

Abstract
The β‐catenin destruction complex is a dynamic cytosolic multiprotein assembly that provides a key node in Wnt signalling regulation. The core components of the destruction complex comprise the scaffold proteins axin and adenomatous polyposis coli and the Ser/Thr kinases casein kinase 1 and glycogen synthase kinase 3. In unstimulated cells, the destruction complex efficiently drives degradation of the transcriptional coactivator β‐catenin, thereby preventing the activation of the Wnt/β‐catenin pathway. Mutational inactivation of the destruction complex is a major pathway in the pathogenesis of cancer. Here, we review recent insights in the regulation of the β‐catenin destruction complex, including newly identified interaction interfaces, regulatory elements and post‐translationally controlled mechanisms. In addition, we discuss how mutations in core destruction complex components deregulate Wnt signalling via distinct mechanisms and how these findings open up potential therapeutic approaches to restore destruction complex activity in cancer cells.
Linked Articles
This article is part of a themed section on WNT Signalling: Mechanisms and Therapeutic Opportunities. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.24/issuetoc
Abbreviations
- APC
adenomatous polyposis coli
- ARC
ankyrin repeat cluster
- Arm
Armadillo
- ASAD
APC‐self‐associating domain
- CID
catenin inhibitory domain
- CK1
casein kinase 1
- CRC
colorectal carcinoma
- DIX
dishevelled‐axin domain
- Dvl
dishevelled
- FZD
frizzled
- GSK3
glycogen synthase kinase 3
- LRP5/6
lipoprotein‐related protein 5/6
- OD 1/2
oligomerization domain 1/2
- PP1
protein phosphatase 1
- PP2A
protein phosphatase 2A
- RegB
region B
- RGS
regulators of G‐protein signalling
- SAMP
Ser‐Ala‐Met‐Pro
- TNKS
tankyrase
- β‐TrCP
β‐transductin repeat‐containing protein
The destruction complex is a key node for Wnt signalling regulation
Central to the signalling events within the Wnt/β‐catenin pathway is the regulation of the dual function protein β‐catenin. In epithelial cells, a continuous supply of β‐catenin is required to secure its role as a stabilizer of adherens junction complexes, while at the same time, its task as a transcriptional coactivator of Wnt target gene expression remains under tight control (Clevers, 2006). Suppression of β‐catenin‐mediated transcription is accomplished by the destruction complex, a large cytosolic multiprotein assembly that mediates the rapid turnover of nonjunctional β‐catenin (Stamos and Weis, 2013). The core components of the destruction complex include the scaffold proteins axin and adenomatous polyposis coli (APC), as well as the Ser/Thr kinases casein kinase 1 (CK1) and glycogen synthase kinase 3 (GSK3) (Figure 1).
Figure 1.
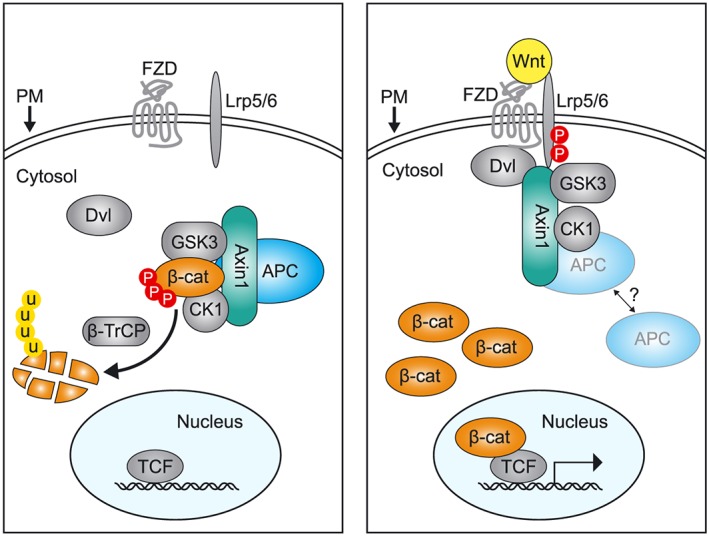
The β‐catenin destruction complex is a central regulatory node in Wnt/β‐catenin signalling. In the absence of Wnt, the β‐catenin destruction complex facilitates continuous degradation of β‐catenin. The destruction complex is comprised of the scaffold proteins APC and axin and the kinases CK1 and GSK3. These proteins act together to mediate phosphorylation (P) of β‐catenin (β‐cat). Phospho‐β‐catenin is recognized and ubiquitinated (U) by the β‐TrCP Skp1‐Cullin F‐box (SCF) E3 ligase, after which the protein undergoes proteasomal degradation. In the presence of Wnt, the membrane‐bound receptors FZD and Lrp5/6 are activated and phosphorylated leading to the recruitment of Dvl. Through subsequent recruitment of axin, the cytosolic β‐catenin destruction complex undergoes inhibitory rearrangements, leading to the accumulation of β‐catenin and its translocation to the nucleus where it acts as a co‐transcription factor in complex with DNA‐bound T‐cell factor (TCF). The cell nucleus, cytosol and plasma membrane (PM) are indicated in the figure.
The importance of a fully functional destruction complex to prevent Wnt pathway activation emerged in the mid 90's with the discovery that inherited and sporadic mutations in APC predispose to the development of colon cancer, due to uncontrolled β‐catenin accumulation and Wnt target gene transcription (Munemitsu et al., 1995; Korinek et al., 1997; Morin et al., 1997; Rubinfeld et al., 1997a; Clevers, 2006). Later studies revealed that the destruction complex captures and phosphorylates β‐catenin at its flexible N‐terminus (Amit et al., 2002; Liu et al., 2002; Marin et al., 2003), earmarking it for recognition by the F‐box protein β‐transductin repeat‐containing protein (β‐TrCP; Orford et al., 1997; Jiang and Struhl, 1998; Wu et al., 2003). Next, the β‐TrCP‐associated Skp1‐Cullin F‐box (SCF) ubiquitin ligase ubiquitinates β‐catenin and delivers it to the proteasome to accomplish its destruction. Together, this proteolysis cascade serves to keep cytosolic β‐catenin levels low and prevent its translocation to the nucleus (Figure 1).
Binding of Wnt to the cell surface frizzled (FZD) receptors and low density lipoprotein‐related protein 5/6 (LRP5/6) interferes with β‐catenin degradation via subcellular redistribution of destruction complex components (Clevers, 2006; MacDonald and He, 2012; Stamos and Weis, 2013). First, the Wnt‐activated FZD receptor recruits the cytosolic effector protein Dishevelled (Dvl), providing an initial docking site for axin at the plasma membrane. In following steps, axin‐bound kinases phosphorylate the cytosolic tail of LRP5/6, which creates additional interaction sites for axin and mediates the formation of stabilized, multimerized Wnt‐receptor–Dvl–axin complexes (MacDonald et al., 2009; MacDonald and He, 2012). As a result, the destruction complex is turned off and β‐catenin accumulates in the cytosol (Figure 1). The molecular basis for Wnt‐mediated destruction complex inactivation remains heavily debated, and for a detailed discussion, we refer to a number of excellent reviews (Metcalfe and Bienz, 2011; MacDonald and He, 2012; Davidson and Niehrs, 2014). Briefly, proposed models include direct blockade of the catalytic site of axin‐bound GSK3 by binding of phosphorylated LRP6 motifs (Cselenyi et al., 2008; Piao et al., 2008; Wu et al., 2009), inhibition of GSK3 via Wnt‐induced dissociation of APC (Valvezan et al., 2012), or sequestration of GSK3 within multivesicular bodies via endocytosis of the receptor complex (Taelman et al., 2010; Vinyoles et al., 2014). In another model, the destruction complex remains intact and becomes saturated with phosphorylated β‐catenin, while downstream ubiquitination is inhibited (Li et al., 2012; Gerlach et al., 2014). Notwithstanding the mechanism, the undisputed outcome of Wnt signalling is the stabilization of β‐catenin and its translocation to the nucleus to associate with DNA‐bound T‐cell factor/Lef proteins and co‐activate Wnt target gene transcription (Behrens et al., 1996; Molenaar et al., 1996).
