

Abstract
Gastric cancer persists as a frequent and deadly disease that claims over 700 000 lives annually. Gastric cancer is a multifactorial disease that is genetically, cytologically and architecturally more heterogeneous than other gastrointestinal cancers, making it therapeutically challenging. As such, and largely attributed to late‐stage diagnosis, gastric cancer patients show only partial response to standard chemo and targeted molecular therapies, highlighting an urgent need to develop new targeted therapies for this disease. Wnt signalling has a well‐documented history in the genesis of many cancers and is, therefore, an attractive therapeutic target. As such, drug discovery has focused on developing inhibitors that target multiple nodes of the Wnt signalling cascade, some of which have progressed to clinical trials. The collective efforts of patient genomic profiling has uncovered genetic lesions to multiple components of the Wnt pathway in gastric cancer patients, which strongly suggest that Wnt‐targeted therapies could offer therapeutic benefits for gastric cancer patients. These data have been supported by studies in mouse models of gastric cancer, which identify Wnt signalling as a driver of gastric tumourigenesis. Here, we review the current literature regarding Wnt signalling in gastric cancer and highlight the suitability of each class of Wnt inhibitor as a potential treatment for gastric cancer patients, in relation to the type of Wnt deregulation observed.
Linked Articles
This article is part of a themed section on WNT Signalling: Mechanisms and Therapeutic Opportunities. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.24/issuetoc
Abbreviations
- M3 receptor
ACh‐muscarinic receptor‐3
- APC
adenomatous polyposis coli
- BCL‐9
B‐cell lymphoma‐9
- CagA
cytotoxin‐associated gene product
- CBP
cAMP response element‐binding protein
- CK1
casein kinase 1
- CRC
colorectal cancer
- CRD
cysteine rich domain
- CSC
cancer stem cell
- CTNNB1
β‐catenin
- DKK
dickkopf
- Dvl
dishevelled
- ER
endoplasmic reticulum
- FZD
frizzled
- GAPPS
gastric adenocarcinoma and proximal polyposis of the stomach
- GC
gastric cancer
- GSK3B
glycogen synthase kinase‐3β
- H. pylori
Helicobacter pylori
- HER2
human epidermal growth factor receptor 2
- IWP‐2
inhibitor of Wnt production‐2
- LEF1
lymphoid enhancer‐binding factor‐1
- LGR5
leucine‐rich repeat‐containing G‐protein‐coupled receptor‐5
- LOF
loss of function
- Mist1
muscle, intestine and stomach expression‐1
- MSI
microsatellite instability
- PARsylation
poly(ADP‐ribosyl)ation
- PUMA
p53 up‐regulated modulator of apoptosis
- RNF43
ring finger 43
- sFRP
secreted frizzled‐related proteins
- TNK1/2
tankyrase 1/2
- TOP
T‐cell factor optimal promoter
- ZNRF3
zinc‐ring finger 3
Introduction
Gastric cancer (GC) is a frequent malignancy and is the third most common form of cancer‐related death world‐wide (Guggenheim and Shah, 2013). Approximately 1 000 000 new cases of GC are diagnosed annually, with a large proportion of cases reported within East Asia, South America and Eastern Europe (Rahman et al., 2014). Wnt signalling regulates many cell functions, including proliferation, migration and cell death, and although it is essential for the development and homeostasis of several tissues, it is also deregulated in many cancers. The link between aberrant Wnt signalling and cancer is well characterized in intestinal, breast and liver cancers; however, its importance in the stomach is less well understood. This review will describe the current position of the field regarding the role of Wnt signalling in GC and how we might target the Wnt pathway to treat GC patients.
Homeostasis of the stomach
The mammalian stomach is divided into two anatomically distinct regions; the corpus and the antrum. The corpus is responsible for the main digestive action of the stomach, releasing a cocktail of acids, enzymes and hormones, whereas the antrum produces large amounts of mucus and gastric hormones (Figure 1A). The stomach is lined by a simple columnar epithelium that is constantly renewed, a process driven by resident adult stem cells (Hoffmann, 2008; Barker et al., 2010a). The gastric epithelium is organized into numerous mucosal invaginations called gastric units. Each gastric unit houses stem cells as well as the various differentiated cell types that perform distinct functions: mucus cells that secrete protective mucus, parietal cells responsible for secreting hydrochloric acid, chief cells that release active pepsin and several types of endocrine cells that secrete an array of hormones that aid and regulate digestion and absorption (Kim and Shivdasani, 2016). The precise architecture, cellular heterogeneity and turnover rate of the gastric units varies markedly between the antrum and corpus (Figure 1B, C) (Karam and Leblond, 1993; Barker et al., 2010b). There are many facets that help regulate gastric epithelial homeostasis including key developmental pathways and interactions from underlying stromal and nerve cells (Hayakawa et al., 2015; Kim and Shivdasani 2016). High Wnt signalling is observed transiently in the developing forestomach (Kim et al., 2005), while in the adult stomach, expression is highest in the antrum where the Wnt target gene leucine‐rich repeat‐containing GPCR (Lgr5) marks a population of stem cells (Barker et al., 2010a). Further functional experiments will be required to elucidate the full requirement for Wnt signalling during gastric homeostasis, as was recently shown for Notch signalling (Kim and Shivdasani, 2011; Demitrack et al., 2015), but its critical inclusion in the culture medium of gastric organoids advocates its importance (Barker et al., 2010a).
Figure 1.
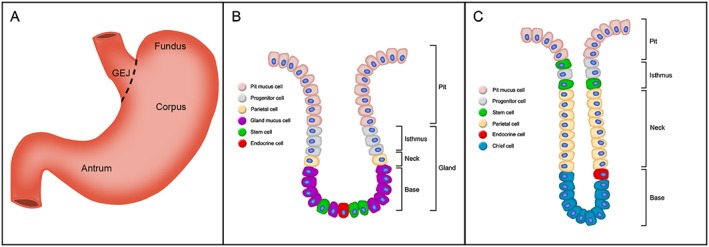
Anatomy of the mammalian stomach and mucosa. (A) Gross anatomy of the stomach, illustrating the gastroesophageal junction (GEJ), fundus, corpus and antrum. (B, C) Schematic of antral (B) and corpal (C) gastric units and their various epithelial cell types. Each unit is divided into a surface pit, isthmus, neck and base regions. Of note, the cellular composition and architecture varies between the antrum and corpus, which reflects functional specificity.
Histopathology of gastric cancer
Gastric cancer can be divided pathologically into broad classes: intestinal‐type and diffuse‐type as classified by Lauren (1965). Of note, each of these classes can be further broken down into sub‐classes based on histopathological, anatomical and genomic characteristics. Proximal intestinal‐type gastric, also referred to as proximal non‐diffuse GC, is defined by tumours located in the gastric cardia, which may extend into the gastroesophageal junction (Figure 1A). Histological analysis of proximal intestinal‐type GC reveals glandular dysplasia, which can be accompanied by chronic inflammation (Shah et al., 2011). However, unlike the chronic inflammation associated with distal intestinal‐type GC, which is often connected to Helicobacter pylori (H. pylori), carcinogenic inflammation of proximal intestinal‐type GC is often linked to gastric acid reflux (Crew and Neugut, 2006). Distal intestinal‐type (non‐diffuse) GCs are primarily located in the antrum but can occasionally be found in the body of the stomach (Wong and Yau, 2013) (Figure 1A). Distal intestinal‐type GCs are well‐differentiated and are composed of neoplastic gland‐forming cells. Furthermore, this type of gastric tumour is often associated with H. pylori infection, the histopathology of which has been well characterized (Wong, 2016). Finally, diffuse GC, which is characterized by a diffuse pattern of cell infiltrate and poorly differentiated signet‐ring cell clusters, is considered to arise de novo and is associated with loss of cell–cell contact via down‐regulation/mutation to CDH1 (E‐cadherin) (Guilford et al., 1998; 1999).
While surgical intervention is the common practice for GC treatment, most patients are often at an advanced stage of disease progression at the time of diagnosis, which limits the value of surgery. As such, chemotherapy is the next appropriate countermeasure for these patients. However, only 20–40% of patients respond to first‐line chemotherapies that target DNA replication and repair mechanisms (folinic acid, fluorouracil and oxaliplatin, and folinic acid, fluorouracil and irinotecan), with a median overall survival of 6–11 months (Jemal et al., 2011). The past decades of fervent research into gastric tumour biology has significantly illuminated our understanding of tumour genomics, heterogeneity, immunoediting and drug resistance, which have culminated in the development of molecular targeted therapies. Targeted therapy not only confers higher anti‐cancer specificity and selectivity than chemotherapy but also reduces unwanted non‐selective toxicity. While targeted therapies such as trastuzamab/pertuzamab [human epidermal growth factor receptor 2 (HER2)], certuximab (EGFR) and ramucirumab (vascular endothelial growth factor receptor‐2) have been utilized in the clinic, often in combination with chemotherapy, they typically yield only a partial response and are prone to resistance (Blackham et al., 2016). Of note, the proportion of Epstein–Barr virus positive GCs that display increased PD‐L1 expression (Cancer Genome Atlas Research, 2014) has prompted several clinical trials testing ‘breakthrough’ immune checkpoint inhibitors (tremelimumab and nivolumab) in patients at various stages of the disease (Kang et al., 2017) Despite the impressive anti‐tumour effects of immune checkpoint inhibitors observed in other solid tumours, GC patients only show a modest increase in survival – 5.32 (nivolumab) versus 4.41 (placebo) months, highlighting the need for fully randomized trials to properly identify GC patients who will benefit from immunotherapies. The limited response and frequent resistance to first‐line chemotherapy and adjuvant molecular therapies is partially attributed to an incomplete understanding of the biology of GC (Zhang and Fan, 2010). As such, the continual discovery of additional signalling pathways that drive gastric tumourigenesis and progression provide novel therapeutic avenues that are desperately needed in the clinic.
To date, there is a large quantity of data demonstrating that Wnt signalling plays a critical role in driving gastric tumourigenesis, invasion and metastasis (Radulescu et al., 2012; Zhao et al., 2013). Here, we review the relevant studies implicating aberrant Wnt signalling in GC and how Wnt‐targeted therapies may offer therapeutic benefit to GC patients.
A brief overview of Wnt signalling
The Wnt signalling pathway is an ancient instructive genetic programme, which is conserved from humans through to Hydra (Phesse et al., 2016). It plays a vital role for orchestrating complex cellular behaviours during development, tissue homeostasis and regeneration where it coordinates cell proliferation, cell fate decisions, cell motility and tissue polarity. Due to its biological pervasiveness, Wnt signalling is implicated in many human cancers and degenerative diseases following pathway deregulation (Clevers and Nusse, 2012). Historically, Wnt signalling is considered to operate in two distinct modalities based on downstream involvement – or lack thereof – of the cytoplasmic protein β‐catenin. These are referred to from hereon in as β‐catenin‐dependent or β‐catenin‐independent Wnt signalling. Of the two Wnt pathway branches, the β‐catenin‐dependent pathway has received the most research interest and is thus better characterized, and as such will be the primary focus of this review.
Wnts are secreted lipid‐modified glycoproteins that act as both short (Farin et al., 2016) and long‐range (Mulligan et al., 2012) ligands to engage with cell surface receptors that can establish complex morphogen gradients, to promote subtle yet sophisticated biological outcomes, depending on cell and tissue context (Clevers, 2006; Clevers and Nusse, 2012; Clevers et al., 2014). The hallmark of β‐catenin‐dependent Wnt signalling is the cytoplasmic accumulation of β‐catenin. In the absence of Wnt activation, cytosolic levels of free β‐catenin are kept to a minimum, despite the gene being continuously transcribed. This pool of free β‐catenin is sequestered in a multi‐protein ‘destruction complex’ that consists of adenomatous polyposis coli (APC) tumour suppressor protein, AXIN, glycogen synthase kinase‐3β (GSK3B) and casein kinase 1 (CK1). Formation of the β‐catenin destruction complex induces phosphorylation of β‐catenin by CK1 at Ser45, which in turn primes GSK3B phosphorylation of β‐catenin on Thr41, Ser37 and Ser33 residues (Liu et al., 2002). Phosphorylated β‐catenin is ubiquitylated by the F‐box‐containing protein β‐TrCP E3 ligase tagging it for proteasomal degradation (Aberle et al., 1997; Kitagawa et al., 1999; Li et al., 2012). In the presence of Wnt, a heterodimeric receptor complex is formed, consisting of the seven‐pass transmembrane protein frizzled (FZD) receptor and low‐density lipoprotein receptor‐related protein 5/6 (LRP5/6). Through an unresolved mechanism involving the adaptor protein Dishevelled (Dvl), both receptor components participate in separate intracellular interactions that trigger both the initiation and amplification of Wnt signalling via inhibition of the β‐catenin destruction complex (Mao et al., 2001; Zeng et al., 2005; Zeng et al., 2008; MacDonald et al., 2009). This allows newly synthesized unphosphorylated β‐catenin to accumulate, stabilize and translocate from the cytoplasm to the nucleus. β‐catenin can then generate a transcriptionally active complex with T‐cell factor/lymphoid enhancing factor (TCF/LEF) family of transcription factors to induce Wnt target gene transcription (for detailed reviews on Wnt signal transduction, see Clevers and Nusse 2012; Niehrs, 2012; Acebron and Niehrs, 2016).
