) and brief (1–2 min) centrifugation, the plunger is pushed upwards toward the opening where MBs form a highly concentrated layer on top of the meniscus (Fig. 4A–B). Because of the propensity of MBs to separate from liquid, they were effectively transferred via capillary effect. In order to harvest cells on a solid support for subsequent immunostaining, MB layer was captured under vacuum on a nitrocellulose membrane disk as a 2x2 mm diameter spot (Fig. 4B). In addition, MBs could be harvested on a Parafilm as a 2–5 μL droplet (Fig. 4B inset). In order to test the efficiency of rare tumor cell isolation, 7.5 mL of washed blood was spiked with exactly 20 μL of DiI labeled breast cancer cells (30–100 cells) and different MBs were applied to isolate and count cells. The same 20 μL of cells was applied to the nitrocellulose membrane and the cells were counted as the loading control (Fig. 5A, top). The fact that isolated cells were focused on a small area of the membrane enabled easy observation and enumeration of isolated cells under low magnification objective (Fig. 5A, bottom). MBs efficiently isolated individual tumor cells as well as cell clusters (Fig. 5A, right). Anti-EpCAM MBs captured over 75% of spiked SKBR3 cells, 87% of spiked MCF7 cells (single cells and clusters) and 79% of spiked MDA-MB-453 cells, while anti-EGFR MBs captured 90% of MDA-MB-231 cells and 75% of BT-549 cells (Fig. 5B). Dual anti-EpCAM/EGFR MBs captured between 73 and 92% of SKBR-3, MCF-7, MDA-MB-453, MDA-MB-231 and BT-549 cells (Fig. 5B).
Figure 5. Isolation of spiked human breast cancer cells.

A) DiI labeled cells (30–100) were spiked into 7.5 mL of potassium EDTA human blood and isolated using anti-human EpCAM MBs as described in Fig. 4. As a control, the same number of cells was applied onto a nitrocellulose membrane and counted. Left panel shows representative whole membrane image of SKBR-3 cells captured under low magnification (2x objective lens). The dimensions of the spot containing the cells are approximately 2x2 mm. Right panel shows the isolated cluster of MCF-7 cells under 200x magnification. Scale bar is 100 μm; B) Isolation efficiency of human breast cancer cell lines spiked into normal donors’ blood. Means ± SD are shown. N=3–5.
c) Isolation of CTCs, clusters of CTCs and tumor microparticles from blood of breast cancer patients
After verifying the isolation efficiency from spiked normal blood, a small-scale clinical study was performed under consent using de-identified blood samples from breast cancer patients. The cohort (n=15) consisted of metastatic breast cancer patients with different metastatic sites and hormone receptor expression status (Table 1). EpCAM was chosen as the primary isolation marker for most patients, and in some patients, EpCAM/EGFR MBs were also tested (Table 1). The reasons for using EpCAM MBs despite concerns about EpCAM as the enrichment marker 15 were the following: a) many CTCs retain epithelial markers despite clinical evidence of EMT23,45; b) presence and number of EpCAM-positive CTCs correlate with the clinical course of the disease 46,47; c) EpCAM positive CTCs provide clinically meaningful information about genetic mutations of main tumors 48. MBs were incubated with blood as described in Fig. 5, and once isolated on the membrane, were stained for pan-cytokeratin (CK), CD45 (leukocyte marker) and Hoechst (nuclear staining) (Fig. 6A). The staining protocol was validated for SKBR-3 cells isolated from blood with anti-EpCAM MBs (Fig. 6B). The results of the study are summarized in Table 1. In samples of breast cancer patients, CK+/CD45− CTCs were detected in in 60% (9/15) of patients. Moreover, in patients 01–005, 01–006, 01–008 and 01–015, there were several CK+/CD45− CTC clusters (2–9 cells each), corresponding to 44% (4/9) of CTC-positive patients. Representative images of CTCs and cluster are shown in Figure 6C. Interestingly, when EpCAM/EGFR MBs were used for isolation, a nucleated CK-/CD45−/Hoechst+ cell was found (Fig. 6C, patient 007, arrow). In the same sample the adjacent cell had a punctate staining suggesting apoptosis.49 Moreover, in some samples (005 and 008) there were CK+/CD45− microparticles (Table 1 and Fig. 6C, arrow), suggesting that these could be tumor-derived microparticles (TMPs) described previously.50 In the second cohort (n=5), patients with non-epithelial malignancies (glioblastoma, meningioma, hemangioblastoma and metastatic melanoma) were recruited (Table S3.). In blood samples from cohort 2, which are non-epithelial cancers, none of CTCs, clusters or TMPs was detected using EpCAM MBs (Table S3). It must be noted that this was not a prospective trial due to limitations of the clinical protocol. However, we recently obtained three consecutive samples from a same patient (#16-1001-002, Table 1) before treatment, 5-week post treatment and 9-week post treatment. The CTC number was 50 at the baseline but increased to 70 after 5 weeks of treatment and decreased to 40 after 9 weeks of treatment. The examples of collected CTCs are in Figure 6D. These data suggests that MBs can be potentially used to monitor the response to treatment.