Thus, the β‐catenin destruction complex provides a critical regulatory node in the Wnt cascade. Not surprisingly, mutational inactivation of key destruction complex components is a frequent occurrence in cancer and, as a consequence, provides a highly attractive target for pharmacological intervention (Polakis, 2012; Zhan et al., 2017). Below, we discuss recent insights in the molecular working mechanisms of the β‐catenin destruction complex in healthy and cancer cells, focusing on the role of inter‐ and intramolecular interactions, post‐translational modifications as well as newly emerging targeting strategies.
CK1 and GSK3 kinase activity initiate β‐catenin destruction
The central activity of the destruction complex is executed by the axin‐bound kinases CK1 and GSK3 (Ikeda et al., 1998; Liu et al., 2002; Xue et al., 2013). CK1 first phosphorylates the flexible β‐catenin N‐terminus at Ser45, which primes it for GSK3 phosphorylation at Thr41, followed by Ser37 and Ser33 (Amit et al., 2002; Liu et al., 2002). The phosphorylation motif generated by Ser37 and Ser33 ultimately mediates the recognition, ubiquitination and proteasomal degradation by β‐TrCP (Orford et al., 1997; Wu et al., 2003). In addition, both kinases phosphorylate other components of the destruction complex, including axin and APC. These modifications are key in the regulation of protein and complex function and will be discussed later in this review.
Mammalian cells express different CK1 isoforms, classified as CK1α, CK1δ, CK1ε and CK1γ. The α, δ and ε isoforms reside in the cytosol, while the γ isoform is membrane‐tethered via C‐terminal lipidation (Amit et al., 2002; Davidson et al., 2005). CK1α is the shortest variant, merely consisting of the catalytic kinase domain. Both the δ and ε isoforms carry an extended C‐terminus that can be auto‐phosphorylated, leading to auto‐inhibition of catalytic activity (Cegielska et al., 1998; Graves et al., 1993). All three cytosolic isoforms are detected in association with axin, and phosphorylation of Ser45 in β‐catenin in vitro was confirmed for CK1δ (Amit et al., 2002). RNAi experiments in mammalian cells as well as Drosophila however suggested that CK1α is the primary kinase responsible for β‐catenin Ser45 phosphorylation in living cells (Liu et al., 2002). Besides their role in Wnt/β‐catenin signalling, CK1 kinases are involved in various cellular processes, including membrane transport, cytoskeleton maintenance, DNA repair and nuclear localization (Cruciat, 2014).
In mammalian cells, two different genes encode for GSK3 isoforms GSK3α and GSK3β (Woodgett, 1990). While GSK3α displays a more extended N‐terminus as compared with GSK3β, both kinases appear to function redundantly in the destruction complex (Doble et al., 2007) and thus will be termed GSK3 throughout this review. Of note, only a small fraction (5–10%) of the total cytosolic pool of GSK3 is bound to axin and dedicated to β‐catenin destruction (Lee et al., 2003; Ng et al., 2009; Kaidanovich‐Beilin and Woodgett, 2011). Such compartmentalization of kinase activity allows GSK3 to control many cellular activities, including glycogen biosynthesis, microtubule stability, cell‐cycle control and the regulation of inflammatory pathways (Ding et al., 2000; Frame and Cohen, 2001).
Axin is the primary coordinator of destruction complex activity
Axin brings together all core components of the destruction complex and is thus regarded as its main organizer. Axin carries folded, structured domains at both its termini that are interconnected by a large, intrinsically disordered central region (Spink et al., 2000; Noutsou et al., 2011) (Figure 2A). The N‐terminal axin regulators of G‐protein signalling (RGS) domain displays homology to the RGS protein family and provides a primary binding site for APC, the second scaffold of the destruction complex (Zeng et al., 1997; Behrens et al., 1998; Kishida et al., 1998; Spink et al., 2000). Details of the axin–APC interaction are discussed below.
Figure 2.
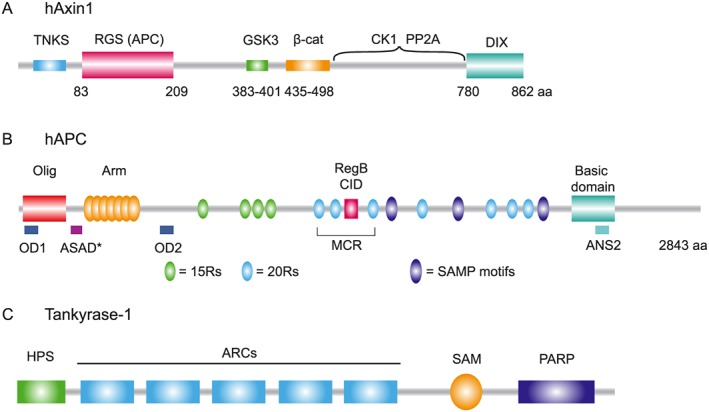
Structural organization of axin, APC and tankyrase (TNKS). (A) Human axin carries two structured domains, indicated as the N‐terminal RGS domain and C‐terminal DIX domain. The N‐terminal region contains a TNKS binding motif. The central intrinsically disordered region of axin contains binding motifs for GSK3, β‐catenin (β‐cat), CK1 and PP2A. (B) Human APC contains multiple domains including the oligomerization domain (Olig, red), Armadillo repeat domain (Arm, yellow), Region B (RegB, pink) or CID and the basic domain (aqua blue). The β‐catenin binding 15‐mer repeats (15Rs; green) and 20‐mer repeats (20Rs; blue) and axin binding SAMP motifs (purple) are indicated. Cancer mutations in APC frequently truncate the APC protein in the mutational cluster region (MCR). Self‐oligomerization of APC is facilitated by N‐terminal OD1 and OD2 and C‐terminal ANS2. Drosophila APC can self‐polymerize via the ASAD domain that shows sequence conservation in human APC, shown here as ASAD*. (C) Human TNKS1 contains five Ankyrin repeat clusters (ARCs; blue), a polymerization domain (SAM, yellow) and a C‐terminal catalytic PARP (purple) domain. The N‐terminus contains an HPS domain (green), a homopolymeric run of histidine, proline and serine of which the function is unknown.