Recent experimental findings have revealed inherent pathway crosstalk and complexity that cannot be accounted for by current linear signal transduction models, with components at virtually every level of Wnt signal transduction being shown to affect both β‐catenin‐dependent and β‐catenin‐independent outputs (Topol et al., 2003; Mikels and Nusse, 2006; Niehrs, 2012). As such, Wnt signalling is beginning to be viewed as a signalling network (van Amerongen and Nusse, 2009). An example of Wnt pathway complexity is the dazzling number of possible ligand–receptor interactions from the vast repertoire of mammalian Wnts (19), FZD receptors (10) and co‐receptors (>6). These combinations influence just one facet of signal output following Wnt pathway activation and will yield different biological outcomes depending on cell/tissue context. This illustrates that despite major breakthroughs over the last several decades, there are still gaps in our understanding of how this pathway operates including precisely how Wnt‐FZD receptor selectivity is achieved.
Duplicitous Wnt signalling – regulator of both normal and cancer stem cell biology
Many adult tissues, such as the skin and gut, undergo constant renewal, meaning, a balance between cell extrusion and replacement by newly born cells. It is now understood that cellular attrition, either through natural exhaustion or injury, within diverse tissues is fuelled by stem cells. Stem cell activity is often controlled by the micro‐environment (niche) so that stem cell output is matched to the homeostatic needs or regenerative demands of the tissue (Clevers et al., 2014). Wnt signalling controls stem cell activity in a variety of tissues such as the intestines, stomach, skin, bone and haematopoietic system (Clevers and Nusse, 2012; Visvader and Clevers, 2016). For example, intestinal stem cells sustain the constant turnover of the intestinal epithelium and express the cell surface receptor LGR5 (Barker et al., 2007). Lgr5 is a Wnt target gene (Barker et al., 2007), and Lgr5 + intestinal stem cells require the Wnt receptor FZD7 to maintain homeostasis in the intestinal epithelium (Flanagan et al., 2015). Indeed, LGR5 is a co‐receptor for Wnts, which are expressed by neighbouring Paneth cells in the small intestine (Sato et al., 2011) and c‐kit+ goblet cells and Reg4+ deep secretory cells in the colon (Rothenberg et al., 2012; Sasaki et al., 2016). Lgr5 + stem cells are also located in the gastric epithelium suggesting that Wnt signalling also regulates gastric homeostasis. This is supported by the observation that deletion of Fzd7 in the gastric epithelium is deleterious and triggers rapid repopulation (Flanagan et al., 2017). The TNF‐family receptor TNFRSF19 (Troy) was recently identified as a marker of a subset of chief cells which act as ‘reserve’ stem cells in the stomach following injury, which are characterized by an elevated expression of Wnt target genes (Stange et al., 2013). Together, these data suggest Wnt signalling regulates gastric stem cell activity; however, the full extent of Wnt‐regulated homeostasis in the gastric epithelium is relatively poorly understood in comparison with that in the intestine.
The cancer stem cell model suggests that tumour growth is driven by a small sub‐population of cells [cancer stem cells (CSCs)] rather than the bulk of the tumour cells (Visvader, 2011). Over several years, this model has been refined as researchers have discovered significant plasticity between CSCs and non‐CSCs within a tumour depending on the exposure of cells to growth factors and cytokines expressed by tumour cells or surrounding cells. Importantly, despite the progress that has led to an improved standard of care, resistance to chemotherapy, whether intrinsic or acquired, is a complex and multifactorial phenomenon and remains the main cause of treatment failure and death in GC patients (Brungs et al., 2016). Gastric CSCs (gCSCs) have shown to be resistant to GC therapy and subsequently responsible for tumour recurrence and metastasis (Mayer et al., 1993; Brungs et al., 2016). Consequently, identifying the mechanisms of CSC regulation and maintenance is crucial to clarifying how these mechanisms influence the development of chemoresistant tumour cells in GC patients. Indeed, the similarities between normal adult somatic stem cells and CSCs suggest that the same signalling pathways that are involved in regulating somatic stem cell maintenance are also involved in the regulation of CSCs. As such, deregulated Wnt signalling has shown to increase ‘stemness’, trigger transformation and influence the development of chemoresistant CSCs (Sansom et al., 2007; Barker et al., 2009; Melo et al., 2017). For instance, an elevated expression of the transcription factor and Wnt target gene SOX9 (Blache et al., 2004) is observed in human GC patients and correlates with decreased patient survival (Santos et al., 2016). Targeted knockdown of SOX9 is sufficient to reduce CSCs viability/formation with concomitant suppression of Wnt signalling and target gene expression, which was phenocopied following β‐catenin knockdown (Santos et al., 2016). Importantly, the increase in SOX9+ resistant cells following exposure to cisplatin was largely prevented in SOX9 knockdown GC cells, demonstrating that agents targeting SOX9‐β‐catenin signalling can overcome chemoresistant cells (Santos et al., 2016). Similarly, Wnt6 expression is positively associated with tumour stage and inversely correlates with response to the anthracycline chemotherapeutics epirubicin and doxorubicin (Yuan et al., 2013). Treatment with epirubicin or doxorubicin increases Wnt6 expression by enhancing the binding of cavolin‐1 to β‐catenin at the Wnt6 promoter, which in turn boosts cell survival. As such, targeted knockdown of Wnt6 reduces GC cell survival via increased caspase‐3 induction (Yuan et al., 2013). While it is likely that Wnt6+‐resistant cells represent gCSCs, this was not formally demonstrated. More recently, investigations to identify cell‐surface proteins that mark chemoresistant and self‐renewing GC cells following chemotherapy (cisplatin) reveal enrichment for the cell‐surface glycoprotein PMP22 (Cai et al., 2017). PMP22, a Wnt target gene, is expressed in patients that had undergone perioperative chemotherapy, highlighting a strong correlation between PMP22 expression and tumour recurrence. GC cells and tumour xenograft sensitivity to cisplatin were significantly increased when combined with pharmacological inhibition of PMP22 (Cai et al., 2017). The cell‐surface water channel protein aquaporin‐3 (AQP3) is overexpressed in GC tissues and promotes invasion and metastasis via EMT (Chen et al., 2014); however, its role in chemoresistance has only recently been investigated. GC cells expressing high levels of AQP3 are refractory to treatment with cisplatin, but when combined with targeted AQP3 knockdown, GC cells show increased sensitivity to chemotherapy (Dong et al., 2016). Of note, an independent study revealed that modulation of β‐catenin‐dependent signalling, via GSK3B inhibition or Axin stabilization, is sufficient to regulate the abundance and behaviour of AQP3+ GC cells (Zhou et al., 2016), which implies that targeted modulation of Wnt signalling may represent a way to combat therapy resistant GC cells. This observation of Wnt‐regulated chemoresistance has also been observed in other cancer types including medulloblastoma and colon cancer, in which Wnt regulation of the DNA repair enzyme O6‐methylguanine‐DNA methyltransferase, restored chemo‐sensitivity in vitro and in tumour xenografts (Wickstrom et al., 2015). Collectively, these data demonstrate that Wnt signalling could be an attractive target to inhibit gastric CSC activity, which will affect tumour growth and recurrence.
Evidence for deregulated Wnt signalling in gastric cancer
The role of β‐catenin‐dependent Wnt signalling in GC is now well established, with approximately 10–30% of human gastric tumours displaying deregulated Wnt signalling (Wang et al., 2014; Cristescu et al., 2015), with the latest TCGA study confirming significant Wnt pathway mutations in GCs (Cancer Genome Atlas Research, 2014). Functional evidence also demonstrates that deregulated Wnt signalling can trigger tumourigenesis in the stomach, as deletion of Gsk3b, a component of the β‐catenin degradation complex, resulted in gastric tumour formation (Radulescu et al., 2012), as does deletion of Apc in muscle, intestine and stomach expression‐1 positive (Mist1+) cells of the gastric epithelium (Hayakawa et al., 2017). Several Wnt genes are up‐regulated in GC including WNT1 (Mao et al., 2014), WNT5A (Boussioutas et al., 2003; Kurayoshi et al., 2006) and WNT6 (Yuan et al., 2013). These recent discoveries provide a timely backdrop to review the potential of targeting the Wnt pathway for the treatment of GC, as resistance to current chemotherapy is common, and the exact role of each component of the pathway and its potential as a therapeutic target is described in the sections below.
Genetic lesions of the β‐catenin destruction complex
Somatic mutation is a common mechanism to facilitate Wnt pathway deregulation in many solid cancers, including GC. Loss‐of‐function (LOF) mutations of multiple downstream Wnt pathway components such as APC, AXIN or activating mutations of CTNNB1 (gene encoding β‐catenin) feature in the initiation and progression in both intestinal‐type and diffuse‐type GCs (Cancer Genome Atlas Research, 2014; Wang et al., 2014). This section will review strategies targeting these three components of the destruction complex in GC.
Axin
GCs positive for microsatellite instability (MSI) comprise one of several molecular subgroups identified by large‐scale molecular characterization studies (Cancer Genome Atlas Research, 2014). Frameshift mutations of cancer‐associated genes with mono‐ or dinucleotide repeats in the coding sequences are a feature of gastric tumours positive for MSI (Simpson et al., 2001). The scaffold protein Axin serves as a critical rate‐limiting protein in the assembly of the β‐catenin destruction complex (Lee et al., 2003), highlighting its role as a negative regulator of Wnt signalling and tumour suppressor protein (Li et al., 2012). As such, approximately 30% of MSI‐high human GCs harbour frameshift mutations in AXIN2 (Kim et al., 2009). The AXIN2 frameshift mutation identified by Kim et al. is predicted to introduce a premature stop of amino acid synthesis in the C‐terminus of Axin2 protein and hence resemble a typical LOF mutation (Kim et al., 2009). This frameshift mutation (p.Gly665Alafs24) would eliminate a C‐terminal half of PP2Ac‐binding domain and the entire Axin‐binding domain. In addition, missense mutations in Axin have been reported in gastric adenocarcinomas (http://www.cbioportal.org/); however, their functional significance has not been properly investigated. Recent work in Drosophila and human cells has identified missense mutations in AXIN1 that disrupt the conserved core of the N‐terminal Axin regulator of G‐protein signalling domain, which is necessary for binding APC (Anvarian et al., 2016). Cells with a non‐functioning Axin scaffold gain paraneoplastic properties by forming protruding nanoscale aggregates, which engage with atypical signal transducers to confer cell‐growth advantages (Anvarian et al., 2016). Indeed, Axin1 null mice develop liver tumours, confirming its role as a tumour suppressor in vivo (Feng et al., 2012). Whether this same mutation‐aggregate mechanism occurs in GCs harbouring missense mutations to AXIN remains to be addressed.
Adenomatous polyposis coli
Since its discovery as a major tumour suppressor in colorectal cancer (Kinzler et al., 1991; Su et al., 1992; Korinek et al., 1997), LOF mutations to APC have been commonly reported in many other epithelial cancers, including GC (Sano et al., 1991). Sequencing of human gastric tumours showed several different mutations to APC; however, the small sample size of this study makes it difficult to conclude any correlation between APC mutation status and tumour sub‐type (Nakatsuru et al., 1992). Recent large‐scale genomic characterization of gastric tumours reveals frequent somatic mutations to APC in non‐hypermutated chromosomal instable GCs (Cancer Genome Atlas Research, 2014), which is further supported by an independent patient dataset reporting an even higher incidence of somatic mutations to APC (Cristescu et al., 2015). Likewise, in gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS) patients, mutation analysis and Sanger sequencing successfully mapped point mutations in APC promoter 1B, which was shown to reduce binding of the Yin Yang 1 (YY1) transcription factor and impaired the activity of APC 1B promoter (Li et al., 2016). Importantly, allelic imbalance of APC is detectable in patient blood and saliva samples, serving as an excellent biomarker for prospective GAPPS patients (Li et al., 2016). In support, GC cell lines reveal mutations at codon 1450 of APC that encodes a truncated form of APC that fails to negatively regulate β‐catenin, which causes constitutive activation of Wnt signalling (Sasaki et al., 2001). Importantly, several reports have demonstrated that conditional deletion or loss‐of‐heterozygosity of wild type Apc is sufficient to drive gastric hyperplasia and subsequent adenoma formation (Tomita et al., 2007; Radulescu et al. 2012; Powell et al., 2014; Sarkar et al., 2016). Collectively, patient genomics and preclinical mouse models highlight the relevance of APC mutations in driving gastric tumourigenesis, which offer an attractive therapeutic target to treat GC patients. As such, the application of tankyrase inhibitors (discussed in detail below) has been shown to be efficacious in colorectal cancer (CRC) cell lines and mouse models with APC mutations, which suggests that a similar therapeutic benefit could be observed in preclinical models of GC harbouring APC mutations (Chen et al., 2009; Huang et al., 2009; Waaler et al., 2011; Table 1).
Table 1.
