

Abstract
There is growing interest in the conservation and utilization of crop wild relatives (CWR) in international food security policy and research. Legumes play an important role in human health, sustainable food production, global food security, and the resilience of current agricultural systems. Pea belongs to the ancient set of cultivated plants of the Near East domestication center and remains an important crop today. Based on genome-wide analysis, P. fulvum was identified as a well-supported species, while the diversity of wild P. sativum subsp. elatius was structured into 5 partly geographically positioned clusters. We explored the spatial and environmental patterns of two progenitor species of domesticated pea in the Mediterranean Basin and in the Fertile Crescent in relation to the past and current climate. This study revealed that isolation by distance does not explain the genetic structure of P. sativum subsp. elatius in its westward expansion from its center of origin. The genetic diversity of wild pea may be driven by Miocene-Pliocene events, while the phylogenetic diversity centers may reflect Pleisto-Holocene climatic changes. These findings help set research and discussion priorities and provide geographical and ecological information for germplasm-collecting missions, as well as for the preservation of extant diversity in ex-situ collections.
Introduction
Legumes represent the second most important family of crop plants after Poaceae, accounting for approximately 27% of the world’s crop production. Legumes play an important role in human health, sustainable food production, global food security, and the resilience of current agricultural systems1,2. There is a growing awareness of the need to ensure the global food supply3,4. One currently underdeveloped option for achieving this goal is a more systematic and targeted use of crop wild relatives (CWR) in crop breeding programs5. CWRs contain a wealth of genetically important traits due to their adaptation to a diverse range of habitats due to not having passed through the genetic bottlenecks of domestication. CWRs are increasingly recognized as a primary reserve of genetic variation, critical to maintaining agricultural productivity in the face of agricultural challenges6,7. CWRs play an important role in resolving fundamental questions concerning the domestication, ecological genetics and diversity of agronomically valuable variation8–11.
Pea is an emblematic plant in the field of biology, as it is linked to Mendel’s discovery (1866) of the laws of inheritance12. Pea belongs to an ancient set of cultivated plants of the Near East domestication center and is an economically important crop today1,13,14. Domesticated about 10,000 years ago15–19, pea, among other grain legumes, accompanied cereals in becoming an important dietary component of early civilizations in the Middle East and the Mediterranean13. The garden pea (Pisum sativum L.) belongs to the tribe Fabeae, which contains five genera, including important grain legumes: Lathyrus (grass pea); Lens (lentils); Pisum (peas) and Vicia (vetches)1,14,20. Two species, P. fulvum Sibth. & Sm. and P. sativum L., are most commonly recognized, the latter of which is divided into two subspecies, the domesticated pea subsp. sativum and the wild form, subsp. elatius (M. Bieb.) Asch. & Graebn.1,14,21. Populations of wild pea (Pisum sativum subsp. elatius) are scattered over the Mediterranean basin, while the distribution of P. fulvum is restricted to the Middle East1,14,20. Although worldwide pea germplasm includes approximately 98,000 accessions, only a small proportion (less than 1%) represent wild pea22. Pea genetic diversity held in collections was assessed using various morphological and molecular tools; however, wild material was largely underrepresented in these studies1,14. Comprehensive analysis of Pisum sp. diversity using retrotransposon markers revealed P. fulvum to be the most distant from cultivated pea, while P. sativum subsp. elatius is the closest23–25. Nevertheless, the diversity and distribution of wild P. sativum have not been explored to the extent of cultivated pea24,25.
In this research, we asked the following questions: 1) Is the genetic diversity of wild pea geographically or environmentally structured? 2) Is there evidence of hybridization between species? 3) Does the center of phylogenetic diversity for Pisum coincide with the genetic diversity centers of these species across the Mediterranean Basin and the Fertile Crescent? 4) How might climate chage impact the species distributions of these two species?
Results
The genetic structure of wild pea
DArTseq analysis performed on 161 wild-origin Pisum samples resulted in 66,910 polymorphic markers, which, upon filtering for missing data (>10%) and minor allele frequency (MAF < = 0.05), resulted in 35,647 SNPs, informative SNPs used for further analysis. Of these, 2,421 SNPs were mapped to the Medicago truncatula v 4.0 genome and were shown to be evenly distributed across the chromosomes (not shown). Allele frequency data readily resolved two groups of wild P. sativum subsp. elatius and the distant relative P. fulvum (Fig. 1a–d). STRUCTURE analysis revealed K = 6 to be the most probable partition of the data using the ad hoc delta K method26. This partioning clearly separates P. fulvum as a group from P elatius samples. Further analysis subdivided the P. elatius samples into 5 lineages. The five lineages vary in their genetic diversity, largely overlapping in their spatial location within the Levant, with the exception of the Q2 lineage, which showed a European location (Fig. 1a). The Q1 lineage consisted of 19 samples, 14 of which originate from Israel, Jordan and Turkey. The Q2 lineage contained 23 samples, mainly from Europe (France, Portugal, Spain, Hungary, Italy), except for two from Turkey and Israel. Q3 had 15 samples, mainly from Israel and Syria. The Q4 lineage had 43 samples of variable origin (Morocco, Georgia, Turkey, Iran and Syria, Crimea, Georgia, Armenia). The Q5 lineage had 40 samples, with 19 collected in situ in their origin of southeastern Turkey, and an additional 13 samples distinct at K = 7 that showed substantial admixture (0.3–0.4 membership coefficient Q).
Figure 1.

Inferred population structure of wild Pisum sp. based on 35,647 DArT seq SNPs. (a) Geographical distribution of five P. sativum subsp. elatius groups (ArcGIS for Desktop 10.4.1 http://desktop.arcgis.com/en). (b) The K value of 6 is shown, and the assignment to respective taxonomical groups is given, (c) Principal component analysis, coloured according to STRUCTURE groups with additional six (violet) group, (d) SplitsTree analysis (SplitTree v4, http://www.splitstree.org).
Relative FST values supported differentiation of the P. fulvum (F) group (0.545 to 0.683), followed by the Q2 lineage (0.439 to 0.561), while weak differentiation was observed between the Q3 and Q4 P. sativum subsp. elatius groups in comparison to Q5 (0.139 and 0.192, respectively).
Twenty nine out of 161 DArTseq samples originated from direct in-situ sampling (Table S1) from eastern Europe and southeastern Turkey. We calculated a percentage of loci heterozygous per individual. In the case of P. fulvum, this percentage was 0.39% to 3.5% (mean of 0.72%); that of P. sativum subsp. elatius was 0.33% to 13.5% (mean of 1.48%). There were no significant differences between germplasm-derived and in-situ samples, although in-situ samples displayed a slightly higher percentage of heterozygous SNPs (0.55% to 13.5%, mean of 1.68%) than germplasm-derived samples (0.33% to 4.18%, mean of 1.45%).