The C‐terminally located DIX domain of axin (or DAX for DIX‐of‐axin) exhibits an ubiquitin‐like fold and can self‐polymerize in a head‐to‐tail manner, nucleating the formation of DIX domain filaments in vitro that merge into higher‐order fibres (Fagotto et al., 1999; Kishida et al., 1999; Schwarz‐Romond et al., 2007a; Fiedler et al., 2011). In cells, axin DIX‐mediated self‐interactions drive the assembly of highly dynamic, spherically shaped cytosolic puncta (Fagotto et al., 1999; Schwarz‐Romond et al., 2007a; Fiedler et al., 2011). While these studies generally rely on axin overexpression, endogenous axin puncta can be observed in conditions where its degradation is inhibited, indicating concentration‐dependent effects (de la Roche et al., 2014). The high local concentrations of axin in these puncta are deemed to mediate enhanced avidity for low‐affinity binding partners, promoting assembly of the β‐catenin destruction complex (Schwarz‐Romond et al., 2007a; Bienz, 2014). As DIX–DIX interactions are relatively weak (mid‐micromolar range), efficient axin multimerization probably depends on additional intermolecular interactions with partner proteins, such as APC (Lee et al., 2003; Pronobis et al., 2015). Besides self‐polymerization, the DIX domain can also mediate heterotypic interactions with the DIX‐containing proteins Dvl and Ccd1 (Kishida et al., 1999; Julius et al., 2000; Shiomi et al., 2003; Liu et al., 2011). These Wnt‐induced interactions interfere with destruction complex activity, thereby regulating pathway activation.
The intrinsically disordered central region of axin harbours short linear binding segments for the kinases CK1α, GSK3, their substrate β‐catenin as well as protein phosphatase 2A (PP2A) and protein phosphatase 1 (PP1) (Figure 2A) (discussed below) (Ikeda et al., 1998, 2000; Hsu et al., 1999; Yamamoto et al., 1999, 2001; Rubinfeld et al., 2001; Liu et al., 2002; Luo et al., 2007). By bringing the kinases and their substrate in close proximity, axin strongly accelerates their chemical interactions (Ikeda et al., 1998; Kikuchi, 1999; Rubinfeld et al., 2001; Liu et al., 2002; Dajani et al., 2003; Ha et al., 2004; Noutsou et al., 2011; Xue et al., 2013). Crystal structures of axin–β‐catenin and axin–GSK3 complexes show that the disordered axin segments involved turn into helices upon binding (Dajani et al., 2003; Xing et al., 2003). Moreover, the GSK3 catalytic domain and flexible N‐terminus of β‐catenin remain available for enzyme–substrate interactions in the bound state. The structure of the CK1α–axin complex has not yet been resolved, probably due to the fact that CK1α interacts with two well‐separated regions in the disordered axin central domain (Zhang et al., 2002; Sobrado et al., 2005). The interaction mode is predicted to involve loop formation of axin segments, which might further enhance colocalization of proteins in the complex (Xue et al., 2013).
Thus, axin coordinates the assembly of a multiprotein complex that brings APC, CK1, GSK3 and β‐catenin in close proximity to facilitate the capturing, phosphorylation and subsequent degradation of β‐catenin. Notably, axin variants in which individual binding domains for APC, GSK3 or β‐catenin are deleted retained a significant level of tumour suppressor activity when tested for their ability to rescue Drosophila axin null mutations in vivo (Oosterveen et al., 2007; Peterson‐Nedry et al., 2008). Moreover, while a double deletion of the RGS‐ and β‐catenin binding domains was deleterious, heteroallelic coexpression of the individual deletion mutants showed functional complementation (Peterson‐Nedry et al., 2008). These findings support a model in which multiple direct and indirect interactions between components redundantly cooperate to enhance robustness of the destruction complex. These redundancy features of the axin complex critically depend on interactions with APC (Pronobis et al., 2017).
Axin2/conductin‐mediated feedback promotes β‐catenin destruction in Wnt‐stimulated cells
Both vertebrate and nematode genomes carry an axin homologous gene, called axin2 or conductin. Both axin and axin2 proteins share key sequence elements, show similar structural organization and are functionally related (Behrens et al., 1998; Fagotto et al., 1999; Chia and Costantini, 2005). However, axin is constitutively expressed, while axin2 is a direct Wnt target gene that is up‐regulated after pathway activation (Jho et al., 2002; Lustig et al., 2002). These findings have implicated axin2 as an important negative feedback regulator of Wnt signalling, by increasing cellular destruction complex concentrations. Interestingly, the activity of axin and axin2 might not be fully redundant, since overexpression of axin2 was unable to compensate for knockdown of axin in skeletal muscle satellite cells (Figeac and Zammit, 2015). Moreover, Wnt pathway activation only seems to drive a modest increase of axin2 levels relative to axin, suggesting that quantitative expression differences do not explain its feedback role. Instead, the interaction of axin2 with Dvl is markedly reduced as compared with axin and, consequently, its role in β‐catenin degradation is relatively insensitive to Dvl‐mediated interference (Bernkopf et al., 2015). Thus, this diminished sensitivity of axin2 for inhibition by upstream signalling provides an elegant explanation for the effective restoration of destruction complex activity by axin2 (Bernkopf et al., 2015). The importance of axin2‐mediated feedback is further illustrated by the clear association between axin2 germline variants with increased cancer risk (Liu et al., 2014; Aristizabal‐Pachon et al., 2015; Rosales‐Reynoso et al., 2016; Bahl et al., 2017) and the occurrence of somatic axin2 frameshift mutations in various types of cancer (Mazzoni and Fearon, 2014; Li et al., 2015).
Essential role of APC in the destruction complex
The second critical scaffold for destruction complex activity is the large 310 kD protein APC. Mammals carry two APC genes, named APC (2843 aa) and the slightly shortened APC2 (2303 aa). The APC N‐terminus contains an oligomerization domain and an armadillo repeat (Arm) domain (Figure 2B). The Arm domain binds a number of cytoskeletal regulators that have not been linked to β‐catenin destruction, as well as B56, an essential regulator subunit of PP2A (Seeling et al., 1999; Kawasaki et al., 2000; Jimbo et al., 2002; Watanabe et al., 2004; Breitman et al., 2008). The remainder of the protein, spanning the entire region between the Arm domain and the C‐terminus, is predicted to be unstructured (Li and Nathke, 2005; Liu et al., 2006; Minde et al., 2013). This region of APC harbours a number of short axin and β‐catenin binding motifs as well as regulatory regions essential for β‐catenin proteolysis, as discussed below. At its very C‐terminus, APC carries a basic domain that promotes actin assembly (Okada et al., 2010) and a microtubule interaction region, both of which are dispensable for β‐catenin degradation (Smits et al., 1999; McCartney and Nathke, 2008; Pronobis et al., 2017). Overall, regulatory interactions of APC with the cytoskeleton are thought to mediate alternative roles of APC in spindle formation, kinetochore attachment, microtubule stability as well as the regulation of cell motility and polarity (Nathke, 2006; Okada et al., 2010).