Table 1.
Results of CTC isolation from cancer patient samples Number in parenthesis after sample ID is the number of tubes collected in the same draw and processed.
| Metastatic Breast Cancer Patients | ||||||
|---|---|---|---|---|---|---|
| Sample ID | Receptor Status | Metastatic Sites | MB Type | CK+CTC | CK+ CTC Cluster | CD45+ |
| 01-001 | ER− PR− HER2+ | Lymph Node, Brain | EpCAM | 0 | No | >200 |
| 01-002 | ER+ PR+ HER2− | Bone | EpCAM | 3 | No | ~10,000 |
| 01-003 | ER+ PR− HER2+ | Bone, Lung, Liver, Lymph Node | EpCAM | 0 | No | >1,000 |
| 01-004 | ER− PR− HER2+ | Bone | EpCAM | 0 | No | >10,000 |
| 01-005 | ER+ PR+ HER2+ | Liver | EpCAM | 50 | Yes | >10,000 |
| 01-006 | ER+ PR+ HER2− | Liver, Skin | EpCAM | 4 | Yes | >10,000 |
| 01-007 | ER− PR− HER2− | Bone, Skin | EpCAM/ EGFR | 1 | No | >10,000 |
| 01-008 | ER− PR− HER2− | Bone, Lung, Lymph Node | EpCAM/ EGFR | 65 | Yes | >10,000 |
| 01-009 (2) | ER+ PR+ HER− | Bone, Lung, Lymph Node | EpCAM | 2 | No | <1,000 |
| 01-010 (2) | ER+ PR+ HER2− | Bone, Lung | EpCAM | 2 | No | <1,000 |
| 01-011 | ER+ PR+ HER2− | Bone | EpCAM | 3 | No | <200 |
| 01-012 | ER+ PR− HER2+ | Bone, Lymph Node | EpCAM | 0 | No | <500 |
| 01-013 (2) | ER+ PR− HER2+ | Bone | EpCAM | 0 | No | <500 |
| 01-014 (2) | ER+ PR+ HER2− | Lymph node, Skin | EpCAM | 0 | No | <500 |
| 01-015 | ER+ PR+ HER2− | Bone, Lung, Liver, Retina | EpCAM | 10 | Yes | <200 |
| Patient 16-1001-002 | ||||||
| Base-line | ER+ PR+ HER2− | N/A | EpCAM | 55 | Yes | <500 |
| 5-week | ER+ PR+ HER2− | N/A | EpCAM | 70 | No | <1,000 |
| 9-week | ER+ PR+ HER2− | N/A | EpCAM | 40 | No | <500 |
Figure 6. Isolation of CTCs, CTC fragments, clusters and microparticles from metastatic breast cancers patients’ blood.

A) Scheme of MB destruction and CTC staining procedure after isolation on the membrane. Isopropanol destroys MBs, whereas the cells stay bound to nitrocellulose; B) SKBR-3 cells spiked into normal human blood and isolated with MBs were stained for CK and CD45 in order to validate the CTC staining procedure and verify that cells remain intact after MB isolation and destruction. The image shows a typical fine fibrillose structure of cytokeratin. The cells are surrounded by CD45+ leukocytes; C) examples of CTCs, CTC clusters and microparticles isolated from patients’ samples. In sample 007, arrow points to a CK-/CD45− cell. CK+ cells in samples 007 and 009 appear apoptotic. In sample 008, arrow points to a CK+/CD45− microparticle next to a CTC cluster. Size bar: 10 μm except for #008 is 20 μm; D) representative CTCs isolated from patient 16-1001-002 before and during treatment. Note highly variable CK staining, and some cells appear to be negative for both CK and CD45 (arrows).