However, there were outliers: P. fulvum JI2527 (3.9%) and JI1796 (1.39%), and particularly two P. sativum subsp. elatius samples, UP_Serbia (17.5%) and IG52532 (17.33%), followed by IG52496 (8.65%) and IG140897 (8.63%), which can be explained by their admixture status, as revealed by STRUCTURE analysis. It is worth noting that in contrast to most of the samples, UP_Serbia is a recent collection, having produced only two generations ex situ. Similarly, IG52532 and IG52496 were collected in Turkey back in 1988, and IG140897 from Armenia was collected in 2004 (www.genesys-pgr.org). Together, these data indicate a very low natural outcrossing rate and a high genetic homogeneity of populations.
Principal component analysis (PCA) separated P. fulvum along the first principal component and explained 4.6% of the variation, with the second principal component explaining 4% of the variation (Fig. 1c) and the four major groups of P. elatius. The ordination analysis also partitioned the P. elatius samples into multiple groups that largely correspond to the STRUCTURE partitions. Some of the known hybrid samples, such as UP_Serbia, were intermediate in the PCA bi plot (Fig. 1c).
Because the individual samples may reflect past hybrization events, a neighbor-net tree was constructed from the Hamming distance between individuals using SplitsTree software. The reticulate dendrogram is useful if the data contain incompatible signals. The incompatible or ambiguous sample placements are represented by splits with cycles or boxes, resulting in several paths between any two samples. The partitioning of the data complimented the STRUCTURE and PCA findings in that they identified a significant partition of P. fulvum from a multiply partitioned, diverse set of P elatius samples (Fig. 1d). The UP-Serbia, IG52532 and JI3271 samples show ambiguous placement in the splits tree network, reflecting their admixture status.
We analysed the spatial distribution with a distance to centroid approach and that the clusters were mostly all overlapping and showed no isolation by distance (not shown). Furthermore we applied spatial autocorrelation analysis (Fig. 2) to P. elatius samples from Turkey and the Near East in order to minimalize the effect of wide geographical variability (mountain ranges, seas, etc.). The results indicated that kinship drops to zero at about 250 km. It is important to note, however, that the kinship realtionships beyond this distance do not monotonically decrease, but rather fluctuate in slope. This supports the hypothesis that isolation by distance does not explain all of the genetic differentiation within this species.
Figure 2.

Spatial autocorrelation analysis profiles for 89 wild pea accessions from Turkey and Near East. Geographic distances on the x-axis are the means of distance classes. Bar colours (green and violet) correspond to high kinship coefficients of accessions pairwise comparisonsat 600 and 900 km peaks in inlay. (SPAGeDiver 1.5, http://ebe.ulb.ac.be/ebe/SPAGeDi.html and Google Maps (https://maps.google.com/).
As genome-wide analysis requires high-quality genomic DNA and is costly, we used a biparentally inherited ITS marker for the entire set of 364 samples. The alignment of 664 bp of ITS locus included 27 bp of 18S rDNA, 238 bp of ITS1, 164 bp of 5.8S rDNA, 213 bp of ITS2 and 22 bp of 26S rDNA, totalling 664 bp. This resulted in 18 SNPs detecting 45 haplotypes altogether (Table S1). 149 samples of P. fulvum had 4 haplotypes distinguished by one mutation step, while these haplotypes are separated from the closest “elatius” samples by 11 mutations (Fig. 3). The haplotypes were partly geographically structured (Fig. 4a,b), with its-ful1 to the north (Syria, Turkey) and its-ful3 and its-ful4 to the south (Israel, Jordan). Wild P. “elatius” (216) samples had 24 haplotypes represented by more than one sample and 17 unique ones (Tables S1, S2). Two large and complex clusters were found, one with its-ela1 (27) and derived (its-ela2 to ela8, 69 altogether). The second cluster had its-ela13 (41), an associated complex network of its-ela10 to ela24, and unique haplotypes (63).
Figure 3.

ITS network based on ITS locus with 18 polymorphic sites detecting 28 major and 17 unique haplotypes (NETWORK v5 http://www.fluxus-engineering.com/sharepub.htm#a10).
Figure 4.

Haplotype distribution of wild pea accessions. (a) Of P. sativum subsp. elatius major ITS haplotypes, (b) P. fulvum ITS haplotypes, (c) of cpDNA trnS-G haplotypes (ArcGIS for Desktop 10.4.1 http://desktop.arcgis.com/en/).
Similarly, uniparentally inherited chloroplast trnS-G locus was analyzed in a set of 364 samples (Table S1). An 855-bp region of the chloroplast trnS-G locus identified seven haplotypes in 364 samples, which differed by five SNPs and one six-bp indel. These defined six haplotypes in P. sativum subsp. elatius and one haplotype in P. fulvum (Table S1). Of the 149 P. fulvum samples, all but six had the typical trnSG-F1 haplotype. The six exceptions were JI2510, JI2521, JI2539, VIR2523 and WL2140, which had “elatius” E6 and IG112136, which had the E3 haplotype (Table S1), suggesting introgression. Among the wild “elatius” samples, E5 (68) and E6 (58), followed by E1 (34), E2 (29) and E3 (21), were the most abundant, while E4 (5) was rare. E4 was identical to E1, except for the six-bp (TACAAA) insertion. Geographically, trnSG-E1 and E6 are the most widespread, spanning the entire geographical range, while trnSG-E3 is restricted to Turkey, Syria, Israel and the Caucasus (Fig. 4c).
Although ITS/cpDNA and DArTseq sets overlapped only partially (94/161 samples), there was no relationship between the nuclear-encoded IT, the uniparentally inherited chloroplast haplotypes, or the clusters identified by genome-wide DARTseq assignment into STRUCTURE, except for P. fulvum (Table S1).