The multiple independent β‐catenin binding motifs in the APC unstructured central region comprise four homologous 15 amino acid repeats (15Rs) and seven 20 amino acid repeats (20Rs) (Rubinfeld et al., 1997a; Eklof Spink et al., 2001). Three short Ser‐Ala‐Met‐Pro (SAMP)‐containing repeats are located, interspersed between the third 20R motif and basic domain, which mediate the interaction with axin (Behrens et al., 1998; Spink et al., 2000). The affinity of the 20Rs for β‐catenin is strongly enhanced (about 300‐fold) by CK1‐ and GSK3‐mediated phosphorylation (Rubinfeld et al., 1996; Ha et al., 2004; Liu et al., 2006). Presumably, phosphorylation occurs within the destruction complex when APC is brought in close proximity to axin‐bound kinases (Figure 3C) (Ikeda et al., 2000; Rubinfeld et al., 2001). Notably, the β‐catenin binding surface of phosphorylated 20R overlaps with that of axin, indicative of a competitive interaction (Xing et al., 2003; 2004; Ha et al., 2004). These data led to a cyclic model in which phosphorylated β‐catenin is transferred from axin to high affinity phosphorylated 20Rs in APC, after which APC facilitates the delivery of β‐catenin to the E3 ligase β‐TrCP (Figure 3C–E). In this model, PP2A‐mediated dephosphorylation of APC resets the system for binding and processing a new β‐catenin (Kimelman and Xu, 2006; Xu and Kimelman, 2007). This attractive model however requires precise timing of APC phosphorylation and dephosphorylation, which is considered an unlikely feature due to the random collisions that mediate interactions between unstructured protein segments in the complex, as discussed previously (Stamos and Weis, 2013; Xue et al., 2013). In an alternative model, high and low affinity binding sites on APC offer a wide dynamic range for efficient sequestration of β‐catenin in the Wnt off (low β‐catenin) and Wnt on (high β‐catenin) state (Figure 3) (Ha et al., 2004). However, in this model the question of when and how β‐catenin is transferred from APC to axin to undergo phosphorylation remains unexplained, leaving room for future investigation.
Figure 3.
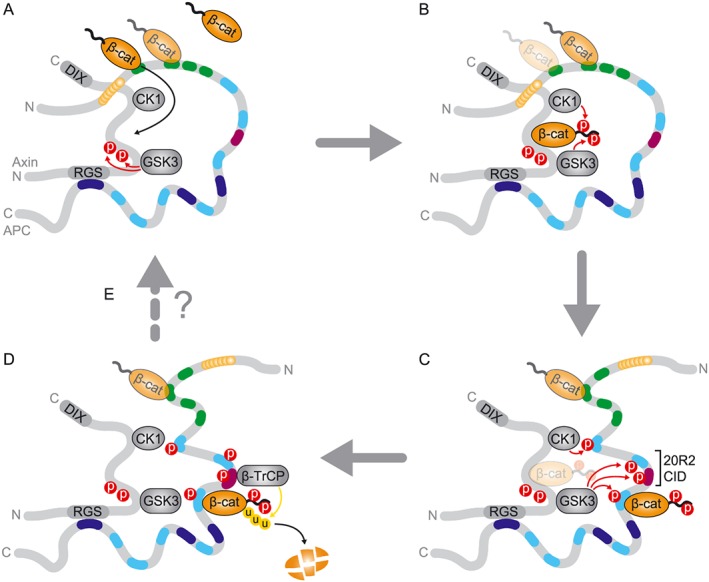
Schematic summary of destruction complex assembly and the molecular steps involved in β‐cat degradation. For APC, Arm domain (yellow), CID domain (pink), 15R (green), 20R (light blue) and SAMP repeats (dark blue) are indicated. (A) Interactions between axin and APC are stabilized via multiple binding sites as well as via self‐oligomerization (not shown for clarity). Due to redundancy in β‐cat binding sites, β‐cat substrate might enter the complex either via APC or axin binding. The initial capturing of free β‐cat from the cytosol by β‐cat binding motifs in APC (15R repeats, green) is shown. Axin‐bound kinases induce phosphorylate the axin central region to keep the protein in an open conformation that allows for efficient β‐cat binding and processing. (B) β‐catenin captured by non‐phosphorylated, low‐affinity binding sites in APC is transferred to axin, followed by CK1‐ and GSK3‐mediated phosphorylation. (C) Phosphorylation of APC 20R repeats in the complex creates high affinity β‐cat binding sites that enables phosphorylated β‐cat to transfer from axin to APC. Phosphorylation of the 20R2‐CID region induces a rearrangement in the complex that leads to the release of the APC Arm repeats from axin. (D) APC shields β‐cat from phosphatases and presents phosphorylated β‐cat to β‐TrCP, followed by ubiquitination and proteasomal degradation. (E) After handing over β‐cat for proteasomal degradation the destruction complex might be recycled for another round of β‐cat destruction. This step possibly involves dephosphorylation by destruction complex‐associated phosphatases.
Another functionally important APC region comprises the 20R repeat 2 (20R2), which does not interact with β‐catenin (Liu et al., 2006; Kohler et al., 2008), and an adjacent conserved sequence called the ‘catenin inhibitory domain’ (CID) or region B (Figure 2B) (Kohler et al., 2009). Based on results obtained with various truncated APC fragments, the 20R2‐CID region was determined to be essential for β‐catenin ubiquitination, independent of β‐catenin binding activity (Kohler et al., 2009; Roberts et al., 2011). Mechanistically, the 20R2‐CID region was proposed to mediate the association with β‐TrCP, protect β‐catenin from PP2A‐mediated dephosphorylation and modulate the interaction of axin and APC, as discussed below (Su et al., 2008; Pronobis et al., 2015) (Figure 3D). The nature of the underlying protein–protein interactions required for these 20R2‐CID‐mediated activities remains unclear, but might involve a functional interaction of the CID domain with α‐catenin, as proposed recently (Choi et al., 2013).
Self‐polymerization of human APC is mediated via its N‐terminal oligomerization domain (OD) 1, OD2 and C‐terminal ANS2 domains, but these interactions are not deemed relevant for Wnt pathway regulation (Figure 2B) (Li et al., 2008; Okada et al., 2010). In contrast, a recent study described an N‐terminal coil in Drosophila APC2, called the APC‐self‐associating domain (ASAD), that increased the size of cytosolic axin–APC puncta and promoted destruction complex efficiency in both Drosophila S2 and SW480 cells (Kunttas‐Tatli et al., 2014). While the predicted coil structure of the ASAD domain appears conserved in all Bilateria APC proteins (Figure 2B) (Kunttas‐Tatli et al., 2014), its role in mammalian Wnt pathway regulation remains to be established.
The axin–APC interaction is highly dynamic
Key interactions between axin and APC are mediated via binding of axin RGS to the SAMP repeat motifs of APC (Figure 3A) (Behrens et al., 1998; Kishida et al., 1998; Spink et al., 2000). However, the role of SAMPs in regulating destruction complex activity might be more complex than initially anticipated as individual SAMP repeats display differential axin binding affinities and are strongly regulated by phosphorylation (Kunttas‐Tatli et al., 2015). These findings suggest that the SAMP repeats possibly mediate functionally distinct yet cooperative roles. Notwithstanding the mechanism, the importance of the SAMP repeat region for β‐catenin proteolysis is evidently shown by APC cancer truncations that have lost all SAMPs and exhibit strong oncogenic effects (Smits et al., 1999; Kohler et al., 2009; Roberts et al., 2011). Surprisingly, however, a Drosophila APC2 variant lacking all SAMPs displayed residual APC–axin binding activity, revealing the existence of alternative interaction sites (Roberts et al., 2011). Indeed, Peifer and colleagues uncovered a second interaction mode in which the Arm domain of Drosophila APC2 binds the central region of axin (Figure 3A) (Pronobis et al., 2015). This interaction is highly dynamic and regulated by GSK3‐mediated APC phosphorylation of the 20R2‐CID region. In the proposed model, APC2 employs multiple interactions with axin to promote multimerization, thereby increasing the size and stability of the destruction complex. In subsequent steps, GSK3 phosphorylates β‐catenin as well as parts of APC, including the 20R2‐CID region. Next, phosphorylated 20R2‐CID induces the release of the weak interaction of APC–Arm with axin, opening up the complex and allowing the transfer of phospho‐β‐catenin to β‐TrCP (Figure 3C) (Pronobis et al., 2015). This model introduces a number of novel regulatory steps and provides an attractive explanation for misregulation by APC cancer truncations by hypophosphorylation or loss of the 20R2‐CID region. However, substantial validation will be required to explain the proposed phosphorylation‐induced rearrangements in APC–axin interactions within the complex as well as the consequences for interactions with the ubiquitin machinery.