DISCUSSION
In this work, multifunctional immunomicrobubbles were developed in order to isolate CTCs, CTCs cluster and TMPs from blood of metastatic breast cancer patients. Compared to our previous reports, 25–27 we were able to conjugate 10 times more antibodies per MBs (100,000 versus over 1,000,000). Due to high-density PEGylated shell and high-density tethered antibody, blood stability of MBs was significantly improved compared to previously described lipid-based MBs or albumin-based MBs.25,26,30 The total isolation procedure was fast (30 min) with minimal blood processing (one centrifugation step to remove 60% of plasma). By using several antibodies immobilized on the same immuno-MB, the ability to isolate cells with variable EpCAM expression was demonstrated. The exact structure and orientation of the antibodies tethered on MBs, the molar ratio between antibodies, and the clustering on the MB surface are some of the parameters that have not been investigated here. Future studies will be needed to relate the nanostructured surface details of the multitargeted MBs to the cell binding and isolation efficiency.
The rate of CTC detection in this study, 62.5% (10/16) having >1 CTC in 7.5 mL blood, is similar to the previously described studies in metastatic breast cancer patients that utilized EpCAM enrichment technologies14,46,51. Both anti-EpCAM and anti-EpCAM/EGFR MBs were able to isolate CTCs, clusters and TMPs, but more studies involving larger patient cohorts are needed to compare between two formulations. Moreover, benchmarking against other methods of CTCs enumeration, including CellSearch, may be necessary in the future. Another limitation of the study is that it is not prospective and therefore the data cannot be used for correlation with the disease. However, the stable number of CTCs during treatment in the patient 16-1001-002 suggests that MBs can at the very least detect lack of response to treatment. Some studies of CTC in lung cancer patients have showed the clinical value of CTC to predict treatment response 52–54. A recent meta-analysis report showed CTCs were significantly decreased after treatment in breast cancer patients55.
Our study found that 41.6% (5/12) of CTC-positive blood contained CTC clusters of 2 and more cells. Previous work detected clusters in blood in around 40% of breast cancer patients 57. The presence of clusters is associated with a more aggressive disease due to the ability of CTCs to survive and lodge in blood vessels and subsequently lead to metastasis 58,59. CTC clusters can be used for testing of new therapies and CTC culture 59,60. In addition to clusters, MBs were able to isolate micron-sized cytokeratin-positive TMPs from blood. Most likely, these particles are cell fragments that originate from necrotic and apoptotic areas in the tumor. TMPs is an emerging prognostic marker as well as a standalone marker of tumor burden50,61–65. The exact content of these particles is not clear, but previous studies on TMPs and exosomes suggest that they may contain membrane proteins and nucleic acids62,65,66. Hence, fast and specific isolation of these structures using immuno-MBs could open an interesting venue for tumor liquid biopsy.
The liquid cancer biopsy field is at the crossroads, and there is a growing sentiment that simple CTC enumeration has limited value as a diagnostic test. Further downstream analysis such as DNA and RNA sequencing, high-resolution and metabolomics imaging, CTC culturing and drug sensitivity testing are being explored to expand the clinical utility of CTCs as a liquid biopsy 67,68. In view of these exciting applications, MBs offer several interesting possibilities. A small final sample volume is critical for downstream molecular analysis. The self-aggregating property of MBs and high malleability 69 helps concentrate the target cells from large-volume complex biological fluids in a microliter volume. The cells are often damaged due to mechanical forces after flow cytometry and magnetic microparticle-based cell sorting 70,71; therefore malleability of MBs could potentially mitigate these problems. Finally, MB isolation method is fast and simple, and combined with immunostaining can deliver the results within a few hours from receiving a blood sample.
Several issues related to MB technology still need to be addressed. While the number of leukocytes isolated from healthy donor blood and most of the patients’ blood was low (on the order of 20–200 cells/7.5 mL blood), larger numbers of contaminating leukocytes were observed in some patients’ samples. Leukocyte contamination is a serious problem for single cell genomics and mutational analysis 72, therefore further optimization of washing steps might be needed to get sufficiently pure CTC samples. Further improvement in sample purity can be achieved by adding an extra washing step or use antibodies without Fc fragment (to prevent recognition by neutrophils). Optimization of the isolation protocol could include the use of non-washed blood to shorten the processing time, the use of bigger tips to accommodate larger sample volume. The major effort is ongoing to develop an inexpensive semi-automated benchtop device for MB separation and harvesting to minimize manual processing steps. The ability to efficiently isolate highly concentrated tumor derived structures from blood with minimal processing could advance new liquid biopsy applications in research and clinical use.