P. fulvum is a clearly separated species, possibly due to divergent evolution in a specific habitat
Predictions of the potential current and past distribution of the two species are demonstrated by the results of niche modelling (Fig. 5a). According to model evaluations, modelling accuracy for the species P. fulvum and P. sativum subsp. elatius was excellent, with all Area Under the receiver operating characteristic Curve (AUC) values above 0.89. These predictions are generally in accordance with the distribution of the occurrence points, with P. fulvum showing a much narrower potential distribution than P. sativum subsp. elatius. In the case of P. fulvum, BIO19 (Precipitation of Coldest Quarter) had the highest permutation importance in the bioclimatic model (53.9%), followed by BIO5 (Max Temperature of Warmest Month) (18.5%) and BIO7 (Temperature Annual Range) (9.9%). For P. sativum subsp. elatius, BIO16 (Precipitation of Wettest Quarter) had the highest permutation importance (34.8%), followed by BIO14 (Precipitation of Driest Month) (15.5%) and BIO8 (Mean Temperature of Wettest Quarter) (15.4%). Additionally, prediction models reveal new areas of potential distribution for the two species. The outcomes of niche similarity tests are shown in Fig. S2. Spatial diversity of the niche patterns for the two wild taxa is indicated by Shannon’s index (Fig. 6b). There is a clear geographical pattern, with high diversity in the southeastern part of the Eastern Mediterranean Basin (Northern Africa, the Near East, Cyprus, the southwestern Mediterranean coasts of Turkey and the southern Aegean islands), where the spatial centers of species diversity was predicted. Of the seven cpDNA haplotypes identified, six occurred in enough locations to be modelled using Maxent (Fig. 5b,c). The model evaluation showed high predictive performance, with AUC values ranging from 0.862 ± 0.252 to 0.992 ± 0.019 (mean ± s.d.). Upon visual inspection, the spatial patterns of certain pairs, such as trnSG-E1 - E2, seem to be following similar patterns, while some haplotypes, such as F1, have a more distinct pattern. The tests of niche similarity (Fig. S3) offer a clearer view of the similarities between the potential niche patterns. As with the species, there was no definite case of divergence, while the pairs E1-E2 and E3-F were found to be statistically significantly similar. The spatial pattern of Shannon’s diversity index, calculated using the six cpDNA haplotypes, can be seen in Fig. 6a. The discrepancy between this pattern and the spatial diversity of the niche patterns of the two wild pea species concerns the western Mediterranean Basin and is expanded in the Balkans and the northern part of the eastern Mediterranean Basin. The projections of the distribution of the species P. fulvum and P. sativum. subsp. elatius and their six cpDNA haplotypes (Fig. 5a–c) during the Last Glacial Maximum (LGM) were similar compared to the current potential distribution. The pattern of Shannon’s diversity index, calculated using the projection of the distribution of the species P. fulvum and P. sativum. subsp. elatius during the LGM (Fig. 6b), was also similar to the current pattern of distribution and only slightly expanded on the North African part of the western Mediterranean Basin. The pattern of Shannon’s diversity index of cpDNA haplotypes (Fig. 6a), based on their predicted distribution during the LGM, was similar to the current distribution in most of the study area. Interestingly, the predicted distribution was highly impoverished in the Balkan region and the central European areas.
Figure 5.
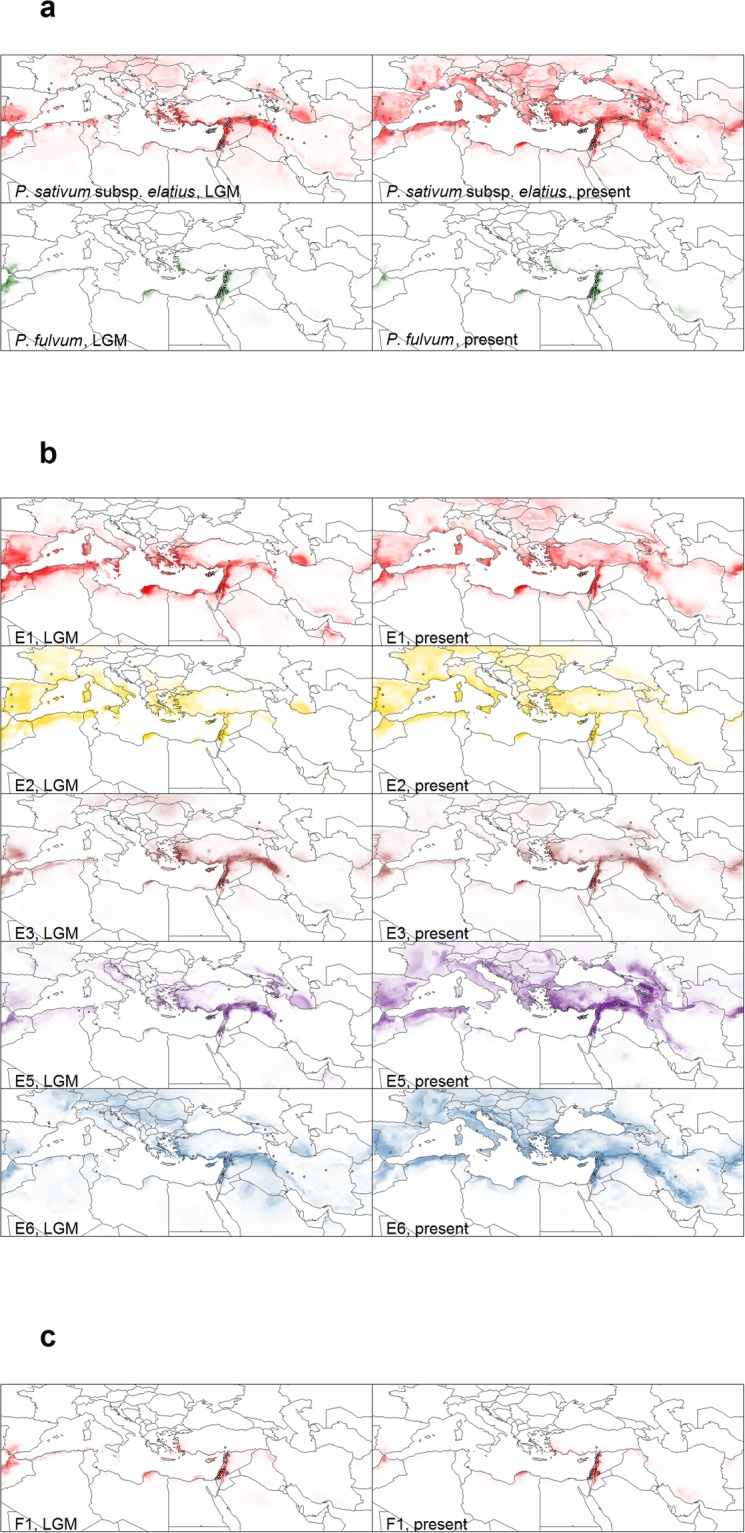
Results of ecological niche models. For (a) P. sativum subsp. elatius and P. fulvum, (b) haplotypes E1, E2, E3, E5 and E6 and (c) haplotype F1, as they resulted from sequencing analysis of cpDNA (trnS-G) region. Lighter colors correspond to lower probabilities of occurrence, while more saturated colors correspond to higher probabilities of occurrence (created with R version 3.2.2. https://cran.r-project.org/bin/windows/base/old/3.2.2/). White dots with black outlines represent the occurrence points that were used in the models.