Due to the presence of multiple binding sites for a single partner as well as overlap in self‐oligomerization capacity, axin and APC appear to partially share redundant functions inside the destruction complex. To identify the essential parts of both scaffold proteins, a recent study compiled a minimal destruction complex by using only five essential regions of axin and APC (Pronobis et al., 2017). For APC, these regions included the self‐associating ASAD domain, Arm repeats and the 20R2‐CID region. These APC regions were coupled to the axin C‐terminus containing the β‐catenin binding domain and DIX domain. The artificial scaffold protein formed cytosolic puncta and allowed full restoration of β‐catenin destruction in APC‐mutant SW480 cells (Pronobis et al., 2017). While these results are highly informative, it should be noted that these experiments relied on overexpression and were performed in the presence of endogenous wild‐type axin as well as truncated APC, both of which could contribute to the formation and activity of the destruction complex. One related and unresolved issue concerns the question of how kinases are recruited to this artificial complex. If and how the minimal complex is susceptible to inhibition by Wnt signals also remains a matter for future investigation.
Regulation by dephosphorylation
PP2A and PP1 both associate with the destruction complex, suggesting they affect a balanced regulation via phosphorylation and dephosphorylation, Their precise modes of interaction and functional roles however remain to be clarified (Hsu et al., 1999; Seeling et al., 1999; Ratcliffe et al., 2000; Yamamoto et al., 2001; Luo et al., 2007). PP1 interacts with axin (Luo et al., 2007), while PP2A was reported to bind both axin and APC (Hsu et al., 1999; Seeling et al., 1999; Yamamoto et al., 2001). Of note, PP2A binding to axin might be indirect, involving the heat shock protein 70 (HSP70) family member HSP105 (Yu et al., 2015). PP2A is composed of a core catalytic subunit (PPP2CA), a structural subunit (PR65/A) and variable regulatory B subunits (Janssens and Goris, 2001). A consistent finding across numerous studies is that PP2A dephosphorylates β‐catenin to prevent its ubiquitination and support the Wnt activation pathway (Su et al., 2008; Zhang et al., 2009; Yu et al., 2015). In the first model, PP2A‐mediated dephosphorylation of the APC 20R region was proposed to mark the end of a destruction complex cycle, allowing phosphorylated β‐catenin to leave the complex and so initiate a new round of β‐catenin modifications (Xu and Kimelman, 2007). However, Weis and colleagues were unable to dephosphorylate the APC 20R region when bound to β‐catenin using the catalytic domain of PP1 in an in vitro setting (Ha et al., 2004). Besides, this assumption is also in conflict with the recently proposed role of the 20R2‐CID region, for which phosphorylation appears to be required to release axin and transfer β‐catenin to β‐TrCP (Pronobis et al., 2015). The precise role of heterotrimeric PP2A inside the destruction complex thus awaits further experimental validation.
The phosphatase PP1 was reported to promote Wnt signalling via intramolecular autoinhibition of axin (Kim et al., 2013). In the suggested model, destruction complex activity strongly depends on the phosphorylation status of axin. In the absence of Wnt, GSK3 phosphorylates axin at Ser497 and Ser500, which retains the protein in an active, ‘open’ state that allows for β‐catenin binding and processing. Wnt‐mediated receptor activation leads to the recruitment of axin (Mao et al., 2001; Cliffe et al., 2003; Tolwinski et al., 2003; Tamai et al., 2004; Zeng et al., 2005; Bilic et al., 2007; Schwarz‐Romond et al., 2007b; MacDonald et al., 2008; Fiedler et al., 2011) and subsequent inhibition of GSK3 via pseudo‐substrate interactions with the phosphorylated LRP6 cytosolic tail (Cselenyi et al., 2008; Piao et al., 2008; Wu et al., 2009; Kim et al., 2013; Stamos et al., 2014). These steps initiate PP1‐dependent axin dephosphorylation, after which the scaffold undergoes a conformational switch. Mechanistically, dephosphorylation of axin promotes an intramolecular interaction between the β‐catenin binding domain and the DIX domain, inducing the protein to adopt an inactive, ‘closed’ conformation and its release into the cytosol (Kim et al., 2013). The resulting drop in destruction complex activity allows the stabilization of β‐catenin and pathway activation. When intracellular levels of β‐catenin rise above a critical concentration, β‐catenin binding might compete with the autoinhibitory interaction, restoring assembly of the axin‐based destruction complex to avoid excessive accumulation. Together, these findings highlight the critical importance of axin phosphorylation in the regulation of β‐catenin turnover. Further studies are needed to elucidate the contribution of other axin phosphorylation sites, shed light on the structural details of different axin conformational states and analyse the consequences for assembly with other binding partners, including APC. Moreover, the question of how the different axin conformational changes depend on axin multimerization deserves further investigation.
Regulation by poly‐ADP‐ribosylation
Over recent years, a major regulatory pathway has emerged that potentiates cellular responses to Wnt via poly‐ADP‐ribosylation (PARylation)‐mediated destabilization of axin. The enzymes responsible are tankyrase (TNKS) 1 and 2, members of the PARP family (Smith et al., 1998). TNKS binds the axin N‐terminus via its large ankyrin repeat cluster (ARC) domain after which the C‐terminal PARP domain catalyses the modification of axin by poly‐ADP‐ribose chains (Huang et al., 2009) (Figure 2C). Next, PARylated axin is recognized and ubiquitinated by the E3 ligase RNF146, which targets axin for proteasomal degradation (Callow et al., 2011; Zhang et al., 2011). Decreased axin levels presumably compromise the activity of the destruction complex, leading to enhanced activation of the Wnt pathway. Thus, TNKS 1 and 2 were identified as positive regulators of the Wnt signalling pathway.
Structural approaches revealed molecular requirements for TNKS‐mediated regulation of Wnt signalling. The axin–TNKS crystal structure divulged two TNKS‐binding motifs in axin, each of which binds to a different ARC domain within the TNKS protein (Figure 2A, C) (Morrone et al., 2012). Notably, the sequence of the second binding motif is considerably different from the agreed TNKS‐binding sequence, and TNKS binding to this region was not detected by standard biochemical protein interaction methods, possibly due to a weaker affinity (Croy et al., 2016). Furthermore, despite the presence of five ARC repeats, the structural properties of these domains limit the interactions with axin only to specific ARC combinations within one TNKS molecule (Eisemann et al., 2016). The overall multiplicity of intermolecular binding sites as well as the polymerizing properties of the TNKS SAM domain promote assembly of higher order complexes that allow for efficient targeting of axin for ADP‐ribosylation and degradation (Figure 2C) (Mariotti et al., 2016).