CONCLUSIONS
In this work, we developed an improved MB formulation for capture of CTCs and tested them in a limited number of patient samples. The apparent advantage of the system is the ability to quickly isolate cells via several surface markers, including cells with low EpCAM expression. On the other hand, further improvement of sample purity, viability, and ability to analyze other molecular markers (beyond cytokeratin) will be necessary in order to advance the technology.
Supplementary Material
Highlights.
Prepared gas microbubbles (MBs) targeted to multiple cell surface markers
MBs show efficient and specific binding to breast cancer cells in vitro
MBs showed efficient recovery of spiked breast cancer cells in 7.5 ml blood
MBs showed fast isolation of CTCs in 5–10μl volume from blood of cancer patients
In addition to CTCs, microparticles and CTCS clusters were observed,
Acknowledgments
The study was funded by Innovative Molecular Analysis Program (IMAT) of the National Cancer Institute (grant R33CA174554) to YTL and Small Business Innovation Research grants from the National Cancer Institute (R43CA176892 & R44CA176892) to GS, and CA194058 to DS. XOMA Corporation is acknowledged for the gift of ING-1 (anti-human EpCAM) antibody.
Footnotes
COI: YTL is a founder and scientific consultant of Diagnologix, LLC. GS is a current employee of Diagnologix, LLC. Other authors do not have any financial interest in the company.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fehm T, Sagalowsky A, Clifford E, Beitsch P, Saboorian H, Euhus D, Meng S, Morrison L, Tucker T, Lane N, Ghadimi BM, Heselmeyer-Haddad K, Ried T, Rao C, Uhr J. Clin Cancer Res. 2002;8:2073–2084. [PubMed] [Google Scholar]
- 2.Steeg PS. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 3.Pantel K, Brakenhoff RH. Nat Rev Cancer. 2004;4:448–456. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 4.Harris L, Fritsche H, Mennel R, Norton L, Ravdin P, Taube S, Somerfield MR, Hayes DF, Bast RC., Jr J Clin Oncol. 2007;25:5287–5312. doi: 10.1200/JCO.2007.14.2364. [DOI] [PubMed] [Google Scholar]
- 5.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. J Cell Biol. 2011;192:373–382. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, Stojanov P, Brachtel E, Sgroi D, Kapur R, Shioda T, Ting DT, Ramaswamy S, Getz G, Iafrate AJ, Benes C, Toner M, Maheswaran S, Haber DA. Science. 2014;345:216–220. doi: 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos-Medina R, Moreno F, Lopez-Tarruella S, Del Monte-Millan M, Marquez-Rodas I, Duran E, Jerez Y, Garcia-Saenz JA, Ocana I, Andres S, Massarrah T, Gonzalez-Rivera M, Martin M. Clin Transl Oncol. 2016;18:749–759. doi: 10.1007/s12094-015-1460-2. [DOI] [PubMed] [Google Scholar]
- 8.Cayrefourcq L, Mazard T, Joosse S, Solassol J, Ramos J, Assenat E, Schumacher U, Costes V, Maudelonde T, Pantel K, Alix-Panabieres C. Cancer Res. 2015;75:892–901. doi: 10.1158/0008-5472.CAN-14-2613. [DOI] [PubMed] [Google Scholar]
- 9.Kang JH, Krause S, Tobin H, Mammoto A, Kanapathipillai M, Ingber DE. Lab Chip. 2012;12:2175–2181. doi: 10.1039/c2lc40072c. [DOI] [PubMed] [Google Scholar]
- 10.McGregor JR, Samlowski WE, Tharkar S, Donepudi S, Ferrone S. 2012 [Google Scholar]
- 11.van der Toom EE, Verdone JE, Gorin MA, Pienta KJ. Oncotarget. 2016 doi: 10.18632/oncotarget.11191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isselbacher KJ, Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Maheswaran S, Haber DA, Toner M. P Natl Acad Sci USA. 2010;107:18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235–1239. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. N Engl J Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 15.Grover PK, Cummins AG, Price TJ, Roberts-Thomson IC, Hardingham JE. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2014;25:1506–1516. doi: 10.1093/annonc/mdu018. [DOI] [PubMed] [Google Scholar]