Wnt mutations and modifications in gastric cancers
| Gene name | Mutation/modification type | Suitable class of drug(s) | Reference |
|---|---|---|---|
| APC | Truncation, point mutation | Tankyrase inhibitors, inhibitors of β‐catenin transcription | Wang et al., 2014, Li et al., 2016 |
| AXIN2 | Missense mutation | Inhibitors of β‐catenin transcription | Kim et al., 2009 |
| CTNNB1 | Exon3 mutation, SNPs | Tankyrase inhibitors, inhibitors of β‐catenin transcription | Radulescu et al., 2012, Wang et al., 2012b |
| DKK1–3 | Promoter methylation | Porcupine inhibitors, FZD‐blocking antibodies | Yu et al., 2009 |
| FZD1, FZD2 and FZD7 receptors | Overexpression | FZD‐blocking antibodies | Zhao et al., 2014 |
| RNF43 | Truncation, missense | Porcupine inhibitors, FZD‐blocking antibodies | Wang et al., 2014 |
| sFRP1–5 | Promoter methylation | Porcupine inhibitors, FZD‐blocking antibodies | Nojima et al., 2007 |
| TCF7 | Missense mutation | Tankyrase inhibitors, inhibitors of β‐catenin transcription | Kim et al., 2009 |
| Wnt‐1, Wnt‐2b, Wnt‐3, Wnt‐5a and Wnt‐6 | Overexpression | Porcupine inhibitors, FZD‐blocking antibodies |
Mao et al., 2014, Zhao et al., 2014
Yuan et al., 2013 |
CTNNB1 (β‐catenin)
Nuclear β‐catenin, a hallmark of active Wnt signalling, is detected in approximately 30% of GC tumours, identifying β‐catenin as a suitable target for therapeutic intervention (Clements et al., 2002). Gastric tumours displaying nuclear β‐catenin frequently harbour mutations at exon3 of CTNNB1 (Clements et al., 2002; Woo et al., 2001). Activating mutations at exon3 alter targeted serine‐threonine phosphorylation sites by GSK3B, which confer resistance to phosphorylation and lead to the accumulation of cytoplasmic and nuclear β‐catenin and subsequent changes in expression of genes that regulate proliferation (cyclin D1, D2 and E) (Akama et al., 1995; Takano et al., 2000; Liang et al., 2003; Arici et al., 2009). Genetic association analysis investigating the correlation between tagged single nucleotide polymorphisms (SNPs) spanning CTNNB1 and GC incidence and survival showed that SNPs rs1880481, rs4135385, rs11564475 and rs2293303 were significantly associated with GC susceptibility (Wang et al., 2012b). In addition, the rs4135385 AG/AA genotypes were associated with a 0.74‐fold reduced adjusted hazard ratio for favourable overall 5 year survival of non‐cardia GC (Chiurillo, 2015; Wang et al., 2012b). A recent proof‐of‐principle study demonstrated that conditional mutation of exon3 in CTNNB1 is sufficient to induce intestinal‐type gastric adenomas in the antral stomach of adult mice and increased activation of Wnt signalling (Radulescu et al., 2012). As such, targeted siRNA knockdown of β‐catenin in human GC cells leads to inhibition of Wnt target gene transcription, decreased cell proliferation and an increase in apoptosis of GC cells (Jiang et al., 2010). The expression of survivin (BIRC5), a Wnt target gene, was deregulated following β‐catenin knockdown, suggesting that elevated Wnt signalling might inhibit apoptosis by regulating survivin during GC (Jiang et al., 2010). A complementary gene‐targeted approach demonstrated that recombinant adenovirus carrying the p53 up‐regulated modulator of apoptosis (PUMA) under the control of β‐catenin/TCF‐responsive promoter (AdTOP‐PUMA) selectively targeted and killed AGS GC cells with active Wnt signalling (Dvory‐Sobol et al., 2007). Synergistic cell killing was observed when AdTOP‐PUMA was used in combination with standard chemotherapeutic agents (5‐florouracil, doxorubicin and paclitaxel) highlighting the potential of adjuvant Wnt‐targeted therapies in GC (Dvory‐Sobol et al., 2007). Of note, even in GCs with no detectable mutations to CTNNB1 or APC, the abundance of β‐catenin mRNA is greatly enhanced (Ebert et al., 2002), and although post‐translational rather than transcriptional regulation of β‐catenin is at the core of active Wnt signalling, increased β‐catenin protein production may suggest that upstream components of the Wnt pathway are deregulated, thereby activating Wnt signalling in GC.
Genetic lesions of the Wnt receptor complex
Aberrant activation of the Wnt pathway can also occur at the level of Wnts and/or FZD receptors. Genetic and/or epigenetic events that alter the function of Wnt regulators, thereby deregulating the expression of Wnt and/or FZD receptors resulting in pathway activation, have been identified in several cancers, including GC, and also represent a target for cancer therapy which is reviewed in this section.
Loss‐of‐function mutations to RNF43
Mammalian tissues that undergo constant renewal, such as the skin, blood and the gut, rely on tightly controlled Wnt signalling to maintain stem cell populations that fuel the replenishment of exhausted cells. For example, LGR5+ intestinal stem cells, which are exquisitely sensitive to Wnt, sustain the constant turnover of the intestinal epithelium (Barker et al., 2007). More recently, the potent Wnt agonist R‐spondin (RSPO1) was shown to be a ligand for the receptor LGR5 and sufficient to potentiate β‐catenin‐dependent signalling (Kazanskaya et al., 2004; Kim et al., 2008; de Lau et al., 2011; Carmon et al., 2012). Following the discovery of R‐spondin as the ligand for LGR5, independent investigations identified two highly related transmembrane E3 ubiquitin ligases, zinc‐ring finger 3 (ZNRF3) and ring finger 43 (RNF43), as negative regulators of Wnt signalling that are integral to R‐spondin/LGR5 Wnt potentiation (Hao et al., 2012; Koo et al., 2012). Extensive biochemical experiments reveal that ZNRF3 and RNF43 regulate Wnt signalling by promoting the ubiquitylation, internalization and degradation of FZD receptor‐LRP5/6 complexes following Wnt activation, thereby limiting the duration and intensity of Wnt signalling (Hao et al., 2012). These findings support the following model: in the absence of R‐spondin, ZNRF3/RNF43 ubiquitylates the FZD receptor via the DEP domain of Dvl and promotes the degradation of the FZD receptor‐LRP5/6 complex (Jiang et al., 2015), thus keeping Wnt signalling to low levels. However, in the presence of R‐spondin, an interaction between LGR5 and ZNRF3/RNF43, via R‐spondin furin domains (1&2) leads to the clearance of ZNRF3/RNF43, allowing FZD receptor/LRP5/6 complexes to accumulate at the membrane to enhance β‐catenin‐dependent Wnt signalling (Hao et al., 2012; Peng et al., 2013).
Recent large‐scale genomic data have identified frequent RNF43 LOF mutations, predominantly truncating or missense alterations, in >50 and 4.8% of MSI and microsatellite stable gastric tumours respectively (Cancer Genome Atlas Research, 2014; Wang et al., 2014). A recent examination of the progressive genomic and transcriptomic alterations from early‐stage gastric adenomas through to later‐stage disease validate the recurrent mutations to RNF43 previously described (Min et al., 2016). Furthermore, the frequency of RNF43 mutations in early‐mid stage gastric tumours identifies deregulated Wnt signalling as a critical driver and potential biomarker of early gastric tumourigenesis. Thus, determining if a gastric tumour has RNF43 mutations will help stratify which patients are more likely to benefit from therapies targeted to the Wnt receptor complex or the production of Wnts (Min et al., 2016). To date, the efficacy of antibodies that block FZD receptors and/or inhibitors of Wnt secretion has not been thoroughly tested in preclinical models of GC. However, one study has shown significant cell growth arrest and Wnt pathway inhibition using an inhibitor of Wnt secretion, IWP‐2 (Chen et al., 2009), on human intestinal‐type gastric adenocarcinoma cells (MKN28); however, there have been no reports of RNF43 mutations in this cell line (Mo et al., 2013).
Encouragingly, other preclinical models of solid cancers harbouring RNF43 mutations such as pancreatic, colon and breast cancer treated with inhibitors directed to the receptor complex (inhibitors of Wnt secretion or FZD receptor‐blocking antibodies) have yielded potent anti‐tumourigenic effects, especially when used in combination with standard chemotherapeutics (Gurney et al., 2012; Jiang et al., 2013; Steinhart et al., 2017).
Epigenetic silencing of Wnt antagonists
Until relatively recently, it was considered that constitutive Wnt activation triggered by mutation to APC, CTNNB1 or AXIN was impervious to further regulation from upstream Wnt components, that is, ligands and receptors. However, it has been demonstrated that the secreted frizzled‐related protein (sFRP) family of Wnt negative regulators are frequently silenced via promoter hypermethylation in a variety of cancers, including GC (Caldwell et al., 2004; Suzuki et al., 2004; Cheng et al., 2007). The family of sFRP glycoproteins is comprised of five family members, which can bind directly to Wnt via its cysteine‐rich domain (CRD), the Wnt‐FZD receptor binding interface (Janda et al., 2012), thereby competing with FZD receptors for Wnt binding. Given that sFRPs can bind directly to Wnt, they are able to inhibit both β‐catenin‐dependent and β‐catenin‐independent signalling. In normal gastric mucosa, the expression of sFRP1, sFRP2 and sFRP5 is readily detected. However, in primary gastric tumours and GC cell lines, the expression of sFRP is absent, which is attributed to significant DNA methylation within the promoter‐associated cytosine‐phosphate‐guanine islands (Cheng et al., 2007; Nojima et al., 2007; Zhao et al., 2009). The silencing of sFRP via methylation is detected in pre‐neoplastic gastric tissue, suggesting that this is a mechanism of tumour initiation in the stomach and thus could be used as a biomarker to screen for patients with an enhanced risk of developing GC (Cheng et al., 2007). Much like the studies performed in CRC cell lines (Caldwell et al., 2004; Suzuki et al., 2004; Caldwell et al., 2006), transfection of sFRP‐1,‐2 or ‐5 successfully suppressed Wnt signalling, which is sufficient to block proliferation and induce apoptosis in GC cell lines harbouring APC or CTNNB1 mutations (Nojima et al., 2007). Xenografts have also been used to demonstrate that transfecting mice with sFRP2 could block tumour growth, thus illustrating the potential for sFRP2 to act as a functional tumour suppressor (Cheng et al., 2007). The ability of sFRP to attenuate Wnt signalling is dependent on a functional Wnt‐binding CRD as sFRP constructs lacking a functional CRD failed to inhibit proliferation and induce apoptosis (Nojima et al., 2007).
Similar to the sFRP family of Wnt negative‐modulators, the dickkopf family of glycoproteins (DKK1–4) are potent inhibitors of Wnt signalling (Niehrs, 2006). However, in contrast to sFRPs, DKK selectively inhibits β‐catenin‐dependent Wnt signalling through interacting with Lrp via the EGF repeat domains within Lrp6 (Mao et al., 2001), thus preventing Wnt and the FZD receptor from forming a ternary complex (MacDonald et al., 2004). DKK can also modulate Wnt by associating with kremen 1 and kremen 2 to form a complex that regulates the internalization of Lrp (Niehrs, 2006). Much like sFRPs, the expression of DKK3 is commonly silenced (70%) in GC tissues via promoter methylation and is associated with poor patient survival, as shown by multivariate analysis (Yu et al., 2009). Reversal of DNA methylation with demethylating agents or ectopic expression of DKK3 is sufficient to restore DKK3 expression and subvert GC cell growth and Wnt signalling (Yu et al., 2009). More recently, conventional adenoviral gene therapy has been utilized to deliver functional DKK1 to inhibit Wnt signalling and attenuate gastric tumourigenesis (Wang et al., 2012a). Following infection with the chimeric Ad5/35‐DKK1 adenovirus, the number of CD44+ GC stem cells and volume of tumour xenografts was significantly reduced (Wang et al., 2012a). While the authors report successful infection of GC cells in vitro, issues surrounding delivery, tissue penetration and immunological response currently limit the feasibility of gene therapy technologies in GC patients (Sutter and Fechner, 2006).
Collectively, the silenced expression of sFRP and/or DKK inhibitors in GC undoubtedly contributes to unrestrained Wnt pathway activation at the level of the receptor complex. As such, patients with DNA‐promoter modifications to sFRP and/or DKK negative modulators, which can be detected in pre‐neoplastic tissue, might benefit from Wnt‐targeted therapies at the level of Wnt receptors and/or Wnt secretion.
Overexpression of Wnt ligands and FZD receptors
While the incidence of genetic lesions to genes that encode for Wnts and FZD receptors is low, deregulated expression of Wnts and FZD receptors is a common feature of GCs, which can be attributed to the Wnt pathway mutations described previously. This suggests that GCs with abundant Wnts and FZD receptors can be targeted therapeutically. For instance, Wnt5a, a prototypical β‐catenin‐independent Wnt known to inhibit β‐catenin‐dependent Wnt signalling and promote cell migration and invasion (Moon, 2002), is overexpressed and correlated with aggressive GC phenotypes (Kurayoshi et al., 2006). Infiltrating macrophages secrete Wnt5a following H. pylori infection, which induces the migration and invasion of GC cells via CXCR4 chemokine receptors (Zhao et al., 2013). In support, a recent preclinical investigation has identified that Wnt5a produced by innate lymphoid cells supports diffuse GC progression by inhibiting anoikis, which enables anchorage‐independent growth of GC cells (Hayakawa et al., 2015). Indeed, chimeric mice transplanted with bone marrow from Wnt5a flox/flox mice (Wnt5a deficient) exhibit significantly fewer signet‐ring foci than wild‐type bone marrow recipient chimeras (Hayakawa et al., 2015). Furthermore, therapeutic targeting of Wnt5a using a novel anti‐Wnt5a polyclonal antibody (pAb5a‐5) has been shown to reduce the migration and invasion of GC cells in vitro and in vivo by blocking receptor complex internalization, which is necessary to activate target gene expression required for cell motility (Hanaki et al., 2012). Originally identified as a gene that preferentially integrates into mouse mammary tumour virus proviral DNA (Nusse and Varmus, 1982), Wnt‐1 is overexpressed in GC tissues and has been shown to induce the expression of CSC genes (Oct4 and Cd44), which is associated with disease progression and poor outcome (Mao et al., 2014). Modified AGS GC cells that overexpress Wnt‐1 confer increased GC cell proliferation and Wnt target gene expression in vitro and in xenograft tumours (Mao et al., 2014). Treatment of AGS‐Wnt‐1 overexpressing cells with the antibacterial potassium ionophore salinomycin, which was shown to inhibit β‐catenin‐dependent signalling by inducing the degradation of LRP6 (Lu et al., 2011), effectively reduces tumour growth and the associated elevated expression of CSC and Wnt target genes (Mao et al., 2014).