Figure 6.

Geographical variation in Shannon’s index (a) of diversity, calculated from the niche model outputs for haplotypes E1, E2, E3, E5, E6 and F and (b) for Pisum sativum subsp. elatius and P. fulvum (created with R version 3.2.2. https://cran.r-project.org/bin/windows/base/old/3.2.2/).
Discussion
Crop wild relatives, including wild Pisum species, present an important source of novel, useful genetic diversity related to adaptive traits that may be of agricultural relevance27. While the genetic diversity of cultivated pea germplasm has been extensively analyzed over the past decade1,14,22,24,25, a limited number of wild pea samples has been studied24,25,28–30. Crop wild relatives have been extensively studied for their diversity and their genetic relationship with derived crop lineages, including legumes31–36. All these studies showed lower diversity of the crop compared to progenitor species. In a few cases, the gene flow between progenitor and crop was documented32. Our study detected a complex ITS network (Fig. 3, Table S1) more extensive than in previous studies28,29. Two major (its-ela1 and its-ela13) P. sativum subsp. elatius ITS haplotypes are separated from each other (Fig. 3) but do not correspond to proposed eco-geographical or taxonomical separation37. Consistent with previous studies20,23–25, P. fulvum was identified as a clearly distinct species (Figs 1 and 3). The P. fulvum ITS haplotypes were geographically structured (not shown) into northern (Syria, Turkey) and southern (Israel, Jordan), as reported earlier23,24. Interestingly, six out of 146 P. fulvum accessions showed common P. sativum subsp. elatius cpDNA hyplotypes (E3, E6), while also having a typical P. fulvum ITS, DArTseq assignment (Table S1). This may be explained by hybridization and backcrosses, as shown in other crop-wild-relative pairs, such as Phaseolus and Oryza38,39. The markers studied identified several P. fulvum and P. sativum subsp. elatius accessions with evidence of past hybridization events40. The level of heterozygosity and the intermediate assignment coefficients estimated in the population structure of two P. fulvum samples (JI2527 and JI1796) suggest possible hybrid origin, perhaps a putative cross with P. sativum subsp. elatius. Unfortunately, the DArTseq analysis did not include P. fulvum samples displaying the cpDNA haplotype of P. sativum subsp. elatius (Supplementary Table S1). The situation is complex due to the possibility of rare bi-parental inheritance of the plastids41.
It is hypothesized that legumes differentiated sometime before the end of Cretaceous in Africa42, while a recent phylogenetic study, together with fossil evidence, suggested that dispersal and vicariance putatively linked to the Tethys seaway is a more likely explaination of present legume distributions43. The tribe Fabeae originated and evolved in the Eastern Mediterranean in the middle Miocene (23–16 Mya) and expanded thereafter20. The stepping-stone hypothesis44 has been proposed, in which shallowly submerged seamounts would emerge during the extreme glacial sea-level minima distributed between the present-day islands and the Iberian Peninsula and North Africa45. The long-distance dispersal events are relatively common in Fabeae20. This also characterizes the Pisum genus, which spread from its center of origin in the Middle East eastwards to the Caucasus, Iran and Afghanistan, and westwards to the Mediterranean1,14.
Our results on the spatial diversity of the niche patterns, as indicated by Shannon’s index, suggest that while the species´ center of diversity is in the Near East, there may be two secondary centers: (1) Northern Africa in the Eastern Mediterranean Basin and (2) the coasts of Turkey, Cyprus and the Aegean islands. These findings suggest that the Northern African route was another hitherto unreported route for the westward expansion of wild pea. Historical records also support this view. For example, Columella, an important writer of the Roman Empire, mentions that “Roman legionaries still gathered wild peas from the sandy soils of Numidia and Palestine, to supplement their rations”46. Numidia was the ancient region of Africa north of the Sahara, with boundaries corresponding roughly to those of modern Tunisia and Algeria. Relatively recently, it has been recognized that gradual expansion would have contributed substantially to westward or eastward colonization along the Mediterranean Basin, either across the northern (European) side or across North Africa, and have been decisive in shaping the current species and genetic diversity of the Mediterranean wild flora47. A gradual expansion of herbaceous Fabaceae in the opposite direction, eastward from the west, has been reported for Anthyllis montana48.
Our results indicated that the spatial structure of genetic diversity of P. elatius (cpDNA and ITS haplotypes) in their westward expansion in the Mediterranean Basin does not correlate with a strict pattern of isolation by distance14,20. The wild pea results are in contradiction to the diversity pattern of many species, in which there is a gradual decrease in diversity running east-west along the Mediterranean Basin49,50. This diversity gradient has been attributed to the role of two interrelated processes around the Pleistocene. Specifically, it has been attributed to the east-west recolonization during the Holocene and the population size contraction under local LGM climate in resident western and low-elevation populations50. Our results are not in agreement with this biogeographic scenario. The discordance between the predicted pea species diversity center in the southern parts of the Eastern Mediterranean Basin and the predicted genetic diversity centers, which are scattered around the Mediterranean Basin and the Balkans, agrees with the view that there is no overall correlation between genetic diversity and species diversity across the Basin50. In the case of pea, the absence of an east-west gradient of genetic diversity suggests a different mechanism of dispersal and colonization. Our results with pea are more closely aligned with the pattern found in Northern African populations of Erophaca (Leguminosae), which are much more diverse genetically than European ones, despite the plant being (currently) relatively rare in North Africa51.
Discordance between predicted species and genetic diversity centers of wild pea was also revealed during the LGM. This pattern is differentiated from the longitudinal decline of genetic diversity in the Mediterranean Basin50. This discordance seems reasonable, taking into consideration that the Pisum genus evolved in the Eastern Mediterranean and spread westward. During the LGM, climate was drastically harsher in the Western Mediterranean (cold and dry) compared to the more favorable climate of the Eastern Mediterranean (wetter and warmer)47,49. Such harsh conditions are likely to have modified the available ecological niches of several species, including pea, causing discontinuities or eliminations of the predicted species diversity centers50. The geographical broad projected areas of high diversity of cpDNA haplotypes during the LGM may have been facilitated by the Messinian crisis of salinity during the late Miocene. In that period, land bridges allowed for the exchange of genetic material and formulated a spatial pattern of high diversity of wild pea throughout the Mediterranean Basin. The expansion during the Messinian is in agreement with scenario concerning the colonization of the western part of the Mediterranean Basin by Irano-Turanian elements20,52. Our results indicated that the predicted genetic diversity centers of Pisum may be driven by Miocene-Pliocene events, while the predicted species diversity centers may reflect recent (Pleisto-Holocene) climatic changes.