- 16.Lustberg MB, Balasubramanian P, Miller B, Garcia-Villa A, Deighan C, Wu Y, Carothers S, Berger M, Ramaswamy B, Macrae ER, Wesolowski R, Layman RM, Mrozek E, Pan X, Summers TA, Shapiro CL, Chalmers JJ. Breast Cancer Res. 2014;16:R23. doi: 10.1186/bcr3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, Wu CC, LeBleu VS, Kalluri R. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janni WJ, Rack B, Terstappen LW, Pierga JY, Taran FA, Fehm T, Hall C, de Groot MR, Bidard FC, Friedl TW, Fasching PA, Brucker SY, Pantel K, Lucci A. Clin Cancer Res. 2016;22:2583–2593. doi: 10.1158/1078-0432.CCR-15-1603. [DOI] [PubMed] [Google Scholar]
- 19.Wicha MS, Hayes DF. J Clin Oncol. 2011;29:1508–1511. doi: 10.1200/JCO.2010.34.0026. [DOI] [PubMed] [Google Scholar]
- 20.de Wit S, van Dalum G, Lenferink AT, Tibbe AG, Hiltermann TJ, Groen HJ, van Rijn CJ, Terstappen LW. Scientific reports. 2015;5:12270. doi: 10.1038/srep12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mikolajczyk SD, Millar LS, Tsinberg P, Coutts SM, Zomorrodi M, Pham T, Bischoff FZ, Pircher TJ. Journal of oncology. 2011;2011:252361. doi: 10.1155/2011/252361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hager G, Cacsire-Castillo Tong D, Schiebel I, Rezniczek GA, Watrowski R, Speiser P, Zeillinger R. Gynecol Oncol. 2005;98:211–216. doi: 10.1016/j.ygyno.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 23.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, Isakoff SJ, Ciciliano JC, Wells MN, Shah AM, Concannon KF, Donaldson MC, Sequist LV, Brachtel E, Sgroi D, Baselga J, Ramaswamy S, Toner M, Haber DA, Maheswaran S. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vishnoi M, Peddibhotla S, Yin W, ATS, George GC, Hong DS, Marchetti D. Scientific reports. 2015;5:17533. doi: 10.1038/srep17533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi G, Cui W, Benchimol M, Liu YT, Mattrey RF, Mukthavaram R, Kesari S, Esener SC, Simberg D. PLoS One. 2013;8:e58017. doi: 10.1371/journal.pone.0058017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi G, Cui W, Mukthavaram R, Liu YT, Simberg D. Methods. 2013;64:102–107. doi: 10.1016/j.ymeth.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simberg D, Mattrey R. J Drug Target. 2009;17:392–398. doi: 10.1080/10611860902902797. [DOI] [PubMed] [Google Scholar]
- 28.Schutt EG, Klein DH, Mattrey RM, Riess JG. Angew Chem Int Ed Engl. 2003;42:3218–3235. doi: 10.1002/anie.200200550. [DOI] [PubMed] [Google Scholar]
- 29.Von Bibra H, Voigt JU, Froman M, Bone D, Wranne B, Juhlin-Dannfeldt A. Echocardiography. 1999;16:733–741. doi: 10.1111/j.1540-8175.1999.tb00143.x. [DOI] [PubMed] [Google Scholar]
- 30.Liou YR, Wang YH, Lee CY, Li PC. PLoS One. 2015;10:e0125036. doi: 10.1371/journal.pone.0125036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruan HH, Scott KR, Bautista E, Ammons WS. Neoplasia. 2003;5:489–494. doi: 10.1016/s1476-5586(03)80033-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang G, Griffin JI, Inturi S, Brenneman B, Banda NK, Holers VM, Moghimi SM, Simberg D. Front Immunol. 2017;8:151. doi: 10.3389/fimmu.2017.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ, Uhr JW, Terstappen LWMM. Clinical Cancer Research. 2004;10:6897–6904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 34.Kabalnov A, Klein D, Pelura T, Schutt E, Weers J. Ultrasound Med Biol. 1998;24:739–749. doi: 10.1016/s0301-5629(98)00034-9. [DOI] [PubMed] [Google Scholar]