Similar to their cognate ligands, the frizzled family of Wnt receptors is frequently deregulated in GCs, which is often associated with poor clinical outcome (Schmuck et al., 2011; Phesse et al., 2016). Of the 10 family members, multiple reports have identified Fzd7 to be overexpressed in GC tissues, which are associated with various stages of disease progression (Kirikoshi et al., 2001; Schmuck et al., 2011; Zhao et al., 2014). The FZD7 receptor is unique among other FZD members as it is one of the few FZD receptors shown to transduce all major signalling branches of the Wnt pathway and is associated with maintaining proliferation in adult stem cell populations (Fernandez et al., 2014; Flanagan et al., 2015; Phesse et al., 2016). This places the FZD7 receptor in a unique position to mediate cell proliferation as well as tumour dissemination and metastasis (Vincan et al., 2005; Vincan et al., 2007; Ueno et al., 2009). A side population (SP) of cells, which display cancer stem cell properties, isolated from human GC cell lines reveal increased expression of Fzd7 and other genes associated with CSCs, which supports a role for Fzd7 in promoting GC (Schmuck et al., 2011). Moreover, our unpublished data demonstrates that targeted molecular inhibition of Fzd7 or conditional deletion of Fzd7 is sufficient to block the growth of human GC cells and in mouse models of intestinal‐type GC. Similarly, siRNA knockdown of the FZD2 receptor, which is structurally related to the FZD1 and FZD7 receptors (Sagara et al., 1998; Fredriksson et al., 2003), reduced cell proliferation and invasion in MKN45 and MKN74 GC cells (Tomizawa et al., 2015). In contrast, GC cell lines (AGS and SGC7901) reveal transcriptional silencing of Fzd6 by miRNA‐21 (Yan et al., 2016). Re‐introduction of Fzd6 expression represses GC cell proliferation by antagonizing β‐catenin‐dependent Wnt signalling, which can be blocked by siRNA‐targeted Fzd6 knockdown or pharmacological inhibition of miRNA‐21 (Yan et al., 2016). Together, these data demonstrate that aberrant expression of Wnts and FZD receptors contributes to gastric tumourigenesis, and thus, they represent a therapeutic target for GC patients with tumours showing elevated Wnt activity.
Infection and innervation influence Wnt activation in gastric cancer
The aetiology of GC is further complicated by complex interactions between bacteria, host and environmental factors (Gravaghi et al., 2008). Infection with the bacterial carcinogen H. pylori is the greatest risk factor for GC (Hardbower et al., 2014), with ~75% of the global GC burden attributed to H. pylori‐induced inflammation and associated tumourigenesis (Parkin et al., 2005). Following successful colonization of the stomach epithelium, H. pylori drives superficial gastritis, which progresses to chronic inflammation (Correa, 1996). H. pylori‐driven inflammation is sufficient to induce the expression of caudal‐related homeobox transcription factor 2 (CDX2) and mucin 2 (MUC2), which enables the transdifferentiation of gastric cells to adopt an intestinal cell phenotype (intestinal metaplasia). This is characterized by the loss of parietal cells and presence of Paneth and goblet cells. The resulting intestinal metaplasia (predominantly in the antrum) or spasmolytic polypeptide‐expressing metaplasia (predominantly in corpus) progresses to dysplasia and ultimately cancer. H. pylori delivers bacterial virulence factors that modulate epithelial biology and inflammatory responses for its own benefit. Of the virulence factors produced by H. pylori associated with GC development, cytotoxin‐associated gene product (CagA) and its associated type IV secretion system have been shown to activate Wnt signalling and promote gastric tumourigenesis and progression (Amieva and Peek, 2016; Neal et al., 2013).
Gerbils infected with a carcinogenic strain of H. pylori develop rapid and highly penetrant changes in the gastric mucosa of recipient animals that progress to gastric adenocarcinoma (Franco et al., 2005). The induction of gastric dysplasia following H. pylori infection was associated with increased expression of nuclear β‐catenin, which was shown to be CagA‐dependent (Franco et al., 2005). Research using transgenic zebrafish has extended previous findings, demonstrating that activation of Wnt and epithelial changes triggered by CagA are downstream or parallel to the β‐catenin destruction complex but upstream of Tcf‐4 (Neal et al., 2013). In addition, gastric cells co‐cultured with H. pylori induce phosphorylation of Lrp6 within 30 min, which is sufficient to stabilize β‐catenin where it can mediate β‐catenin target gene transcription (Gnad et al., 2010). The success of H. pylori to induce changes to the gastric epithelium hinge on its capacity to attach to and transform gastric stem cells. Adult gastric stem cells, marked by the Wnt target gene Lgr5, are not only reliant on Wnt signalling for their maintenance but are also the cell‐of‐origin in gastric tumourigenesis following Lgr5 +‐targeted Wnt activation (Barker et al., 2010a). Adult murine Lgr5 + gastric stem cells infected with H. pylori increase proliferation and the expression of Wnt target genes, which is sufficient to transform Lgr5 + gastric stem cells and their progeny (Sigal et al., 2015). Strains of H. pylori with defects in chemotaxis or that are CagA‐deficient are unable to induce transformation of Lgr5 + gastric stem cells and subsequent hyperplastic changes (Sigal et al., 2015). A complementary approach using ex vivo human gastric organoids also demonstrated the transformation of human gastric stem cells following infection with H. pylori (Bartfeld et al., 2015). Taken together, these studies suggest that small molecule inhibitors targeted to the interaction between β‐catenin and Tcf or other co‐factors required for Wnt‐driven transcription might abolish the increased activation of Wnt signalling observed following infection and thus have therapeutic value in patients infected with carcinogenic strains of H. pylori.
Several recent studies from Timothy Wang's group have elegantly shown that a functional nervous system innervation is required at all stages of GC in a Wnt‐dependent manner. The studies collectively reveal a nerve growth factor/M3 receptor signalling axis that activates Wnt signalling via promoting YAP/β‐catenin complexes (Hayakawa et al., 2017) in gastric stem cells, which in turn fuels gastric tumour growth (Hayakawa et al., 2015; Zhao et al., 2014). Furthermore, pharmacological inhibition of cholinergic signalling with botulinum toxin was sufficient to block tumour growth in mouse models of GC, associated with down‐regulation of Wnt target genes (Zhao et al., 2014).
These data suggest that Wnt signalling is rate limiting for cholinergic signalling‐associated GC. Indeed, gastric tumours that developed in the antrum following Apc truncation in Mist1+ cells of Mist1CreERT2; Apc flox/flox mice were significantly reduced when the M3 receptor was co‐deleted (Hayakawa et al., 2017). In addition to demonstrating that Wnt signalling is rate limiting for M3 receptor‐driven gastric tumours, these data also highlight that the antrum is more sensitive to Wnt‐driven tumourigenesis than the corpus, since Mist1+ cells are found in both areas of the stomach (Hayakawa et al., 2017).
Suitable inhibitors of Wnt signalling for gastric cancers
Aberrant Wnt signalling in GC can be achieved through either mutational or non‐mutational alterations. As such, molecular blockade of Wnt signalling is sufficient to inhibit tumour growth in several preclinical models of solid cancers, prompting the recent development of Wnt‐targeted inhibitors, several of which are in early‐stage clinical trials. However, it is clear that the advancement of anti‐Wnt drug candidates is slow both in numbers of candidates entering the trials and in the stage of advancement through the phase 1–2–3 clinical trials. Pharmacological modulation of Wnt signalling can be divided into compounds that modulate the ligand/receptor interface, stabilize the degradation complex or interfere with β‐catenin‐dependent gene transcription (Figure 2A, B) (Tai et al., 2015). Note that the pharmacology and anti‐tumour effects of Wnt inhibitors described below have not been validated in preclinical models of GC, although there is now sufficient biological evidence to suggest they may be of therapeutic benefit for the treatment of GC in the future.
Figure 2.
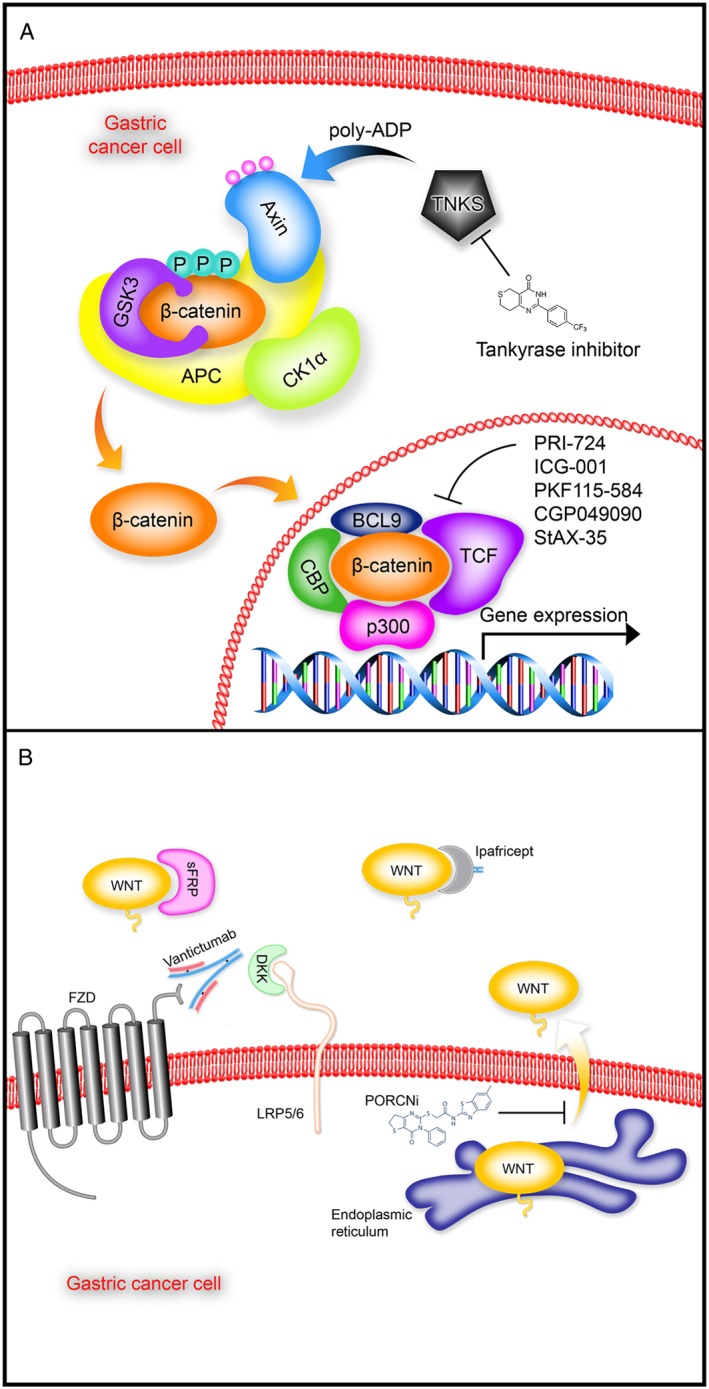
Inhibitors of Wnt signalling. (A) Following Wnt/FZD receptor binding (not shown), recruitment of the scaffolding protein AXIN to the receptor complex leads to inhibition of GSK3B and the β‐catenin destruction complex, which allows newly synthesized β‐catenin to accumulate and translocate to the nucleus (orange arrows), where it binds with co‐factors to form a transcriptionally active complex. Indicated are some of the inhibitors that target various intracellular nodes of Wnt signal transduction that could have therapeutic benefit for gastric cancer patients with APC, AXIN or CTNNB1 mutations. (B) Wnt signalling (WNT) can be inhibited at the cell surface by various pathway inhibitors such as sFRPs and DKK, which bind to Wnt and LRP5/6 respectively. Other approaches target Wnt signalling using small molecules that inhibit porcupine (PORCN), which prevent the secretion of Wnt from the endoplasmic reticulum (yellow arrow). In addition, FZD receptor‐blocking antibodies (vantictumab) and decoy FZD receptors (ipafricept) inhibit Wnt signalling by blocking FZD receptors and sequestering Wnts respectively. Gastric cancer patients with RNF43 mutations or overexpression of Wnt and/or FZD receptors would probably benefit from these types of Wnt inhibitors.