Research on the potential effects of climate change projections on pea production is limited. However, pea production is very likely to be affected by rising CO2 levels and temperatures, impacting important traits such as flowering time, mycorrhizal colonization, water use and photosynthesis53. There is no related research done on wild pea or on the question of how plastic it can be in its natural habitat.
In future studies, biotic interactions (including endophytes) may be a critical factor in understanding both range alterations and responses to climate change in pea. Although we did not explore this interaction, evidence among other legume species such as Medicago suggest that their endophytic diversity impacts their colonization success. For example, it has been hypothesized that the geographic expansion of Medicago was directly influenced by the geographical diversity of rhizobia symbionts54.
These results describing the genetic diversity of wild Pisum and their spatial and environmental structure suggest that these important genetic resources are under pressure from climate change and may need additional conservation planning. The genetic data also suggest that while species identities are intact, the diversity within these species is impacted by changes in the environment. The spatial analysis in these species can be a useful tool in developing comprehensive conservation strategies that include both in-situ and ex-situ elements. In combination with the International Treaty on Plant Genetic Resources for Food and Agriculture (ITPGRFA, FAO 2009), these pose the urgent need for the development of specific conservation strategies to consider the effects of climate change. In Article 5 of the ITPGRFA on Conservation, Exploration, Collection, Characterization, Evaluation and Documentation of Plant Genetic Resources for Food and Agriculture4, it is reported that each contracting party shall subject to national legislation, and in cooperation with other contracting parties, where appropriate support an integrated approach to promoting in-situ conservation of wild crop relatives and wild plants for food production. Predicted current spatial patterns resulting from niche modelling can also contribute to discovering new populations of the target species55. Geographical and ecological information has been key to many successful germplasm-collecting missions, as well as to the preservation of extant diversity in ex-situ collections, including legumes49,56.
Conclusions
This is the first comprehensive study of wild pea diversity. The analysis utilizing classical phylogenetic markers (ITS and cpDNA) supports that hybridization between wild peas is not an extensive phenomenon. Ecological niche modelling results support that the predicted genetic diversity centers of wild pea in the Mediterranean area may have been driven by Miocene-Pliocene events. These findings set conservation priorities/needs in the implementation of Article 5 of the International Treaty on Plant Genetic Resources for Food and Agriculture and provide geographical and ecological information for germplasm-collecting missions, as well as for the preservation of extant diversity in ex-situ collections.
Methods
Plant material
Samples for this study were drawn from two discrete, complementary sources. In order to obtain the cp DNA and ITS haplotypes, we used 364 samples of P. sativum subsp. elatius (216) and P. fulvum (148) (Table S1). We employed the taxonomical classification of Ambrose and Maxted21. A subset of 161 samples (143 P. sativum subsp. elatius and 18 P. fulvum), which included 29 samples from in-situ sampling, were used for genome-wide DArTseq (Fig. 1, Table S1) analysis. This material was collected between 1970 and 1990, largely prior to the use of GPS collection site identification. Thus, for some accessions with early collection dates, latitudes and longitudes used in the current study are based on the estimates of passport site description data. These accessions are therefore likely to be less precisely located. The samples were retrieved based on reliable origin22, tested for possible duplication (by passport data), cultivated in greenhouses and analyzed for possible misidentification (by morphological assessment of wild traits, namely pod dehiscence, seed dormancy and typical phenotype of wild forms)57. Notably, most of the material underwent germplasm multiplication; due to the predominant selfing, it is expected to be highly homozygous and therefore resistant to the effects of genetic drift. Moreover, important herbaria were inspected and samples taken from 109 vouchers (Table S1).
DNA isolation, PCR amplification and sequence analysis
Genomic DNA used for DArTseq analysis was isolated from single-plant samples. The DArTseq methodology requires high-molecular-weight DNA, typically obtained only from fresh material, while ITS and trnSG regions were PCR amplified and sequenced; therefore, herbarium samples could be used. PCR reactions were performed, using primers for ITS and trnSG regions20,58. PCR products were treated with Exonuclease-Alkaline Phosphatase (Thermo Scientific) and sequenced (BigDye Terminator v3.1 kit) at Macrogene. Haplotype network analysis was performed with PopART using a median-joining algorithm59.
DArTseq analysis
Genomic DNA was subjected to the standardized next-generation sequencing technique called DArTseq analysis at Diversity Arrays Technology Ltd. using proprietary methodology60. Approximately 2,500,000 (+/−7%) sequences per barcode/sample were used for marker calling using DArT PL’s proprietary DArTseq (SNP data) and SilicoDArT (binary presence/absence data) algorithms (DArTsoft14).
Molecular Data Analysis
Bayesian model-based clustering was performed using STRUCTURE26,61, which has been widely used on cultivated and wild pea germplasm14,24,25. Population structure was assessed using 161 accessions (P. sativum subsp. elatius & P. fulvum) with 66,910 polymorphic markers to infer genetic structure and to define the number of clusters using the STRUCTURE software version 2.3.4. The number of presumed populations (K) was evaluated from 3 to 16. The length of the burn-in period was set to 10,000, after which 200,000 iterations of the Monte Carlo Markov Chain (MCMC) were used for data collection. We ran 4 replicate MCMC chains for each value of K to evaluate the posterior likelihood using the ad hoc delta K method26. Principal component analysis was performed using the eigen function of R software (R Core Team) after applying a normalization technique62. Spatial autocorrelation analysis using SPAGeDi63was performed to assess the relationship between individual genetic identities and their geographic distance. We selected samples from Turkey and the Near East only in order to exclude the influence of seas and prohibitively large distances. Ritland´s kinship coefficient64 was employed to quantify average pairwise genetic identity based on 20 distance groups in each group with 200 pairwise comparisons. Randomization testing with 100 permutations was conducted to assess whether individual kinship values differed from expectations.The first 15 pairwise comparisons with the highest kinship coefficient from two potentionally interesting distance groups with a mean distance of 617 km and 888 km were depicted using Google Maps (https://maps.google.com/). Pairwise estimation of population Fst was done using the hierfstat package in R. The heterozygosity of the detected SNPs within the DArTseq dataset was calculated as a percentage of loci heterozygous per individual. Furthermore, the heterozygosity of putative interspecies hybrids was calculated for sets of SNPs associated (P-value of < 5 × 10−8) with respective parental species. To visualize the diversity and structure of the the individual samples in a complementary way, an unrooted split decomposition tree was rendered with the unfiltered DArTsilico data containing 187,298 binary characters using SplitsTree65.