- 35.Podell S, Burrascano C, Gaal M, Golec B, Maniquis J, Mehlhaff P. Biotechnol Appl Biochem. 1999;30(Pt 3):213–223. [PubMed] [Google Scholar]
- 36.Shen Y, Powell RL, Longo ML. J Colloid Interface Sci. 2008 doi: 10.1016/j.jcis.2008.07.047. [DOI] [PubMed] [Google Scholar]
- 37.Myrset AH, Fjerdingstad HB, Bendiksen R, Arbo BE, Bjerke RM, Johansen JH, Kulseth MA, Skurtveit R. Ultrasound in Medicine and Biology. 2011;37:136–150. doi: 10.1016/j.ultrasmedbio.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Kwan JJ, Borden MA. Advances in colloid and interface science. 2012;183–184:82–99. doi: 10.1016/j.cis.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 39.Kucerka N, Nieh MP, Katsaras J. Biochimica et biophysica acta. 2011;1808:2761–2771. doi: 10.1016/j.bbamem.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 40.Schneck H, Gierke B, Uppenkamp F, Behrens B, Niederacher D, Stoecklein NH, Templin MF, Pawlak M, Fehm T, Neubauer H. Disseminated Cancer Cell Network, D. PLoS One. 2015;10:e0144535. doi: 10.1371/journal.pone.0144535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martowicz A, Spizzo G, Gastl G, Untergasser G. BMC cancer. 2012;12:501. doi: 10.1186/1471-2407-12-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subik K, Lee JF, Baxter L, Strzepek T, Costello D, Crowley P, Xing L, Hung MC, Bonfiglio T, Hicks DG, Tang P. Breast cancer : basic and clinical research. 2010;4:35–41. [PMC free article] [PubMed] [Google Scholar]
- 43.Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gostner JM, Fong D, Wrulich OA, Lehne F, Zitt M, Hermann M, Krobitsch S, Martowicz A, Gastl G, Spizzo G. BMC cancer. 2011;11:45. doi: 10.1186/1471-2407-11-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ, Garcia-Blanco MA. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Konigsberg R, Obermayr E, Bises G, Pfeiler G, Gneist M, Wrba F, de Santis M, Zeillinger R, Hudec M, Dittrich C. Acta oncologica. 2011;50:700–710. doi: 10.3109/0284186X.2010.549151. [DOI] [PubMed] [Google Scholar]
- 47.Wit S, Dalum G, Lenferink AT, Tibbe AG, Hiltermann TJ, Groen HJ, van Rijn CJ, Terstappen LW. Scientific reports. 2015;5:12270. doi: 10.1038/srep12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carter L, Rothwell DG, Mesquita B, Smowton C, Leong HS, Fernandez-Gutierrez F, Li Y, Burt DJ, Antonello J, Morrow CJ, Hodgkinson CL, Morris K, Priest L, Carter M, Miller C, Hughes A, Blackhall F, Dive C, Brady G. Nat Med. 2016 doi: 10.1038/nm.4239. [DOI] [PubMed] [Google Scholar]
- 49.Miller MC, Doyle GV, Terstappen LW. Journal of oncology. 2010;2010:617421. doi: 10.1155/2010/617421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Coumans FA, Doggen CJ, Attard G, de Bono JS, Terstappen LW. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2010;21:1851–1857. doi: 10.1093/annonc/mdq030. [DOI] [PubMed] [Google Scholar]
- 51.Hvichia GE, Parveen Z, Wagner C, Janning M, Quidde J, Stein A, Muller V, Loges S, Neves RP, Stoecklein NH, Wikman H, Riethdorf S, Pantel K, Gorges TM. Int J Cancer. 2016;138:2894–2904. doi: 10.1002/ijc.30007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Punnoose EA, Atwal S, Liu W, Raja R, Fine BM, Hughes BG, Hicks RJ, Hampton GM, Amler LC, Pirzkall A, Lackner MR. Clin Cancer Res. 2012;18:2391–2401. doi: 10.1158/1078-0432.CCR-11-3148. [DOI] [PubMed] [Google Scholar]