There is also a strong rational for the repositioning of existing FDA approved drugs for the treatment of diseases with deregulated Wnt signalling such as cancer. For example, the anthelmintic agent niclosamide was originally approved for clinical use in treating tapeworm infections but has been shown to inhibit the growth of several cancer cell lines including colon, breast, pancreas, lung and prostate (Mook et al., 2015). Niclosamide is a multi‐functional drug and has been shown to inhibit several oncogenic signalling pathways including Notch, mTOR, Stat‐3, NFĸB and Wnt. Indeed, niclosamide treatment significantly inhibited the growth of APC mutant colon cancer xenografts, independently of mTOR and NFĸB signalling, which was associated with reduced Wnt signalling. As the stomach is more easily accessible than other organs such as the pancreas and brain, the problem of delivery for anti‐cancer treatments is less of an issue and, therefore, drugs such as niclosamide which have a poor bioavailability have a greater chance of success in GCs.
Inhibitors of Wnt signalling at the receptor complex
The gaps in our understanding pertaining to intrinsic Wnt pathway complexity pose technical concerns when considering how best to inhibit β‐catenin‐dependent and β‐catenin‐independent Wnt signalling. One approach to block both β‐catenin‐dependent and β‐catenin‐independent Wnt signalling is to focus on inhibiting the interaction between Wnts and their cognate FZD receptors, which can be achieved using monoclonal antibodies and decoy receptors. Vantictumab (OMP‐18R5, Oncomed Pharmaceuticals) is an anti‐FZD receptor blocking antibody, originally identified to bind the FZD7 receptor, which can functionally bind to five out of 10 mammalian FZD receptors (1, 2, 5, 7 and 8) by binding a series of highly conserved residues spanning the receptors’ extracellular cleft (Gurney et al., 2012). Vantictumab can successfully inhibit the ability of several Wnts to activate β‐catenin‐dependent Wnt signalling, which is accompanied by decreased Wnt transcription (Gurney et al., 2012). Furthermore, when tested on tumour xenografts, vantictumab significantly inhibited the growth of several types of solid human tumours including breast, pancreatic and colon. Significant synergy was observed when vantictumab was combined with standard‐of‐care chemotherapies, such as paclitaxel and taxol (Gurney et al., 2012). Vantictumab is currently in phase 1b clinical trials for HER2‐negative breast cancer (NCT01973309) and advanced pancreatic cancers (NCT02005315) and was well tolerated up to the current dose of 15 mg·kg−1 every 3 weeks. Reported side effects include fatigue, abdominal pain, constipation and nausea. An interim efficacy assessment has demonstrated an overall response rate of 48% for patients given vantictumab and chemotherapy. Patient‐free survival and overall survival data are not yet available for these clinical studies. Of note, significant bone turnover was observed in a subset of patients, which resulted in a temporary hold on three phase 1b clinical trials (Tai et al., 2015). The temporary hold has been lifted after reviewing substantial clinical safety and efficacy data and revised protocols submitted by Oncomed. Other strategies employ soluble decoy receptors that sequester Wnts, akin to sFRPs, thus preventing Wnt–FZD receptor interactions. Ipafricept (OMP‐54F28, Oncomed Pharmaceuticals) is a soluble Fc fusion protein that consists of the CRD of the FZD8 receptor fused to the Fc domain of human IgG1 (http://www.oncomed.com/). Ipafricept inhibits the growth of patient‐derived xenografts, which is characterized by reduced CSC frequency and proliferation, as well as promoting tumour cell differentiation. As such, ipafricept has progressed to phase 1b clinical trials and is tested in combination with chemotherapy for the treatment of various solid tumours such as hepatocellular carcinoma, ovarian and pancreatic cancer (clinicaltrials.gov). Interim safety data demonstrate that the combination of ipafricept with chemotherapy was well tolerated and an overall response rate of 39% was observed (Tai et al., 2015).
An alternative approach to inhibit Wnt signalling is to block the biogenesis and secretion of Wnt proteins. Wnts undergo two types of known lipid modification. The first is a palmitate to cysteine 77, which is conserved among all Wnt family members (Willert et al., 2003). The second reported modification is the addition of a mono‐unsaturated palmitoleate moiety to serine 209, which is required for release of Wnt from the endoplasmic reticulum (ER) and binding to the FZD receptor (Takada et al., 2006). Porcupine (PORC) is an essential non‐redundant enzyme that is responsible for the serine 209 modification, and as such, PORC inhibition causes Wnt retention in the ER and thus blocks their secretion and subsequent pathway activation (Takada et al., 2006). However, there are some cells, including CD8+ T‐cells and human astrocytes that have a PORC‐independent mechanism of Wnt secretion, as treatment with the PORC inhibitor IWP‐2 did not prevent Wnt secretion in these cells (Richards et al., 2014). Several small molecule PORC inhibitors have been developed in recent years and have shown promising anti‐tumour effects in preclinical models with minimal off‐target effects (Chen et al., 2009; Jiang et al., 2013; Madan et al., 2016), which have been extended to phase 1 clinical trials for Wnt ligand‐dependent malignancies such as pancreatic adenocarcinoma and proto‐oncogene B‐Raf‐mutant colorectal cancer which are currently ongoing (NCT01351103).
Stabilizers of the β‐catenin destruction complex
The scaffolding protein AXIN is the rate‐limiting component of the β‐catenin destruction complex (Lee et al., 2003) with the levels of AXIN1 and AXIN2 proteins being constantly surveyed and regulated by tankyrases, TNK1 and TNK2. Tankyrases regulate the stability of AXIN1 and AXIN2 through poly(ADP‐ribosyl)ation (PARsylation) (Huang et al., 2009), which directs AXIN ubiquitylation by RNF146 and proteasomal degradation (Callow et al., 2011; Zhang et al., 2011).
Recent efforts using cell‐based screens have discovered several small‐molecule tankyrase inhibitors, which successfully stabilize AXIN1 and AXIN2 by preventing PARsylation, thus promoting β‐catenin destruction complex stability and functionality (Huang et al., 2009). First generation tankyrase inhibitors, inhibitors of Wnt response and XAV939, were independently discovered, and both were shown to inhibit β‐catenin‐dependent signalling and cell viability in APC or CTNNB1 mutant cancer cell lines, highlighting their therapeutic promise for the inhibition of Wnt‐dependent cancers (Chen et al., 2009; Huang et al., 2009). Second generation tankyrase inhibitors have been optimized for potency, selectivity and tested in preclinical mouse models of intestinal cancer, which demonstrate stabilization of AXIN to reduce β‐catenin‐dependent signalling and subsequent tumour burden (Lau et al. 2013; Waaler et al., 2012). The translation of this class of drugs into the clinic has been frustrating as significant intestinal toxicity is observed in preclinical models (Lau et al., 2013; Kahn, 2014). Next generation tankyrase inhibitors are being developed to reduce off‐target effects on genes such as PARP1 and are currently being tested in preclinical models (Tai et al., 2015; Nathubhai et al., 2016). As such, it will be of interest to see if these new compounds can be optimized to balance inhibiting Wnt activity while reducing toxicity, which may advance their progression into the clinic.
Inhibitors of β‐catenin interaction partners and transcription
A key mechanism of β‐catenin‐dependent Wnt signalling is the dynamic regulation, and subsequent activation, of a β‐catenin‐centric complex where β‐catenin interacts with and recruits a series of nuclear co‐activator proteins such as TCF/LEF, cAMP response element‐binding protein (CBP), B‐cell chronic lymphocytic leukaemia/lymphoma‐9 (BCL‐9), histone acetyltransferase p300 and others, to activate target gene transcription (Valenta et al., 2012). Thus, drugging such protein–protein interfaces offers an attractive route for blocking Wnt signalling downstream of common pathway mutations such as APC, which would limit the risk of mutational circumvention leading to drug resistances. However, this therapeutic approach is not suitable for oncogenic mutations identified in genes responsible for β‐catenin transcriptional activity, such as TCF7L2 (Kim et al., 2009). To date, a number of small‐molecule inhibitors have been identified via cell‐based high throughput screens that successfully inhibit β‐catenin interactions necessary for transcriptional activation, which significantly limit Wnt signalling and subsequent tumourigenesis in preclinical models.
An early approach to detect β‐catenin–TCF interactions utilized a high‐throughput ELISA screen. Subsequent screening of extensive compound libraries (>45 000 compounds) identified eight compounds that dose‐dependently inhibited β‐catenin–TCF complex formation (Lepourcelet et al., 2004). Functional inhibition of β‐catenin–TCF interaction was confirmed via decreased activation of Wnt‐specific reporter assays (TOPflash), colon cancer cell proliferation/viability and β‐catenin‐induced axis duplication in Xenopus laevis embryos (Lepourcelet et al., 2004). Of the eight compounds, two structurally related compounds, PKF115‐584 and CGP049090, proved the most potent and were also found to disrupt the β‐catenin–APC interaction, implicating potential direct binding to β‐catenin (Lepourcelet et al., 2004). However, the exact molecular mechanism by which these two compounds inhibit Wnt signalling remains unresolved (Lepourcelet et al., 2004; Kahn, 2014).
Using a similar approach, colon cancer cell lines challenged with a secondary structure‐templated chemical library revealed a novel low‐molecular weight inhibitor of β‐catenin‐dependent signalling, ICG‐001. Characterization of ICG‐001 demonstrated potent suppression of TOPflash assays and Wnt target gene transcription. ICG‐001 suppressed the Wnt pathway by directly binding to CBP, thus disrupting the interaction between CBP and β‐catenin (Emami et al., 2004). ICG‐001 selectively induces apoptosis in transformed cells but not in normal colon cells as seen in the Apc Min/+ mouse model and xenograft models of colon cancer (Emami et al., 2004). A second generation class of CBP‐β‐catenin inhibitors, PRI‐724, have progressed into phase II and Ib clinical trials for metastatic CRC (NCI‐2015‐00436 and NCT02413853) and advanced pancreatic adenocarcinoma (NCT01764477) respectively and are currently ongoing.
Implementing ‘peptide stapling’ technology, which involves the introduction of a synthetic hydrocarbon bridge into an α‐helical peptide, both the β‐catenin–BCL‐9 (Takada et al., 2012) and the β‐catenin–T‐cell factor‐4 (Grossmann et al., 2012) interfaces have been targeted, yielding potent in vitro inhibitors (IC50 ~10–20 nM), which displayed low micromolar cell‐based inhibition. The clinical utility of the stapled peptide class of molecules has yet to be established (Grossmann et al., 2012; Takada et al., 2012; Kahn, 2014). As β‐catenin‐dependent Wnt signalling is deregulated in GC, with mutations in APC and RNF43 observed (Cancer Genome Atlas Research, 2014; Wang et al., 2014), it will be interesting to see if this class of drugs is also effective in gastric tumours displaying high Wnt signalling.
Concerns surrounding drugging the Wnt pathway
As mechanisms of Wnt deregulation during cancer have been slowly exposed, this has been mirrored by considerable research effort into discovering Wnt‐targeted therapeutics (Barker and Clevers, 2006; Kahn, 2014). However, drugging developmental pathways can have potentially devastating effects on embryonic development, adult stem cell populations and the regenerative response following injury (Kahn, 2014). A common concern relating to Wnt pathway therapeutics is the potential for acute toxicity in adult tissues that are maintained by adult stem cells (including intestine, haematopoietic system, skin, bone and hair) regulated by Wnt signalling (Clevers et al., 2014). With respect to the intestinal epithelium, it is encouraging that targeted ablation of intestinal Lgr5+ stem cells, which display exquisite sensitivity to Wnt, does not disrupt intestinal homeostasis and a full recovery of the stem cell compartment is driven by substantial cellular plasticity displayed by differentiated intestinal cell types (Tian et al., 2011; van Es et al., 2012; Buczacki et al., 2013; Tetteh et al., 2016). Additional side effects may include metabolic changes due to the impact of Wnt on liver zonation, bone toxicity and potential neurological effects given the role of Wnt signalling in synapse formation and maintenance in the CNS and PNS (Burke et al., 2009; Liu et al., 2011; Harrison‐Uy et al., 2013). Therefore, when considering the targeting of crucial developmental pathways that are utilized by both CSCs as well as normal somatic stem cells, the potential ‘Jekyll and Hyde’‐like behaviour of this class of therapeutic agents remains an ever‐present issue (Kahn, 2014).
In addition, the appropriate dose of a Wnt pathway inhibitor must be taken into account given the pleiotropic effects and varying thresholds of Wnt signalling across many tissues. For example, the level of β‐catenin‐dependent Wnt signalling in liver and melanoma oncogenesis is lower than intestinal tumourigenesis (Buchert et al., 2010). Therefore, therapeutic agents that alter Wnt signalling activity may reach an efficacious threshold in one tissue but fail to do so in another. Moreover, within the intestinal epithelium, different Wnt threshold levels distinguish stem and progenitor cells, which could be useful for exclusively targeting CSCs without damaging the renewal capability of the intestine (Tao et al., 2015).