Niche Analysis
Using the location data for 409 P. sativum subsp. elatius and 106 P. fulvum accessions (Table S1), the potential climatic niches were modelled using Maxent version 3.3.3k66. Samples that were removed earlier as duplicates, misidentified or otherwise inappropriate, as well as those that had dubious or inaccurate coordinates, were not included in the modelling. A threshold value of 50 km has been used as the maximum accepted distance, and the validation process took place using free available scripts (http://www.movable-type.co.uk/scripts/latlong.html). All the rejected sites have been omitted from the analyses, and validation tests were applied67. The environmental predictors used (19 bioclimatic variables)68 were from www.worldclim.org. The potential niches of the species were projected in past (Last Glacial Maximum, LGM ~22.000 ybp, http://worldclim.org) and future climatic conditions, following in the latter case the Representative Concentration Pathway (RCP) 6.0 scenario using bioclimatic data created by the Global Climate Model CCSM (Community Climate System Model) 4.0. In order to assess the importance of niche differences between the three species, we performed pairwise niche similarity tests69. These tests compare the “observed” niche overlap of the species in question with the “expected” overlap based on the species’ environmental backgrounds. The “observed” overlap, calculated using the metrics D and I, refers to the overlap of the species’ potential niches as they were estimated by Maxent70. The “expected” overlap results from substituting the species’ occurrence points with random points from their backgrounds and from calculating D and I for the resulting species/background pair. This random substitution process is iterated a set number of times (100 in our case) in order to obtain a statistical distribution for the two overlapping metrics, against which the “observed” values are tested. The background for each species was derived from its actual occurrence points using a Gaussian filter67. Niche similarity tests were performed in ENMTools version 1.4.371. Niche diversity among species, as well as their genotypic groups, was investigated with the use of Shannon’s index of diversity. Typically, this index is expressed as
where H′ is Shannon’s diversity index, and pi is the proportion of individuals (or cover) of the ith species in the dataset of interest. In our case, pi is the probability of occurrence of the ith species, and thus H′ can be calculated on a per-cell basis. The index has been calculated separately for the species using the modelling results of each taxon, as well as for the cpDNA haplotypes that were found during the genetic analysis, using the modelling results of each haplotype. Our quantitative analysis is one of the first to apply Shannon’s diversity index with probabilities of Maxent output to a niche modelling approach72. The index was calculated for each cell of the study area using a custom R script. For the manipulation and plotting of spatial data, as well as for the creation of figures, the packages sp, SDMTools and plotrix were employed73–75.
Data availability
Sequences of ITS and trnSG regions were deposited in the NCBI database, and accession numbers are listed in Table S1.
Electronic supplementary material
Acknowledgements
Petr Smýkal’s research is funded by the Grant Agency of the Czech Republic, 14-11782 S and 16-21053 S projects. O.T. is supported by partial institutional funding on long-term conceptual development of Agricultural Research, Ltd. organisation.
Author Contributions
P.S. conceived and managed the project. P.S., C.C., S.P. wrote the article. I.H., D.B. performed genotyping analysis, O.T., P.S., A.K. has performed DARTseq analysis, M.B. and S.P. performed GIS and niche-modelling. O.T. and I.H. analyzed genotyping and sequencing data. A.R., D.R., O.T., P.S., C.R. performed statistical analysis. All authors edited the manuscript and all authors read and approved the article.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
8/29/2018
A correction to this article has been published and is linked from the HTML and PDF versions of this paper. The error has been fixed in the paper.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-17623-4.
References
- 1.Smýkal P, Coyne C, Ambrose MJ, et al. Legume crops phylogeny and genetic diversity for science and breeding. Crit. Rev. Plant Sci. 2015;34:43–104. doi: 10.1080/07352689.2014.897904. [DOI] [Google Scholar]
- 2.Foyer CH, Lam HM, Nguyen HT, et al. Neglecting legumes has compromised human health and sustainable food production. Nature Plants. 2016;2:1–10. doi: 10.1038/nplants.2016.112. [DOI] [PubMed] [Google Scholar]
- 3.Godfray HC, et al. Food security: the challenge of feeding 9 billion people. Science. 2010;327:812–818. doi: 10.1126/science.1185383. [DOI] [PubMed] [Google Scholar]
- 4.International Treaty on Plant Genetic Resources for Food and Agriculture, FAO, Roma, Italy (2009)
- 5.Vincent H, et al. A prioritized crop wild relative inventory to help underpin global food security. Biol. Conserv. 2013;167:265–275. doi: 10.1016/j.biocon.2013.08.011. [DOI] [Google Scholar]
- 6.McCouch S, Baute GJ, Bradeen J, et al. Agriculture: feeding the future. Nature. 2013;499:23–24. doi: 10.1038/499023a. [DOI] [PubMed] [Google Scholar]
- 7.Dempewolf H, et al. agriculture to climate change: a global initiative to collect, conserve, and use crop wild relatives. Agroecol.Sust.Food Systems. 2014;38:369–377. doi: 10.1080/21683565.2013.870629. [DOI] [Google Scholar]
- 8.Ellis, T. H. N. Wild Crop Relatives, Genomic and Breeding Resources(ed. Kole C.), 237–248 (Berlin-Heidelberg, Springer-Verlag 2011).
- 9.Castañeda-Álvarez NP, Khoury CK, Achicanoy HA, et al. Global conservation priorities for crop wild relatives. Nature Plants. 2016;2:16022. doi: 10.1038/nplants.2016.22. [DOI] [PubMed] [Google Scholar]
- 10.Ford-Lloyd BV, Schmidt M, Armstrong SJ, et al. Crop wild relatives - undervalued, underutilized and under threat? BioScience. 2001;61:559–565. doi: 10.1525/bio.2011.61.7.10. [DOI] [Google Scholar]
- 11.Hajjar R, Hodgkin T. The use of wild relatives in crop improvement: a survey of developments over the last 20 years. Euphytica. 2007;156:1–13. doi: 10.1007/s10681-007-9363-0. [DOI] [Google Scholar]
- 12.Smýkal PPea. Pisum sativum L.) in biology prior and after Mendel’s discovery. Czech J: Genet. Plant Breed. 2014;50:52–64. [Google Scholar]
- 13.Abbo S, Lev-Yadun S, &Gopher A. Agricultural origins, centers and noncenters; a Near Eastern reappraisal. Crit. Rev. Plant Sci. 2010;29:317–328. doi: 10.1080/07352689.2010.502823. [DOI] [Google Scholar]
- 14.Smýkal P, Kenicer G, Flavell A, et al. Phylogeny, phylogeography and genetic diversity of the Pisum genus. Plant Genet.Res. 2011;9:4–18. doi: 10.1017/S147926211000033X. [DOI] [Google Scholar]
- 15.De Candolle, A. Origin of cultivated plants. (1884) Whitefish, Kessinger Publishing (2006).