Interestingly, an additional role of TNKS‐mediated PARylation of axin in promoting Wnt pathway activation was recently reported. In this study, Wnt stimulation resulted in a rapid increase in the pool of PARylated axin in both Drosophila and human cells (Yang et al., 2016). Mechanistically, PARylation induced the recruitment of axin to the plasma membrane via an enhanced interaction with phosphorylated LRP6, thereby promoting Wnt signalling (Yang et al., 2016). These findings raise a number of important questions that deserve further investigation, including how Wnt signals alter TNKS activity towards axin, which protein domains promote the interaction of PARylated axin with LRP6 and what are the molecular consequences for signalosome assembly.
Impact of cancer mutations on destruction complex activity
Mutational inactivation of destruction complex activity is a prevalent occurrence in cancer. The most prominent example involves mutations in APC that are found in 80–90% of both inherited and sporadic colorectal cancers (CRC) (Clements et al., 2003; Polakis, 2007; Kandoth et al., 2013; Brannon et al., 2014). Loss of function of both alleles induces inappropriate activation of β‐catenin‐mediated transcription in individual cells, leading to the growth of adenomas or polyps (Polakis, 2007; Polakis, 2012). Additional mutations in genes like KRAS, TP53 and SMAD4 are required subsequently to induce these polyps to progress toward malignancy (Kinzler and Vogelstein, 1996; Conlin et al., 2005; Drost et al., 2015; Matano et al., 2015; Fumagalli et al., 2017; Melo et al., 2017). Unlike other tumour suppressors, APC mutants CRCs do not carry homozygous null mutations but usually keep at least one allele encoding a truncated APC protein. Truncations are generated through frameshift mutations that occur in the so‐called mutational cluster region, generating shortened APC proteins that preserve the Arm domain and some of the 20Rs while lacking all of the SAMP repeats (Figure 2B) (Beroud and Soussi, 1996; Kohler et al., 2008). Notably, truncated APC cancer variants retain a residual ability to target β‐catenin for degradation (Albuquerque et al., 2002; McCartney et al., 2006; Gaspar et al., 2009; Voloshanenko et al., 2013). These findings have led to the ‘just‐right’ hypothesis in which low levels of destruction activity are retained by tumour cells to prevent apoptosis induced by excessive β‐catenin‐mediated signalling (Albuquerque et al., 2002). Mechanistically, the weak suppressor activity of truncated APC might be mediated via cytoplasmic retention of β‐catenin (Roberts et al., 2011), weak interactions with axin through the recently described interactions between APC Arm repeats and the axin central domain as well as the residual ubiquitination‐promoting activity of the 20R2‐CID region (Voloshanenko et al., 2013; Pronobis et al., 2015). Strikingly, the invasive growth of malignant APC depleted, KRAS and TP53 mutant CRC cells could be reversed by restoring the expression of APC, which triggered differentiation and re‐establishment of tissue homeostasis (Dow et al., 2015). These findings provide strong support for the continuation of the intense search for Wnt pathway inhibitors as potential therapeutics for CRC.
Another well‐known class of mutations leading to uncontrolled Wnt pathway activity comprise activating mutations in β‐catenin. These mutations occur in about 5% of CRC patients and are mutually exclusive with APC mutations (Luchtenborg et al., 2005; Thorstensen et al., 2005) [cBioportal.org (Cerami et al., 2012; Gao et al., 2013)]. Moreover, in contrast to APC mutations, β‐catenin mutations are found in many other types of cancer types, including hepatocellular carcinoma, endometrioid ovarian cancer and medulloblastoma (Rubinfeld et al., 1997b; Bell, 2005; Polakis, 2007). Oncogenic β‐catenin mutations predominantly hit the phosphorylation sites in the flexible N‐terminus, masking recognition sites for destruction complex‐mediated phosphorylation, thereby preventing β‐catenin proteolysis.
Mutations in AXIN1 also associate with a diverse set of human tumours, including hepatocellular carcinoma, medulloblastoma and colorectal carcinoma (Salahshor and Woodgett, 2005). Missense mutations are prevalent within the AXIN1 mutational spectrum, but mechanistic information on associated tumourigenic roles is largely lacking. Recently, missense mutations in the axin N‐terminal RGS domain were shown to disrupt Wnt signalling and promote tumour growth in vivo by an unprecedented molecular mechanism (Anvarian et al., 2016). Relevant point mutations in cancer destabilized the structure of the axin RGS domain, driving the formation of soluble, small‐sized axin oligomers. Non‐aggregating unstructured regions of axin were found to protrude from the oligomer as ‘molecular tentacles’ that aberrantly engage key regulators. Collectively, the altered interactions of the mutant axin rewired its signalling network to activate β‐catenin‐mediated transcription. Of note, blocking aggregation partially restored the tumour suppressor activity of the mutant protein, providing a potential new avenue in the search for Wnt pathway inhibitors.
Targeting the destruction complex in cancer
Due to its key role in the regulation of β‐catenin activity, the destruction complex provides an attractive target for therapeutic manipulation. Over recent years, a number of small molecules were identified that enhance the activity of the destruction complex, and have potential as anti‐cancer drugs.
Inhibitors of TNKS are a major class of novel destruction complex regulators that were first discovered in 2009 to potently inhibit Wnt/β‐catenin signalling in APC‐mutant cancer cells (Huang et al., 2009). The small molecule XAV939 has been found to bind and inhibit the catalytic activity of TNKS, leading to stabilization of axin and the subsequent down‐regulation of β‐catenin levels (Karlberg et al., 2010; Kirby et al., 2012). Following this initial finding, numerous studies have applied alternative screening approaches, which have led to the identification of additional TNKS inhibitors with distinct structural properties, further highlighting the potential of this therapeutic approach. Notably, cells treated with the TNKS inhibitors XAV939 and G007‐LK display a rapid induction of enlarged cytosolic puncta called degradasomes, to which all components of the endogenous β‐catenin complex are recruited, including phosphorylated β‐catenin and β‐TrCP (Thorvaldsen et al., 2015). Subsequent studies revealed that treatment with TNKS inhibitors actually promotes and stabilizes TNKS–axin interactions, further boosting oligomerization and the assembly of functional destruction complexes (Martino‐Echarri et al., 2016). Furthermore, close examination revealed that treatment of SW480 CRC cells with a TNKS inhibitor strongly and selectively increases the levels of axin2, indicating that degradasome formation largely depends on axin2 stabilization in these cells (Thorvaldsen et al., 2017). Of note, SW480 cells are APC mutant and display constitutive Wnt pathway activation, leading to permanent expression of target genes including axin2. In line with these findings, a recent study demonstrated that CRC cancer cells with short truncated APC variants lacking all seven 20Rs were dependent on high β‐catenin levels and responded best to TNKS inhibitors. These results suggest that short APC truncations might provide a biomarker for TNKS inhibitor sensitivity (Tanaka et al., 2017). Despite these promising results, prolonged Wnt stimulation may render cells unresponsive to treatment with TNKS inhibitors and thus potentially put constraints on their use in clinical applications (de la Roche et al., 2014).
Another class of compounds reported to regulate destruction complex activity targets alterations in kinase activity. One example is pyrvinium, a small molecule that binds and activates the kinase CK1α, thus promoting β‐catenin phosphorylation and proteolysis (Thorne et al., 2010). In subsequent work, pyrvinium was reported to inhibit the proliferation of lung cancer cells in vitro at a dose below 10 nM (Zhang et al., 2015). Another small molecule, Wnt inhibitor is KYA1797 that was shown to bind directly to the axin RGS domain. Through its activation of GSK3, it promotes destruction complex‐mediated β‐catenin phosphorylation and degradation (Cha et al., 2016). However, further investigations are required to determine the suitability of these inhibitors as future anti‐cancer drugs.