Another question to consider is whether it is best to treat gastric malignancy through antagonizing or activating the Wnt pathway? As mentioned earlier, restoration of Fzd6 expression in GC cells is sufficient to inhibit β‐catenin‐dependent Wnt signalling and associated tumour‐promoting phenotypes (Yan et al., 2016). Similarly, hyperactivation of Wnt signalling in Cited1‐deficient mice reduced tumourigenesis in the intestine (Meniel et al., 2013). In contrast, inhibition of Wnt signalling at several different levels of the pathway has been demonstrated to provide anti‐tumour effects in several cancers (Polakis, 2007; Anastas and Moon, 2013). Therefore, a greater understanding of how Wnt signalling is controlled in GC tissues is needed to guide which, if any, inhibitors of Wnt signalling should be applied. Another parameter to consider is the complex inter‐ and intra‐tumour heterogeneity observed in GCs (Zhong et al., 2016), which can confound drug responsiveness and lead to acquired resistance. It is therefore imperative to have a greater understanding of individual patient tumour biology and to identify predictive biomarkers of efficacy to aid therapeutic strategy.
Concluding remarks
When taken together, the increasing amount of proof‐of‐principle studies strongly identify that aberrant Wnt signalling contributes to GC and suggest that interfering with this signal could be an effective treatment for this disease. Although promising therapeutic leads have been advanced and tested in combination with other chemotherapeutics in several solid tumours, the same Wnt‐targeted therapy combinations have not been tested in preclinical models of GC. While the potential safety concerns associated with Wnt‐targeted therapies are legitimate, similar complications are also relevant with all drugs. If used and integrated correctly (Cancer Genome Atlas Research, 2017), the recent deluge of genomic (Cancer Genome Atlas Research, 2014), epigenomic (Ooi et al., 2016) and functional profiles from cancer patients (van de Wetering et al., 2015) will pave a clear path forward to identify the appropriate patients for a specific Wnt inhibitor.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors wish to thank Gavan Mitchell for helping generate the figures in this manuscript. Funding is gratefully acknowledged from the following; National Health and Medical Research Council of Australia (NHMRC) project grants (566679 and APP1099302 awarded to E.V.); Melbourne Health project grants (605030 and PG‐002 awarded to E.V./T.J.P.), and an Early Career Researcher grant (GIA‐033) awarded to D.J.F.; Cancer Council of Victoria project grants (CCV, APP1020716) awarded to E.V. and T.J.P., CCV Fellowship awarded to D.J.F. and Cardiff University Research Fellowship awarded to T.J.P.
Flanagan, D. J. , Vincan, E. , and Phesse, T. J. (2017) Winding back Wnt signalling: potential therapeutic targets for treating gastric cancers. British Journal of Pharmacology, 174: 4666–4683. doi: 10.1111/bph.13890.
Contributor Information
Elizabeth Vincan, Email: e.vincan@unimelb.edu.au.
Toby J Phesse, Email: phesset@cardiff.ac.uk.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R (1997). Beta‐catenin is a target for the ubiquitin‐proteasome pathway. EMBO J 16: 3797–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acebron SP, Niehrs C (2016). Beta‐catenin‐independent roles of Wnt/LRP6 signaling. Trends Cell Biol 26: 956–967. [DOI] [PubMed] [Google Scholar]
- Akama Y, Yasui W, Yokozaki H, Kuniyasu H, Kitahara K, Ishikawa T et al. (1995). Frequent amplification of the cyclin E gene in human gastric carcinomas. Jpn J Cancer Res 86: 617–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The concise guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amieva M, Peek RM Jr (2016). Pathobiology of Helicobacter pylori‐induced gastric cancer. Gastroenterology 150: 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastas JN, Moon RT (2013). Wnt signalling pathways as therapeutic targets in cancer. Nat Rev 13: 11–26. [DOI] [PubMed] [Google Scholar]
- Anvarian Z, Nojima H, van Kappel EC, Madl T, Spit M, Viertler M et al. (2016). Axin cancer mutants form nanoaggregates to rewire the Wnt signaling network. Nat Struct Mol Biol 23: 324–332. [DOI] [PubMed] [Google Scholar]
- Arici DS, Tuncer E, Ozer H, Simek G, Koyuncu A (2009). Expression of retinoblastoma and cyclin D1 in gastric carcinoma. Neoplasma 56: 63–67. [DOI] [PubMed] [Google Scholar]
- Barker N, Bartfeld S, Clevers H (2010b). Tissue‐resident adult stem cell populations of rapidly self‐renewing organs. Cell Stem Cell 7: 656–670. [DOI] [PubMed] [Google Scholar]
- Barker N, Clevers H (2006). Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov 5: 997–1014. [DOI] [PubMed] [Google Scholar]
- Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH et al. (2010a). Lgr5(+ve) stem cells drive self‐renewal in the stomach and build long‐lived gastric units in vitro. Cell Stem Cell 6: 25–36. [DOI] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M et al. (2009). Crypt stem cells as the cells‐of‐origin of intestinal cancer. Nature 457: 608–611. [DOI] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M et al. (2007). Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449: 1003–1007. [DOI] [PubMed] [Google Scholar]
- Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P et al. (2015). In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 148: 126–136 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blache P, van de Wetering M, Duluc I, Domon C, Berta P, Freund JN et al. (2004). SOX9 is an intestine crypt transcription factor, is regulated by the Wnt pathway, and represses the CDX2 and MUC2 genes. J Cell Biol 166: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackham AU, Greenleaf E, Yamamoto M, Hollenbeak C, Gusani N, Coppola D et al. (2016). Tumor regression grade in gastric cancer: predictors and impact on outcome. J Surg Oncol 114: 434–439. [DOI] [PubMed] [Google Scholar]
- Boussioutas A, Li H, Liu J, Waring P, Lade S, Holloway AJ et al. (2003). Distinctive patterns of gene expression in premalignant gastric mucosa and gastric cancer. Cancer Res 63: 2569–2577. [PubMed] [Google Scholar]
- Brungs D, Aghmesheh M, Vine KL, Becker TM, Carolan MG, Ranson M (2016). Gastric cancer stem cells: evidence, potential markers, and clinical implications. J Gastroenterol 51: 313–326. [DOI] [PubMed] [Google Scholar]
- Buchert M, Athineos D, Abud HE, Burke ZD, Faux MC, Samuel MS et al. (2010). Genetic dissection of differential signaling threshold requirements for the Wnt/beta‐catenin pathway in vivo. PLoS Genet 6 e1000816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczacki SJ, Zecchini HI, Nicholson AM, Russell R, Vermeulen L, Kemp R et al. (2013). Intestinal label‐retaining cells are secretory precursors expressing Lgr5. Nature 495: 65–69. [DOI] [PubMed] [Google Scholar]
- Burke ZD, Reed KR, Phesse TJ, Sansom OJ, Clarke AR, Tosh D (2009). Liver zonation occurs through a beta‐catenin‐dependent, c‐Myc‐independent mechanism. Gastroenterology 136: 2316–2324. [DOI] [PubMed] [Google Scholar]
- Cai W, Chen G, Luo Q, Liu J, Guo X, Zhang T et al. (2017). PMP22 regulates self‐renewal and chemoresistance of gastric cancer cells. Mol Cancer Ther 16: 1187–1198. [DOI] [PubMed] [Google Scholar]
- Caldwell GM, Jones C, Gensberg K, Jan S, Hardy RG, Byrd P et al. (2004). The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res 64: 883–888. [DOI] [PubMed] [Google Scholar]
- Caldwell GM, Jones CE, Taniere P, Warrack R, Soon Y, Matthews GM et al. (2006). The Wnt antagonist sFRP1 is downregulated in premalignant large bowel adenomas. Br J Cancer 94: 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN et al. (2011). Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS One 6 e22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513: 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N (2017). Integrated genomic and molecular characterization of cervical cancer. Nature 16; 543(7645). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmon KS, Lin Q, Gong X, Thomas A, Liu Q (2012). LGR5 interacts and cointernalizes with Wnt receptors to modulate Wnt/beta‐catenin signaling. Mol Cell Biol 32: 2054–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Dodge ME, Tang W, Lu J, Ma Z, Fan CW et al. (2009). Small molecule‐mediated disruption of Wnt‐dependent signaling in tissue regeneration and cancer. Nat Chem Biol 5: 100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wang T, Zhou YC, Gao F, Zhang ZH, Xu H et al. (2014). Aquaporin 3 promotes epithelial‐mesenchymal transition in gastric cancer. J Exp Clin Cancer Res 33: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YY, Yu J, Wong YP, Man EP, To KF , Jin VX et al. (2007). Frequent epigenetic inactivation of secreted frizzled‐related protein 2 (SFRP2) by promoter methylation in human gastric cancer. Br J Cancer 97: 895–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiurillo MA (2015). Role of the Wnt/beta‐catenin pathway in gastric cancer: an in‐depth literature review. World J Exp Med 5: 84–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio‐Preiser C et al. (2002). Beta‐catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res 62: 3503–3506. [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta‐catenin signaling in development and disease. Cell 127: 469–480. [DOI] [PubMed] [Google Scholar]
- Clevers H, Loh KM, Nusse R (2014). Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346 1248012. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R (2012). Wnt/beta‐catenin signaling and disease. Cell 149: 1192–1205. [DOI] [PubMed] [Google Scholar]
- Correa P (1996). Helicobacter pylori and gastric cancer: state of the art. Cancer Epidemiol Biomarkers Prev 5: 477–481. [PubMed] [Google Scholar]
- Crew KD, Neugut AI (2006). Epidemiology of gastric cancer. World J Gastroenterol: WJG 12: 354–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS et al. (2015). Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med 21: 449–456. [DOI] [PubMed] [Google Scholar]
- de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H et al. (2011). Lgr5 homologues associate with Wnt receptors and mediate R‐spondin signalling. Nature 476: 293–297. [DOI] [PubMed] [Google Scholar]
- Demitrack ES, Gifford GB, Keeley TM, Carulli AJ, VanDussen KL, Thomas D et al. (2015). Notch signaling regulates gastric antral LGR5 stem cell function. EMBO J 34: 2522–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Wang Y, Zhou Y, Wen J, Wang S, Shen L (2016). Aquaporin 3 facilitates chemoresistance in gastric cancer cells to cisplatin via autophagy. Cell Death Dis 2: 16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvory‐Sobol H, Sagiv E, Liberman E, Kazanov D, Arber N (2007). Suppression of gastric cancer cell growth by targeting the beta‐catenin/T‐cell factor pathway. Cancer 109: 188–197. [DOI] [PubMed] [Google Scholar]
- Ebert MP, Fei G, Kahmann S, Muller O, Yu J, Sung JJ et al. (2002). Increased beta‐catenin mRNA levels and mutational alterations of the APC and beta‐catenin gene are present in intestinal‐type gastric cancer. Carcinogenesis 23: 87–91. [DOI] [PubMed] [Google Scholar]
- Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M et al. (2004). A small molecule inhibitor of beta‐catenin/CREB‐binding protein transcription [corrected]. Proc Natl Acad Sci U S A 101: 12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farin HF, Jordens I, Mosa MH, Basak O, Korving J, Tauriello DV et al. (2016). Visualization of a short‐range Wnt gradient in the intestinal stem‐cell niche. Nature 530: 340–343. [DOI] [PubMed] [Google Scholar]
- Feng GJ, Cotta W, Wei XQ, Poetz O, Evans R, Jarde T et al. (2012). Conditional disruption of Axin1 leads to development of liver tumors in mice. Gastroenterology 143: 1650–1659. [DOI] [PubMed] [Google Scholar]
- Fernandez A, Huggins IJ, Perna L, Brafman D, Lu D, Yao S et al. (2014). The Wnt receptor Fzd7 is required for maintenance of the pluripotent state in human embryonic stem cells. Proc Natl Acad Sci U S A 111: 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan DJ, Phesse TJ, Barker N, Schwab RH, Amin N, Malaterre J et al. (2015). Frizzled7 functions as a Wnt receptor in intestinal epithelial Lgr5(+) stem cells. Stem Cell Reports 4: 759–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan DJ, Barker N, Nowell C, Clevers H, Ernst M, Phesse T et al. (2017). Loss of the Wnt Receptor Frizzled7 in the Gastric Epithelium is Deleterious and Triggers Rapid Repopulation in vivo . Disease Models and Mechanisms. https://doi.org/10.1242/dmm.029876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB et al. (2005). Activation of beta‐catenin by carcinogenic Helicobacter pylori . Proc Natl Acad Sci U S A 102: 10646–10651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB (2003). The G‐protein‐coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol 63: 1256–1272. [DOI] [PubMed] [Google Scholar]
- Gnad T, Feoktistova M, Leverkus M, Lendeckel U, Naumann M (2010). Helicobacter pylori‐induced activation of beta‐catenin involves low density lipoprotein receptor‐related protein 6 and dishevelled. Mol Cancer 9: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravaghi C, Bo J, Laperle KM, Quimby F, Kucherlapati R, Edelmann W et al. (2008). Obesity enhances gastrointestinal tumorigenesis in Apc‐mutant mice. Int J Obes (Lond) 32: 1716–1719. [DOI] [PubMed] [Google Scholar]