- 16.Vavilov NI. The origin, variation, immunity and breeding of cultivated plants.Translated from the Russian by K. Starchester. Chronica Botanica1. 1951;3:1–364. [Google Scholar]
- 17.Kislev ME, Bar-Yosef O. The Legumes, The Earliest Domesticated Plants in the Near East? Curr.Anthrol. 1988;29:175–179. doi: 10.1086/203623. [DOI] [Google Scholar]
- 18.Smartt, J. Grain Legumes, Evolution and Genetic Resources. Cambridge, Cambridge University Press (1990).
- 19.Zohary, D. & Hopf, M. Domestication of Plants in the Old World. Oxford University Press, Oxford.(2000).
- 20.Schaefer H, Hechenleitner P, Santos-Guerra A, et al. Systematics, biogeography, and character evolution of the legume tribe Fabeae with special focus on the middle-Atlantic island lineages. BMC Evol.Biol. 2012;12:250. doi: 10.1186/1471-2148-12-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maxted, N. & Ambrose, M. Peas (Pisum L.). In, Maxted N., & Bennett, S.J., eds Plant Genetic Resources of Legumes in the Mediterranean.Dordrecht, Kluwer Academic Publishers, 181–190 (2001).
- 22.Smýkal, P., Coyne, C., Redden R. & Maxted, N. Peas. In, Singh, M., Upadhyaya,H.D., Bisht, I.S., eds. Genetic and Genomic Resources of Grain Legume Improvement. Amsterdam, Elsevier (2013).
- 23.Jing R, Johnson R, Seres A, et al. Gene-based sequence diversity analysis of field pea (Pisum) Genetics. 2007;177:2263–2275. doi: 10.1534/genetics.107.081323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jing R, Vershinin A, Grzebyta J, et al. The genetic diversity and evolution of field pea (Pisum) studied by high throughput retrotransposon based insertion polymorphism (RBIP) marker analysis. BMC Evol.Biol. 2010;10:44. doi: 10.1186/1471-2148-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holdsworth WL, Gazave E, Cheng P, et al. A community resource for exploring and utilizing genetic diversity in the USDA pea single plant plus collection. Hort. Res. 2017;4:17017. doi: 10.1038/hortres.2017.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Earl DA, von Holdt BM. STRUCTURE HARVESTER, a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv.Genet.Resources. 2012;4:359–361. doi: 10.1007/s12686-011-9548-7. [DOI] [Google Scholar]
- 27.Warschefsky E, Penmetsa RV, Cook DR, von Wettberg EJ. Back to the wilds, Tapping evolutionary adaptations for resilient crops through systematic hybridization with crop wild relatives. Am. J. Bot. 2014;101:1791–1800. doi: 10.3732/ajb.1400116. [DOI] [PubMed] [Google Scholar]
- 28.Palmer JD, Jorgensen RA, Thompson WF. Chloroplast DNA variation and evolution in Pisum, Patterns of change and phylogenetic analysis. Genetics. 1985;109:195–213. doi: 10.1093/genetics/109.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polans NO, Moreno RR. Microsatellite and ITS sequence variation in wild species and cultivars of pea. Pisum Genet. 2009;41:3–6. [Google Scholar]
- 30.Kosterin OE, Bogdanova VS. Relationship of wild and cultivated forms of Pisum L. as inferred from an analysis of three markers, of the plastid, mitochondrial and nuclear genomes. Genet.Resour. Crop Evol. 2008;55:735–755. doi: 10.1007/s10722-007-9281-y. [DOI] [Google Scholar]
- 31.Kujur A, Bajaj D, Upadhyaya HD, et al. Employing genome-wide SNP discovery and genotyping strategy to extrapolate the natural allelic diversity and domestication patterns in chickpea. Front. Plant Sci. 2015;6:162. doi: 10.3389/fpls.2015.00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Oss R, et al. Genetic relationship in Cicer sp. expose evidence for geneflow between the cultigen and its wild progenitor. PLoS ONE. 2015;10:e0139789. doi: 10.1371/journal.pone.0139789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saxena RK, von Wettberg E, Upadhyaya HD, et al. Genetic diversity and demographic history of Cajanus spp. illustrated from genome-wide SNPs. PLoS ONE. 2014;9:e88568. doi: 10.1371/journal.pone.0088568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez M, et al. Landscape genetics, adaptive diversity and population structure in Phaseolus vulgaris. New Phytol. 2015;209:1781–1794. doi: 10.1111/nph.13713. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Z, Yu Jiang Y, Wang Z, Gou Z, et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nature Biotech. 2015;33:408–416. doi: 10.1038/nbt.3096. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, et al. Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Sci. Rep. 2016;6:20728. doi: 10.1038/srep20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ladizinsky, G., Abbo, S. The search for wild relatives of cool season legumes.The Pisum genus.pp. 55-71 (Springer 2015).
- 38.Desiderio F, et al. Chloroplast microsatellite diversity in Phaseolus vulgaris. Front. Plant Sci. 2013;3:312. doi: 10.3389/fpls.2012.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim, K., Lee, S. C., Lee, J., Yu, Y. et al. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci Rep5, 15655, 10.1038/srep15655. [DOI] [PMC free article] [PubMed]
- 40.Vershinin AV, Allnutt TR, Knox MR, Ambrose MJ, Ellis THN. Transposable elements reveal the impact of introgression, rather than transposition, in Pisum diversity, evolution, and domestication. Mol. Biol. Evol. 2003;20:2067–2075. doi: 10.1093/molbev/msg220. [DOI] [PubMed] [Google Scholar]
- 41.Bogdanova VS, Kosterin OE. Hybridization barrier between Pisum fulvum Sibth.et Smith and P. sativum L. is partly due to nuclear-chloroplast incompatibility. Pisum. Genetics. 2007;39:8–9. [Google Scholar]
- 42.Raven, P. H. & Polhill, R. M. Biogeography of the Leguminosae. In: Polhill, R. M. & Raven P. H., eds Advances in Legume Systematics Kew, RBG, 27–34 (1981).
- 43.Schrire BD, Lavin M, Lewis GP. Global distribution patterns of the Leguminosae: insights from recent phylogenies. BiologiskeSkrifter. 2005;55:375–422. [Google Scholar]
- 44.Axelrod DI. Evolution and biogeography of Madrean-Tethyansclerophyll vegetation. Ann. Missouri Bot. Garden. 1975;62:280–334. doi: 10.2307/2395199. [DOI] [Google Scholar]
- 45.Fernández-Palacios JM, et al. A reconstruction of Palaeo-Macaronesia, with particular reference to the long-term biogeography of the Atlantic island laurel forests. J. Biogeog. 2011;38:226–246. doi: 10.1111/j.1365-2699.2010.02427.x. [DOI] [Google Scholar]
- 46.Toussaint-Samat, M. A History of Food. John Wiley & Sons (2009).