Concluding remarks
Even though it is now 22 years since its discovery, a unifying theory of the inner workings of the β‐catenin destruction complex has not been accomplished. Emerging evidence shows that numerous molecular activities are shared between axin and APC, securing the robustness and adaptability of destruction complex activity under different cellular conditions. Although it is clear that both scaffolds co‐operate, the exact role of APC remains poorly defined. Progress is expected to come from high resolution structural information on the intra‐ and intermolecular interactions at the core of the complex, although flexible protein segments and dynamic interactions complicate this endeavour. Recent studies have also emphasized the importance of post‐translational modifications in the regulation of destruction complex activity. A precise balance between phosphorylation, ADP‐ribosylation, ubiquitination and other possible modifications presumably regulate intra‐ and intermolecular interactions within the complex. An increased understanding of the timing and order in which these modifications take place will be important to resolve outstanding mechanistic issues. Finally, current knowledge is largely based on studies in which components of the destruction complex are overexpressed, which alters the relative ratio of protein concentrations in the cell that are deemed important for precise pathway regulation. Novel technologies such as CRISPR/Cas9 genome editing are likely to provide the appropriate tools to modify and analyse destruction complex components at their endogenous levels in the cell. In addition, recently emerged organoid technologies provide a controllable environment where different cell types form and grow in organized structures similar to complex tissues (Clevers, 2016). Combining organoid culture with endogenous genome editing thus provides advanced test systems for concepts in Wnt pathway regulation as well as the evaluation of newly generated therapeutic compounds.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHAMRMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This work was supported by the European Research Council (ERC starting grant 242958 to M.M.M.), Boehringer Ingelheim Fonds (to E.v.K.), the European Union (FP7 Marie‐Curie ITN 608180 ‘WntsApp’ to M.M.M.) and the Netherlands Organization for Scientific Research NWO (VICI grant 91815604 and ECHO grant 711.013.012 to M.M.M).
van Kappel, E. C. , and Maurice, M. M. (2017) Molecular regulation and pharmacological targeting of the β‐catenin destruction complex. British Journal of Pharmacology, 174: 4575–4588. doi: 10.1111/bph.13922.
References
- Albuquerque C, Breukel C, van der Luijt R, Fidalgo P, Lage P, Slors FJ et al. (2002). The ‘just‐right’ signaling model: APC somatic mutations are selected based on a specific level of activation of the beta‐catenin signaling cascade. Hum Mol Genet 11: 1549–1560. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben‐Shushan E, Mann M et al. (2002). Axin‐mediated CKI phosphorylation of beta‐catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16: 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anvarian Z, Nojima H, van Kappel EC, Madl T, Spit M, Viertler M et al. (2016). Axin cancer mutants form nanoaggregates to rewire the Wnt signaling network. Nat Struct Mol Biol 23: 324–332. [DOI] [PubMed] [Google Scholar]
- Aristizabal‐Pachon AF, Carvalho TI, Carrara HH, Andrade J, Takahashi CS (2015). AXIN2 Polymorphisms, the beta‐catenin destruction complex expression profile and breast cancer susceptibility. Asian Pac J Cancer Prev 16: 7277–7284. [DOI] [PubMed] [Google Scholar]
- Bahl C, Sharma S, Singh N, Behera D (2017). Association study between genetic variations in axin2 gene and lung cancer risk in North Indian population: a multiple interaction analysis. Tumour Biol 39 1010428317695533. [DOI] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R et al. (1998). Functional interaction of an axin homolog, conductin, with beta‐catenin, APC, and GSK3beta. Science 280: 596–599. [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R et al. (1996). Functional interaction of beta‐catenin with the transcription factor LEF‐1. Nature 382: 638–642. [DOI] [PubMed] [Google Scholar]
- Bell DA (2005). Origins and molecular pathology of ovarian cancer. Mod Pathol 18 (Suppl 2): S19–S32. [DOI] [PubMed] [Google Scholar]
- Bernkopf DB, Hadjihannas MV, Behrens J (2015). Negative‐feedback regulation of the Wnt pathway by conductin/axin2 involves insensitivity to upstream signalling. J Cell Sci 128: 33–39. [DOI] [PubMed] [Google Scholar]
- Beroud C, Soussi T (1996). APC gene: database of germline and somatic mutations in human tumors and cell lines. Nucleic Acids Res 24: 121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz M (2014). Signalosome assembly by domains undergoing dynamic head‐to‐tail polymerization. Trends Biochem Sci 39: 487–495. [DOI] [PubMed] [Google Scholar]
- Bilic J, Huang YL, Davidson G, Zimmermann T, Cruciat CM, Bienz M et al. (2007). Wnt induces LRP6 signalosomes and promotes dishevelled‐dependent LRP6 phosphorylation. Science 316: 1619–1622. [DOI] [PubMed] [Google Scholar]
- Brannon AR, Vakiani E, Sylvester BE, Scott SN, McDermott G, Shah RH et al. (2014). Comparative sequencing analysis reveals high genomic concordance between matched primary and metastatic colorectal cancer lesions. Genome Biol 15: 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitman M, Zilberberg A, Caspi M, Rosin‐Arbesfeld R (2008). The armadillo repeat domain of the APC tumor suppressor protein interacts with Striatin family members. Biochim Biophys Acta 1783: 1792–1802. [DOI] [PubMed] [Google Scholar]
- Callow MG, Tran H, Phu L, Lau T, Lee J, Sandoval WN et al. (2011). Ubiquitin ligase RNF146 regulates tankyrase and axin to promote Wnt signaling. PLoS One 6: e22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegielska A, Gietzen KF, Rivers A, Virshup DM (1998). Autoinhibition of casein kinase I epsilon (CKI epsilon) is relieved by protein phosphatases and limited proteolysis. J Biol Chem 273: 1357–1364. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha PH, Cho YH, Lee SK, Lee J, Jeong WJ, Moon BS et al. (2016). Small‐molecule binding of the axin RGS domain promotes beta‐catenin and Ras degradation. Nat Chem Biol 12: 593–600. [DOI] [PubMed] [Google Scholar]
- Chia IV, Costantini F (2005). Mouse axin and axin2/conductin proteins are functionally equivalent in vivo. Mol Cell Biol 25: 4371–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Estaras C, Moresco JJ, Yates JR 3rd, Jones KA (2013). alpha‐Catenin interacts with APC to regulate beta‐catenin proteolysis and transcriptional repression of Wnt target genes. Genes Dev 27: 2473–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements WM, Lowy AM, Groden J (2003). Adenomatous polyposis coli/beta‐catenin interaction and downstream targets: altered gene expression in gastrointestinal tumors. Clin Colorectal Cancer 3: 113–120. [DOI] [PubMed] [Google Scholar]
- Clevers H (2006). Wnt/beta‐catenin signaling in development and disease. Cell 127: 469–480. [DOI] [PubMed] [Google Scholar]
- Clevers H (2016). Modeling development and disease with organoids. Cell 165: 1586–1597. [DOI] [PubMed] [Google Scholar]
- Cliffe A, Hamada F, Bienz M (2003). A role of dishevelled in relocating axin to the plasma membrane during wingless signaling. Curr Biol 13: 960–966. [DOI] [PubMed] [Google Scholar]