- Grossmann TN, Yeh JT, Bowman BR, Chu Q, Moellering RE, Verdine GL (2012). Inhibition of oncogenic Wnt signaling through direct targeting of beta‐catenin. Proc Natl Acad Sci U S A 109: 17942–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggenheim DE, Shah MA (2013). Gastric cancer epidemiology and risk factors. J Surg Oncol 107: 230–236. [DOI] [PubMed] [Google Scholar]
- Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P et al. (1998). E‐cadherin germline mutations in familial gastric cancer. Nature 392: 402–405. [DOI] [PubMed] [Google Scholar]
- Guilford PJ, Hopkins JB, Grady WM, Markowitz SD, Willis J, Lynch H et al. (1999). E‐cadherin germline mutations define an inherited cancer syndrome dominated by diffuse gastric cancer. Hum Mutat 14: 249–255. [DOI] [PubMed] [Google Scholar]
- Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L et al. (2012). Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A 109: 11717–11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaki H, Yamamoto H, Sakane H, Matsumoto S, Ohdan H, Sato A et al. (2012). An anti‐Wnt5a antibody suppresses metastasis of gastric cancer cells in vivo by inhibiting receptor‐mediated endocytosis. Mol Cancer Ther 11: 298–307. [DOI] [PubMed] [Google Scholar]
- Hao HX, Xie Y, Zhang Y, Charlat O, Oster E, Avello M et al. (2012). ZNRF3 promotes Wnt receptor turnover in an R‐spondin‐sensitive manner. Nature 485: 195–200. [DOI] [PubMed] [Google Scholar]
- Hardbower DM, Peek RM Jr, Wilson KT (2014). At the bench: Helicobacter pylori, dysregulated host responses, DNA damage, and gastric cancer. J Leukoc Biol 96: 201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison‐Uy SJ, Siegenthaler JA, Faedo A, Rubenstein JL, Pleasure SJ (2013). CoupTFI interacts with retinoic acid signaling during cortical development. PLoS One 8 e58219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Ariyama H, Stancikova J, Sakitani K, Asfaha S, Renz BW et al. (2015). Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell 28: 800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H et al. (2017). Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 31: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann W (2008). Regeneration of the gastric mucosa and its glands from stem cells. Curr Med Chem 15: 3133–3144. [DOI] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA et al. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461: 614–620. [DOI] [PubMed] [Google Scholar]
- Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC (2012). Structural basis of Wnt recognition by Frizzled. Science 337: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011). Global cancer statistics. CA Cancer J Clin 61: 69–90. [DOI] [PubMed] [Google Scholar]
- Jiang K, Shen Z, Ye Y, Yang X, Wang S (2010). A novel molecular marker for early detection and evaluating prognosis of gastric cancer: N‐myc downstream regulated gene‐1 (NDRG1). Scand J Gastroenterol 45: 898–908. [DOI] [PubMed] [Google Scholar]
- Jiang X, Charlat O, Zamponi R, Yang Y, Cong F (2015). Dishevelled promotes Wnt receptor degradation through recruitment of ZNRF3/RNF43 E3 ubiquitin ligases. Mol Cell 58: 522–533. [DOI] [PubMed] [Google Scholar]
- Jiang X, Hao HX, Growney JD, Woolfenden S, Bottiglio C, Ng N et al. (2013). Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 110: 12649–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn M (2014). Can we safely target the Wnt pathway? Nat Rev Drug Discov 13: 513–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y‐K, Saroh T, Ryu M‐H, Chao Y, Kato K, Chung HC et al. (2017). Nivolumab (ONO‐4538/BMS‐936558) as salvage treatment after second or later‐line chemotherapy for advanced gastric or gastro‐esophageal junction cancer (AGC): a double‐blinded, randomized, phase III trial. J Clin Oncol 35 (Suppl): 2. [Google Scholar]
- Karam SM, Leblond CP (1993). Dynamics of epithelial cells in the corpus of the mouse stomach. I. Identification of proliferative cell types and pinpointing of the stem cell. Anat Rec 236: 259–279. [DOI] [PubMed] [Google Scholar]
- Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W (2004). R‐Spondin2 is a secreted activator of Wnt/beta‐catenin signaling and is required for Xenopus myogenesis. Dev Cell 7: 525–534. [DOI] [PubMed] [Google Scholar]
- Kim BM, Buchner G, Miletich I, Sharpe PT, Shivdasani RA (2005). The stomach mesenchymal transcription factor Barx1 specifies gastric epithelial identity through inhibition of transient Wnt signaling. Dev Cell 8: 611–622. [DOI] [PubMed] [Google Scholar]
- Kim KA, Wagle M, Tran K, Zhan X, Dixon MA, Liu S et al. (2008). R‐Spondin family members regulate the Wnt pathway by a common mechanism. Mol Biol Cell 19: 2588–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Kim SS, Ahn CH, Yoo NJ, Lee SH (2009). Frameshift mutations of Wnt pathway genes AXIN2 and TCF7L2 in gastric carcinomas with high microsatellite instability. Hum Pathol 40: 58–64. [DOI] [PubMed] [Google Scholar]
- Kim TH, Shivdasani RA (2011). Notch signaling in stomach epithelial stem cell homeostasis. J Exp Med 208: 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Shivdasani RA (2016). Stomach development, stem cells and disease. Development 143: 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB et al. (1991). Identification of FAP locus genes from chromosome 5q21. Science 253: 661–665. [DOI] [PubMed] [Google Scholar]
- Kirikoshi H, Sekihara H, Katoh M (2001). Up‐regulation of frizzled‐7 (Fzd7) in human gastric cancer. Int J Oncol 19: 111–115. [PubMed] [Google Scholar]
- Kitagawa M, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, Hattori K et al. (1999). An F‐box protein, FWD1, mediates ubiquitin‐dependent proteolysis of beta‐catenin. EMBO J 18: 2401–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M et al. (2012). Tumour suppressor RNF43 is a stem‐cell E3 ligase that induces endocytosis of Wnt receptors. Nature 488: 665–669. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW et al. (1997). Constitutive transcriptional activation by a beta‐catenin‐Tcf complex in APC−/− colon carcinoma. Science 275: 1784–1787. [DOI] [PubMed] [Google Scholar]
- Kurayoshi M, Oue N, Yamamoto H, Kishida M, Inoue A, Asahara T et al. (2006). Expression of Wnt‐5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res 66: 10439–10448. [DOI] [PubMed] [Google Scholar]
- Lau T, Chan E, Callow M, Waaler J, Boggs J, Blake RA et al. (2013). A novel tankyrase small‐molecule inhibitor suppresses APC mutation‐driven colorectal tumor growth. Cancer Res 73: 3132–3144. [DOI] [PubMed] [Google Scholar]
- Lauren P (1965). The two histological main types of gastric carcinoma: diffuse and so‐called intestinal‐type carcinoma. An attempt at a histo‐clinical classification. Acta Pathol Microbiol Scand 64: 31–49. [DOI] [PubMed] [Google Scholar]
- Lee E, Salic A, Kruger R, Heinrich R, Kirschner MW (2003). The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol 1: E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F et al. (2004). Small‐molecule antagonists of the oncogenic Tcf/beta‐catenin protein complex. Cancer Cell 5: 91–102. [DOI] [PubMed] [Google Scholar]
- Li J, Woods SL, Healey S, Beesley J, Chen X, Lee JS et al. (2016). Point mutations in exon 1B of APC reveal gastric adenocarcinoma and proximal polyposis of the stomach as a familial adenomatous polyposis variant. Am J Hum Genet 98: 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP et al. (2012). Wnt signaling through inhibition of beta‐catenin degradation in an intact Axin1 complex. Cell 149: 1245–1256. [DOI] [PubMed] [Google Scholar]
- Liang B, Wang S, Yang X, Ye Y, Yu Y, Cui Z (2003). Expressions of cyclin E, cyclin dependent kinase 2 and p57(KIP2) in human gastric cancer. Chin Med J (Engl) 116: 20–23. [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y et al. (2002). Control of beta‐catenin phosphorylation/degradation by a dual‐kinase mechanism. Cell 108: 837–847. [DOI] [PubMed] [Google Scholar]
- Liu C, Tu Y, Sun X, Jiang J, Jin X, Bo X et al. (2011). Wnt/beta‐catenin pathway in human glioma: expression pattern and clinical/prognostic correlations. Clin Exp Med 11: 105–112. [DOI] [PubMed] [Google Scholar]
- Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y (2011). Niclosamide suppresses cancer cell growth by inducing Wnt co‐receptor LRP6 degradation and inhibiting the Wnt/beta‐catenin pathway. PLoS One 6 e29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Adamska M, Meisler MH (2004). Hypomorphic expression of Dkk1 in the doubleridge mouse: dose dependence and compensatory interactions with Lrp6. Development 131: 2543–2552. [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X (2009). Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J et al. (2016). Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene 35: 2197–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Fan S, Ma W, Fan P, Wang B, Zhang J et al. (2014). Roles of Wnt/beta‐catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis 5: e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Wang J, Liu B, Pan W, Farr GH 3rd, Flynn C et al. (2001). Low‐density lipoprotein receptor‐related protein‐5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell 7: 801–809. [DOI] [PubMed] [Google Scholar]
- Mayer B, Jauch KW, Gunthert U, Figdor CG, Schildberg FW, Funke I et al. (1993). De‐novo expression of CD44 and survival in gastric cancer. Lancet 342: 1019–1022. [DOI] [PubMed] [Google Scholar]
- Melo FS, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J et al. (2017). A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543: 676–680. [DOI] [PubMed] [Google Scholar]
- Meniel V, Song F, Phesse T, Young M, Poetz O, Parry L et al. (2013). Cited1 deficiency suppresses intestinal tumorigenesis. PLoS Genet 9 e1003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikels AJ, Nusse R (2006). Purified Wnt5a protein activates or inhibits beta‐catenin‐TCF signaling depending on receptor context. PLoS Biol 4: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min BH, Hwang J, Kim NK, Park G, Kang SY, Ahn S et al. (2016). Dysregulated Wnt signalling and recurrent mutations of the tumour suppressor RNF43 in early gastric carcinogenesis. J Pathol 240: 304–314. [DOI] [PubMed] [Google Scholar]
- Mo ML, Li MR, Chen Z, Liu XW, Sheng Q, Zhou HM (2013). Inhibition of the Wnt palmitoyltransferase porcupine suppresses cell growth and downregulates the Wnt/beta‐catenin pathway in gastric cancer. Oncol Lett 5: 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mook RA Jr, Wang J, Ren XR, Chen M, Spasojevic I, Barak LS et al. (2015). Structure‐activity studies of Wnt/beta‐catenin inhibition in the niclosamide chemotype: identification of derivatives with improved drug exposure. Bioorg Med Chem 23: 5829–5838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT (2002). An introduction to non‐canonical Wnt and frizzled signaling. Semin Cell Dev Biol 13: 215. [Google Scholar]
- Mulligan KA, Fuerer C, Ching W, Fish M, Willert K, Nusse R (2012). Secreted Wingless‐interacting molecule (Swim) promotes long‐range signaling by maintaining Wingless solubility. Proc Natl Acad Sci U S A 109: 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuru S, Yanagisawa A, Ichii S, Tahara E, Kato Y, Nakamura Y et al. (1992). Somatic mutation of the APC gene in gastric cancer: frequent mutations in very well differentiated adenocarcinoma and signet‐ring cell carcinoma. Hum Mol Genet 1: 559–563. [DOI] [PubMed] [Google Scholar]
- Nathubhai A, Haikarainen T, Hayward PC, Munoz‐Descalzo S, Thompson AS, Lloyd MD et al. (2016). Structure‐activity relationships of 2‐arylquinazolin‐4‐ones as highly selective and potent inhibitors of the tankyrases. Eur J Med Chem 118: 316–327. [DOI] [PubMed] [Google Scholar]
- Neal JT, Peterson TS, Kent ML, Guillemin K (2013). H. pylori virulence factor CagA increases intestinal cell proliferation by Wnt pathway activation in a transgenic zebrafish model. Dis Model Mech 6: 802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C (2006). Function and biological roles of the dickkopf family of Wnt modulators. Oncogene 25: 7469–7481. [DOI] [PubMed] [Google Scholar]
- Niehrs C (2012). The complex world of Wnt receptor signalling. Nat Rev Mol Cell Biol 13: 767–779. [DOI] [PubMed] [Google Scholar]
- Nojima M, Suzuki H, Toyota M, Watanabe Y, Maruyama R, Sasaki S et al. (2007). Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 26: 4699–4713. [DOI] [PubMed] [Google Scholar]
- Nusse R, Varmus HE (1982). Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 31: 99–109. [DOI] [PubMed] [Google Scholar]
- Ooi WF, Xing M, Xu C, Yao X, Ramlee MK, Lim MC et al. (2016). Epigenomic profiling of primary gastric adenocarcinoma reveals super‐enhancer heterogeneity. Nat Commun 7: 12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P (2005). Global cancer statistics, 2002. CA Cancer J Clin 55: 74–108. [DOI] [PubMed] [Google Scholar]
- Peng WC, de Lau W, Madoori PK, Forneris F, Granneman JC, Clevers H et al. (2013). Structures of Wnt‐antagonist ZNRF3 and its complex with R‐spondin 1 and implications for signaling. PLoS One 8 e83110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phesse T, Flanagan D, Vincan E (2016). Frizzled7: a promising Achilles' heel for targeting the Wnt receptor complex to treat cancer. Cancers (Basel) 8 [Epub ahead of print]. https://doi.org/10.1007/7651_2016_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P (2007). The many ways of Wnt in cancer. Curr Opin Genet Dev 17: 45–51. [DOI] [PubMed] [Google Scholar]