- 47.Nieto Feliner G. Patterns and processes in plant phylogeography in the Mediterranean Basin. A review. Persp. Plant Ecol. Evol. Syst. 2014;16:265–278. doi: 10.1016/j.ppees.2014.07.002. [DOI] [Google Scholar]
- 48.Kropf M, Kadereit JW, Comes HP. Late Quaternary distributional stasis in the submediterranean mountain plant Anthyllismontana L. (Fabaceae) inferred from ITS sequences and amplified fragment length polymorphism markers. Mol. Ecol. 2002;11:447–463. doi: 10.1046/j.1365-294X.2002.01446.x. [DOI] [PubMed] [Google Scholar]
- 49.Russell J, et al. Genetic Diversity and Ecological Niche Modelling of Wild Barley: Refugia, Large-Scale Post-LGM Range Expansion and Limited Mid-Future Climate Threats? PLoS ONE. 2014;9:e86021. doi: 10.1371/journal.pone.0086021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fady B, Conord C. Macroecological patterns of species and genetic diversity in vascular plants of the Mediterranean basin. Diver.Distr. 2010;16:53–64. doi: 10.1111/j.1472-4642.2009.00621.x. [DOI] [Google Scholar]
- 51.Casimiro-Soriguer R, et al. Phylogeny and genetic structure of Erophaca (Leguminosae), a East–West Mediterranean disjunct genus from the Tertiary. Mol. Phyl. Evol. 2010;56:441–450. doi: 10.1016/j.ympev.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 52.Manafzadeh, S., Staedler, Y. M. & Conti, E. Visions of the past and dreams of the future in the Orient: the Irano‐Turanian region from classical botany to evolutionary studies. Biol. Rev, 10.1111/brv.12287 (2016). [DOI] [PubMed]
- 53.Coyne C. J. et al. Chapter 8. Genetic Adjustment to Changing Climates: Pea In: S. S. Yadav, B. Redden, J. L. Hatfield, H. Lotze-Campen, Editors. Crop Adaptation to Climate Change.Wiley-Blackwell, Ames, IA. pp. 238–249 (2011).
- 54.Bena G, Lyet A, Huguet T, Olivier I. Medicago–Sinorhizobium symbiotic specificity evolution and the geographic expansion of Medicago. J. Evol. Biol. 2005;18:1547–1558. doi: 10.1111/j.1420-9101.2005.00952.x. [DOI] [PubMed] [Google Scholar]
- 55.Guisan A, et al. Using niche‐based models to improve the sampling of rare species. Conserv.Biol. 2006;20:501–511. doi: 10.1111/j.1523-1739.2006.00354.x. [DOI] [PubMed] [Google Scholar]
- 56.Ramirez-Villegas J, Khoury C, Jarvis A, Debouck DG, Guarino L. A gap analysis methodology for collecting crop gene pools: A case study with Phaseolus beans. PLoS ONE. 2010;5:e13497. doi: 10.1371/journal.pone.0013497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pavelková A, Moravec J, Hájek D, Baren I, Sehnalová J. Descriptor list genus Pisum L. RICP Prague. GenovéZdroje. 1986;32:46. [Google Scholar]
- 58.Smýkal P, et al. Genetic diversity and population structure of pea (Pisum sativum L.) varieties derived from combined retrotransposon, microsatellite and morphological marker analysis. Theor. Appl. Genet. 2008;117:413–424. doi: 10.1007/s00122-008-0785-4. [DOI] [PubMed] [Google Scholar]
- 59.Bandelt H, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 60.Kilian A, Wenzl P, Huttner E, et al. Diversity Arrays Technology, a generic genome profiling technology on open platforms. Methods Mol. Biol. 2012;888:67–89. doi: 10.1007/978-1-61779-870-2_5. [DOI] [PubMed] [Google Scholar]
- 61.Pritchard JK, Stephens M, Donnelly PJ. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patterson N, Price AL, Reich D. Population Structure and Eigenanalysis. PLoS Genetic. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes. 2002;2:618–620. doi: 10.1046/j.1471-8286.2002.00305.x. [DOI] [Google Scholar]
- 64.Ritland K. Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res. 1996;67:175–185. doi: 10.1017/S0016672300033620. [DOI] [Google Scholar]
- 65.Huson DH, Bryant D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 66.Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modeling of species geographic distributions. Ecol. Model. 2006;190:231–259. doi: 10.1016/j.ecolmodel.2005.03.026. [DOI] [Google Scholar]
- 67.Alsos IG, Alm T, Normand S, Brochmann C. Past and future range shifts and loss of diversity in dwarf willow (Salix herbacea L.) inferred from genetics, fossils and modelling. Glob.Ecol. Biog. 2009;18:223e239. [Google Scholar]
- 68.Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. Very high resolution interpolated climate surfaces for global land areas. Inter. J. Climatol. 2005;25:1965–1978. doi: 10.1002/joc.1276. [DOI] [Google Scholar]
- 69.Warren DL, Glor RE, Turelli M. Environmental niche equivalency versus conservatism, quantitative approaches to niche evolution. Evolution. 2008;62:2868–2883. doi: 10.1111/j.1558-5646.2008.00482.x. [DOI] [PubMed] [Google Scholar]
- 70.Allouche O, Steinitz O, Rotem D, Rosenfeld A, Kadmon R. Incorporating distance constraints into species distribution models. J. Appl. Ecol. 2008;45:599–609. doi: 10.1111/j.1365-2664.2007.01445.x. [DOI] [Google Scholar]
- 71.Warren DL, Glor RE, Turelli M. ENMTools, a toolbox for comparative studies of environmental niche models. Ecography. 2010;33:607–611. doi: 10.1111/j.1600-0587.2009.06041.x. [DOI] [Google Scholar]
- 72.Vorsino AE, et al. Modeling Hawaiian ecosystem degradation due to invasive plants under current and future climates. PLoS One. 2014;9:e95427. doi: 10.1371/journal.pone.0095427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pebesma EJ, Bivand RS. Classes and methods for spatial data in R. R News. 2005;5:9–13. [Google Scholar]
- 74.Van Der Wal, J., Falconi, L., Januchowski, S., Shoo, L. & Storlie, C. SDMTools, Species Distribution Modelling Tools, Tools for processing data associated with species distribution modelling exercises. http,//CRAN.R-project.org/package=SDMTools. (2014).
- 75.Lemon J, Plotrix a. package in the red light district of R. R News. 2006;6:8–12. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequences of ITS and trnSG regions were deposited in the NCBI database, and accession numbers are listed in Table S1.