- 53.Muinelo-Romay L, Vieito M, Abalo A, Nocelo MA, Baron F, Anido U, Brozos E, Vazquez F, Aguin S, Abal M, Lopez RL. Cancers (Basel) 2014;6:153–165. doi: 10.3390/cancers6010153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirose T, Murata Y, Oki Y, Sugiyama T, Kusumoto S, Ishida H, Shirai T, Nakashima M, Yamaoka T, Okuda K, Ohnishi T, Ohmori T. Oncol Res. 2012;20:131–137. doi: 10.3727/096504012x13473664562583. [DOI] [PubMed] [Google Scholar]
- 55.Wen-Ting Yan XC, Chen Qing, Li Ya-Fei, Cui You-Hong, Wang Yan, Jiang Jun. Scientific reports. 2017;7:12. doi: 10.1038/srep43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krebs MG, Metcalf RL, Carter L, Brady G, Blackhall FH, Dive C. Nat Rev Clin Oncol. 2014;11:129–144. doi: 10.1038/nrclinonc.2013.253. [DOI] [PubMed] [Google Scholar]
- 57.Sarioglu AF, Aceto N, Kojic N, Donaldson MC, Zeinali M, Hamza B, Engstrom A, Zhu H, Sundaresan TK, Miyamoto DT, Luo X, Bardia A, Wittner BS, Ramaswamy S, Shioda T, Ting DT, Stott SL, Kapur R, Maheswaran S, Haber DA, Toner M. Nat Methods. 2015;12:685–691. doi: 10.1038/nmeth.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, Brannigan BW, Kapur R, Stott SL, Shioda T, Ramaswamy S, Ting DT, Lin CP, Toner M, Haber DA, Maheswaran S. Cell. 2014;158:1110–1122. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Au SH, Storey BD, Moore JC, Tang Q, Chen YL, Javaid S, Sarioglu AF, Sullivan R, Madden MW, O'Keefe R, Haber DA, Maheswaran S, Langenau DM, Stott SL, Toner M. Proc Natl Acad Sci U S A. 2016;113:4947–4952. doi: 10.1073/pnas.1524448113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi JW, Kim JK, Yang YJ, Kim P, Yoon KH, Yun SH. Cancer Res. 2015;75:4474–4482. doi: 10.1158/0008-5472.CAN-15-0684. [DOI] [PubMed] [Google Scholar]
- 61.Toth B, Liebhardt S, Steinig K, Ditsch N, Rank A, Bauerfeind I, Spannagl M, Friese K, Reininger AJ. Thromb Haemost. 2008;100:663–669. [PubMed] [Google Scholar]
- 62.Liebhardt S, Ditsch N, Nieuwland R, Rank A, Jeschke U, Von Koch F, Friese K, Toth B. Anticancer Res. 2010;30:1707–1712. [PubMed] [Google Scholar]
- 63.Rank A, Liebhardt S, Zwirner J, Burges A, Nieuwland R, Toth B. Anticancer Res. 2012;32:2009–2014. [PubMed] [Google Scholar]
- 64.Roca E, Lacroix R, Judicone C, Laroumagne S, Robert S, Cointe S, Muller A, Kaspi E, Roll P, Brisson AR, Tantucci C, Astoul P, Dignat-George F. Oncotarget. 2016;7:3357–3366. doi: 10.18632/oncotarget.6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willms A, Muller C, Julich H, Klein N, Schwab R, Gusgen C, Richardsen I, Schaaf S, Krawczyk M, Krawczyk M, Lammert F, Schuppan D, Lukacs-Kornek V, Kornek M. Oncotarget. 2016;7:30867–30875. doi: 10.18632/oncotarget.9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Nat Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Joosse SA, Gorges TM, Pantel K. EMBO molecular medicine. 2015;7:1–11. doi: 10.15252/emmm.201303698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ignatiadis M, Lee M, Jeffrey SS. Clin Cancer Res. 2015;21:4786–4800. doi: 10.1158/1078-0432.CCR-14-1190. [DOI] [PubMed] [Google Scholar]
- 69.Ophir J, Parker KJ. Ultrasound Med Biol. 1989;15:319–333. doi: 10.1016/0301-5629(89)90044-6. [DOI] [PubMed] [Google Scholar]
- 70.Sieben S, Bergemann C, Lubbe A, Brockmann B, Rescheleit D. J Magn Magn Mater. 2001;225:175–179. [Google Scholar]
- 71.Garner DL. Theriogenology. 2006;65:943–957. doi: 10.1016/j.theriogenology.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 72.Sieuwerts AM, Kraan J, Bolt-de Vries J, van der Spoel P, Mostert B, Martens JW, Gratama JW, Sleijfer S, Foekens JA. Breast Cancer Res Treat. 2008 doi: 10.1007/s10549-008-0290-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.