- Powell AE, Vlacich G, Zhao ZY, McKinley ET, Washington MK, Manning HC et al. (2014). Inducible loss of one Apc allele in Lrig1‐expressing progenitor cells results in multiple distal colonic tumors with features of familial adenomatous polyposis. Am J Physiol Gastrointest Liver Physiol 307: G16–G23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulescu S, Ridgway RA, Cordero J, Athineos D, Salgueiro P, Poulsom R et al. (2012). Acute Wnt signalling activation perturbs differentiation within the adult stomach and rapidly leads to tumour formation. Oncogene 32: 2048–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman R, Asombang AW, Ibdah JA (2014). Characteristics of gastric cancer in Asia. World J Gastroenterol 20: 4483–4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards MH, Seaton MS, Wallace J, Al‐Harthi L (2014). Porcupine is not required for the production of the majority of Wnts from primary human astrocytes and CD8+ T cells. PLoS One 9 e92159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg ME, Nusse Y, Kalisky T, Lee JJ, Dalerba P, Scheeren F et al. (2012). Identification of a cKit(+) colonic crypt base secretory cell that supports Lgr5(+) stem cells in mice. Gastroenterology 142: 1195–1205 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagara N, Toda G, Hirai M, Terada M, Katoh M (1998). Molecular cloning, differential expression, and chromosomal localization of human frizzled‐1, frizzled‐2, and frizzled‐7. Biochem Biophys Res Commun 252: 117–122. [DOI] [PubMed] [Google Scholar]
- Sano T, Tsujino T, Yoshida K, Nakayama H, Haruma K, Ito H et al. (1991). Frequent loss of heterozygosity on chromosomes 1q, 5q, and 17p in human gastric carcinomas. Cancer Res 51: 2926–2931. [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR et al. (2007). Myc deletion rescues Apc deficiency in the small intestine. Nature 446: 676–679. [DOI] [PubMed] [Google Scholar]
- Santos JC, Carrasco‐Garcia E, Garcia‐Puga M, Aldaz P, Montes M, Fernandez‐Reyes M et al. (2016). SOX9 elevation acts with canonical Wnt signaling to drive gastric cancer progression. Cancer Res 76: 6735–6746. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Huebner AJ, Sulahian R, Anselmo A, Xu X, Flattery K et al. (2016). Sox2 suppresses gastric tumorigenesis in mice. Cell Rep 16: 1929–1941. [DOI] [PubMed] [Google Scholar]
- Sasaki N, Sachs N, Wiebrands K, Ellenbroek SI, Fumagalli A, Lyubimova A et al. (2016). Reg4+ deep crypt secretory cells function as epithelial niche for Lgr5+ stem cells in colon. Proc Natl Acad Sci U S A 113: E5399–E5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Morimoto I, Kusano M, Hosokawa M, Itoh F, Yanagihara K et al. (2001). Mutational analysis of the beta‐catenin gene in gastric carcinomas. Tumour Biol 22: 123–130. [DOI] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M et al. (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469: 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmuck R, Warneke V, Behrens HM, Simon E, Weichert W, Rocken C (2011). Genotypic and phenotypic characterization of side population of gastric cancer cell lines. Am J Pathol 178: 1792–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MA, Khanin R, Tang L, Janjigian YY, Klimstra DS, Gerdes H et al. (2011). Molecular classification of gastric cancer: a new paradigm. Clin Cancer Res 17: 2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal M, Rothenberg ME, Logan CY, Lee JY, Honaker RW, Cooper RL et al. (2015). Helicobacter pylori activates and expands Lgr5(+) stem cells through direct colonization of the gastric glands. Gastroenterology 148: 1392–1404.e21. [DOI] [PubMed] [Google Scholar]
- Simpson AJ, Caballero OL, Pena SD (2001). Microsatellite instability as a tool for the classification of gastric cancer. Trends Mol Med 7: 76–80. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange DE, Koo BK, Huch M, Sibbel G, Basak O, Lyubimova A et al. (2013). Differentiated Troy+ chief cells act as reserve stem cells to generate all lineages of the stomach epithelium. Cell 155: 357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhart Z, Pavlovic Z, Chandrashekhar M, Hart T, Wang X, Zhang X et al. (2017). Genome‐wide CRISPR screens reveal a Wnt‐Fzd5 signaling circuit as a druggable vulnerability of RNF43‐mutant pancreatic tumors. Nat Med 23: 60–68. [DOI] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C et al. (1992). Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256: 668–670. [DOI] [PubMed] [Google Scholar]
- Sutter AP, Fechner H (2006). Gene therapy for gastric cancer: is it promising? World J Gastroenterol 12: 380–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD et al. (2004). Epigenetic inactivation of SFRP genes allows constitutive Wnt signaling in colorectal cancer. Nat Genet 36: 417–422. [DOI] [PubMed] [Google Scholar]
- Tai D, Wells K, Arcaroli J, Vanderbilt C, Aisner DL, Messersmith WA et al. (2015). Targeting the Wnt signaling pathway in cancer therapeutics. Oncologist 20: 1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada K, Zhu D, Bird GH, Sukhdeo K, Zhao JJ, Mani M et al. (2012). Targeted disruption of the BCL9/beta‐catenin complex inhibits oncogenic Wnt signaling. Sci Transl Med 4 148ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H et al. (2006). Monounsaturated fatty acid modification of Wnt protein: its role in Wnt secretion. Dev Cell 11: 791–801. [DOI] [PubMed] [Google Scholar]
- Takano Y, Kato Y, van Diest PJ, Masuda M, Mitomi H, Okayasu I (2000). Cyclin D2 overexpression and lack of p27 correlate positively and cyclin E inversely with a poor prognosis in gastric cancer cases. Am J Pathol 156: 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao S, Tang D, Morita Y, Sperka T, Omrani O, Lechel A et al. (2015). Wnt activity and basal niche position sensitize intestinal stem and progenitor cells to DNA damage. EMBO J 34: 624–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetteh PW, Basak O, Farin HF, Wiebrands K, Kretzschmar K, Begthel H et al. (2016). Replacement of lost Lgr5‐positive stem cells through plasticity of their enterocyte‐lineage daughters. Cell Stem Cell 18: 203–213. [DOI] [PubMed] [Google Scholar]
- Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD et al. (2011). A reserve stem cell population in small intestine renders Lgr5‐positive cells dispensable. Nature 478: 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita H, Yamada Y, Oyama T, Hata K, Hirose Y, Hara A et al. (2007). Development of gastric tumors in Apc(Min/+) mice by the activation of the beta‐catenin/Tcf signaling pathway. Cancer Res 67: 4079–4087. [DOI] [PubMed] [Google Scholar]
- Tomizawa M, Shinozaki F, Motoyoshi Y, Sugiyama T, Yamamoto S, Ishige N (2015). Gastric cancer cell proliferation is suppressed by frizzled‐2 short hairpin RNA. Int J Oncol 46: 1018–1024. [DOI] [PubMed] [Google Scholar]
- Topol L, Jiang X, Choi H, Garrett‐Beal L, Carolan PJ, Yang Y (2003). Wnt‐5a inhibits the canonical Wnt pathway by promoting GSK‐3‐independent beta‐catenin degradation. J Cell Biol 162: 899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno K, Hazama S, Mitomori S, Nishioka M, Suehiro Y, Hirata H et al. (2009). Down‐regulation of frizzled‐7 expression decreases survival, invasion and metastatic capabilities of colon cancer cells. Br J Cancer 101: 1374–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenta T, Hausmann G, Basler K (2012). The many faces and functions of beta‐catenin. EMBO J 31: 2714–2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Amerongen R, Nusse R (2009). Towards an integrated view of Wnt signaling in development. Development 136: 3205–3214. [DOI] [PubMed] [Google Scholar]
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A et al. (2015). Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 161: 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es JH, Sato T, van de Wetering M, Lyubimova A, Nee AN, Gregorieff A et al. (2012). Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 14: 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincan E, Darcy PK, Farrelly CA, Faux MC, Brabletz T, Ramsay RG (2007). Frizzled‐7 dictates three‐dimensional organization of colorectal cancer cell carcinoids. Oncogene 26: 2340–2352. [DOI] [PubMed] [Google Scholar]
- Vincan E, Darcy PK, Smyth MJ, Thompson EW, Thomas RJ, Phillips WA et al. (2005). Frizzled‐7 receptor ectodomain expression in a colon cancer cell line induces morphological change and attenuates tumor growth. Differentiation; research in biological diversity 73: 142–153. [DOI] [PubMed] [Google Scholar]
- Visvader JE (2011). Cells of origin in cancer. Nature 469: 314–322. [DOI] [PubMed] [Google Scholar]
- Visvader JE, Clevers H (2016). Tissue‐specific designs of stem cell hierarchies. Nat Cell Biol 18: 349–355. [DOI] [PubMed] [Google Scholar]
- Waaler J, Machon O, Tumova L, Dinh H, Korinek V, Wilson SR et al. (2012). A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res 72: 2822–2832. [DOI] [PubMed] [Google Scholar]
- Waaler J, Machon O, von Kries JP, Wilson SR, Lundenes E, Wedlich D et al. (2011). Novel synthetic antagonists of canonical Wnt signaling inhibit colorectal cancer cell growth. Cancer Res 71: 197–205. [DOI] [PubMed] [Google Scholar]
- Wang B, Liu J, Ma LN, Xiao HL, Wang YZ, Li Y et al. (2012a). Chimeric 5/35 adenovirus‐mediated dickkopf‐1 overexpression suppressed tumorigenicity of CD44(+) gastric cancer cells via attenuating Wnt signaling. J Gastroenterol 48: 798–808. [DOI] [PubMed] [Google Scholar]
- Wang S, Tian Y, Wu D, Zhu H, Luo D, Gong W et al. (2012b). Genetic variation of CTNNB1 gene is associated with susceptibility and prognosis of gastric cancer in a Chinese population. Mutagenesis 27: 623–630. [DOI] [PubMed] [Google Scholar]
- Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST et al. (2014). Whole‐genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 46: 573–582. [DOI] [PubMed] [Google Scholar]
- Wickstrom M, Dyberg C, Milosevic J, Einvik C, Calero R, Sveinbjornsson B et al. (2015). Wnt/beta‐catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat Commun 6: 8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T et al. (2003). Wnt proteins are lipid‐modified and can act as stem cell growth factors. Nature 423: 448–452. [DOI] [PubMed] [Google Scholar]
- Wong H, Yau T (2013). Molecular targeted therapies in advanced gastric cancer: does tumor histology matter? Therap Adv Gastroenterol 6: 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J (2016). Impact of pathologic tumor response in the treatment of gastric cancer. Transl Gastroenterol Hepatol 1: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo DK, Kim HS, Lee HS, Kang YH, Yang HK, Kim WH (2001). Altered expression and mutation of beta‐catenin gene in gastric carcinomas and cell lines. Int J Cancer 95: 108–113. [DOI] [PubMed] [Google Scholar]
- Yan J, Liu T, Zhou X, Dang Y, Yin C, Zhang G (2016). Fzd6, targeted by miR‐21, represses gastric cancer cell proliferation and migration via activating non‐canonical wnt pathway. Am J Transl Res 8: 2354–2364. [PMC free article] [PubMed] [Google Scholar]
- Yu J, Tao Q, Cheng YY, Lee KY, Ng SS, Cheung KF et al. (2009). Promoter methylation of the Wnt/beta‐catenin signaling antagonist Dkk‐3 is associated with poor survival in gastric cancer. Cancer 115: 49–60. [DOI] [PubMed] [Google Scholar]
- Yuan G, Regel I, Lian F, Friedrich T, Hitkova I, Hofheinz RD et al. (2013). Wnt6 is a novel target gene of caveolin‐1 promoting chemoresistance to epirubicin in human gastric cancer cells. Oncogene 32: 375–387. [DOI] [PubMed] [Google Scholar]
- Zeng X, Huang H, Tamai K, Zhang X, Harada Y, Yokota C et al. (2008). Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development 135: 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R et al. (2005). A dual‐kinase mechanism for Wnt co‐receptor phosphorylation and activation. Nature 438: 873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Fan D (2010). New insights into the mechanisms of gastric cancer multidrug resistance and future perspectives. Future Oncol 6: 527–537. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA et al. (2011). RNF146 is a poly(ADP‐ribose)‐directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13: 623–629. [DOI] [PubMed] [Google Scholar]
- Zhao C, Bu X, Zhang N, Wang W (2009). Downregulation of SFRP5 expression and its inverse correlation with those of MMP‐7 and MT1‐MMP in gastric cancer. BMC Cancer 9: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Ma H, Bu X, Wang W, Zhang N (2013). SFRP5 inhibits gastric epithelial cell migration induced by macrophage‐derived Wnt5a. Carcinogenesis 34: 146–152. [DOI] [PubMed] [Google Scholar]
- Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT et al. (2014). Denervation suppresses gastric tumorigenesis. Science Translational Medicine 6 250ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q, Ruschoff JH, Guo T, Gabrani M, Schuffler PJ, Rechsteiner M et al. (2016). Image‐based computational quantification and visualization of genetic alterations and tumour heterogeneity. Sci Rep 6: 24146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang Y, Wen J, Zhao H, Dong X, Zhang Z et al. (2016). Aquaporin 3 promotes the stem‐like properties of gastric cancer cells via Wnt/GSK‐3beta/beta‐catenin pathway. Oncotarget 7: 16529–16541. [DOI] [PMC free article] [PubMed] [Google Scholar]