- Conlin A, Smith G, Carey FA, Wolf CR, Steele RJ (2005). The prognostic significance of K‐ras, p53, and APC mutations in colorectal carcinoma. Gut 54: 1283–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croy HE, Fuller CN, Giannotti J, Robinson P, Foley AV, Yamulla RJ et al. (2016). The poly(ADP‐ribose) polymerase enzyme tankyrase antagonizes activity of the beta‐catenin destruction complex through ADP‐ribosylation of axin and APC2. J Biol Chem 291: 12747–12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruciat CM (2014). Casein kinase 1 and Wnt/beta‐catenin signaling. Curr Opin Cell Biol 31: 46–55. [DOI] [PubMed] [Google Scholar]
- Cselenyi CS, Jernigan KK, Tahinci E, Thorne CA, Lee LA, Lee E (2008). LRP6 transduces a canonical Wnt signal independently of axin degradation by inhibiting GSK3's phosphorylation of beta‐catenin. Proc Natl Acad Sci U S A 105: 8032–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V et al. (2003). Structural basis for recruitment of glycogen synthase kinase 3beta to the axin‐APC scaffold complex. EMBO J 22: 494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson G, Niehrs C (2014). Wnt signaling at the membrane In: Wnt Signaling in Development and Disease. John Wiley & Sons, Inc.: Hoboken, New Jersey, USA, pp. 15–32. [Google Scholar]
- Davidson G, Wu W, Shen J, Bilic J, Fenger U, Stannek P et al. (2005). Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature 438: 867–872. [DOI] [PubMed] [Google Scholar]
- Ding VW, Chen RH, McCormick F (2000). Differential regulation of glycogen synthase kinase 3beta by insulin and Wnt signaling. J Biol Chem 275: 32475–32481. [DOI] [PubMed] [Google Scholar]
- Doble BW, Patel S, Wood GA, Kockeritz LK, Woodgett JR (2007). Functional redundancy of GSK‐3alpha and GSK‐3beta in Wnt/beta‐catenin signaling shown by using an allelic series of embryonic stem cell lines. Dev Cell 12: 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE, O'Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H et al. (2015). Apc Restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 161: 1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J, van Jaarsveld RH, Ponsioen B, Zimberlin C, van Boxtel R, Buijs A et al. (2015). Sequential cancer mutations in cultured human intestinal stem cells. Nature 521: 43–47. [DOI] [PubMed] [Google Scholar]
- Eisemann T, McCauley M, Langelier MF, Gupta K, Roy S, Van Duyne GD et al. (2016). Tankyrase‐1 ankyrin repeats form an adaptable binding platform for targets of ADP‐ribose modification. Structure 24: 1679–1692. [DOI] [PubMed] [Google Scholar]
- Eklof Spink K, Fridman SG, Weis WI (2001). Molecular mechanisms of beta‐catenin recognition by adenomatous polyposis coli revealed by the structure of an APC‐beta‐catenin complex. EMBO J 20: 6203–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagotto F, Jho E, Zeng L, Kurth T, Joos T, Kaufmann C et al. (1999). Domains of axin involved in protein‐protein interactions, Wnt pathway inhibition, and intracellular localization. J Cell Biol 145: 741–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler M, Mendoza‐Topaz C, Rutherford TJ, Mieszczanek J, Bienz M (2011). Dishevelled interacts with the DIX domain polymerization interface of axin to interfere with its function in down‐regulating beta‐catenin. Proc Natl Acad Sci U S A 108: 1937–1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figeac N, Zammit PS (2015). Coordinated action of axin1 and axin2 suppresses beta‐catenin to regulate muscle stem cell function. Cell Signal 27: 1652–1665. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P (2001). GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli A, Drost J, Suijkerbuijk SJ, van Boxtel R, de Ligt J, Offerhaus GJ et al. (2017). Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. Proc Natl Acad Sci U S A 114: E2357–E2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar C, Franken P, Molenaar L, Breukel C, van der Valk M, Smits R et al. (2009). A targeted constitutive mutation in the APC tumor suppressor gene underlies mammary but not intestinal tumorigenesis. PLoS Genet 5: e1000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach JP, Emmink BL, Nojima H, Kranenburg O, Maurice MM (2014). Wnt signalling induces accumulation of phosphorylated beta‐catenin in two distinct cytosolic complexes. Open Biol 4: 140120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves PR, Haas DW, Hagedorn CH, DePaoli‐Roach AA, Roach PJ (1993). Molecular cloning, expression, and characterization of a 49‐kilodalton casein kinase I isoform from rat testis. J Biol Chem 268: 6394–6401. [PubMed] [Google Scholar]
- Ha NC, Tonozuka T, Stamos JL, Choi HJ, Weis WI (2004). Mechanism of phosphorylation‐dependent binding of APC to beta‐catenin and its role in beta‐catenin degradation. Mol Cell 15: 511–521. [DOI] [PubMed] [Google Scholar]
- Hsu W, Zeng L, Costantini F (1999). Identification of a domain of axin that binds to the serine/threonine protein phosphatase 2A and a self‐binding domain. J Biol Chem 274: 3439–3445. [DOI] [PubMed] [Google Scholar]
- Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA et al. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461: 614–620. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida M, Matsuura Y, Usui H, Kikuchi A (2000). GSK‐3beta‐dependent phosphorylation of adenomatous polyposis coli gene product can be modulated by beta‐catenin and protein phosphatase 2A complexed with axin. Oncogene 19: 537–545. [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A (1998). Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK‐3beta and beta‐catenin and promotes GSK‐3beta‐dependent phosphorylation of beta‐catenin. EMBO J 17: 1371–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens V, Goris J (2001). Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 353: 417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F (2002). Wnt/beta‐catenin/Tcf signaling induces the transcription of axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22: 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Struhl G (1998). Regulation of the Hedgehog and Wingless signalling pathways by the F‐box/WD40‐repeat protein Slimb. Nature 391: 493–496. [DOI] [PubMed] [Google Scholar]
- Jimbo T, Kawasaki Y, Koyama R, Sato R, Takada S, Haraguchi K et al. (2002). Identification of a link between the tumour suppressor APC and the kinesin superfamily. Nat Cell Biol 4: 323–327. [DOI] [PubMed] [Google Scholar]
- Julius MA, Schelbert B, Hsu W, Fitzpatrick E, Jho E, Fagotto F et al. (2000). Domains of axin and disheveled required for interaction and function in Wnt signaling. Biochem Biophys Res Commun 276: 1162–1169. [DOI] [PubMed] [Google Scholar]
- Kaidanovich‐Beilin O, Woodgett JR (2011). GSK‐3: functional insights from cell biology and animal models. Front Mol Neurosci 4: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C et al. (2013). Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlberg T, Markova N, Johansson I, Hammarstrom M, Schutz P, Weigelt J et al. (2010). Structural basis for the interaction between tankyrase‐2 and a potent Wnt‐signaling inhibitor. J Med Chem 53: 5352–5355. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, Iwayama Y et al. (2000). Asef, a link between the tumor suppressor APC and G‐protein signaling. Science 289: 1194–1197. [DOI] [PubMed] [Google Scholar]
- Kikuchi A (1999). Modulation of Wnt signaling by axin and Axil. Cytokine Growth Factor Rev 10: 255–265. [DOI] [PubMed] [Google Scholar]
- Kim SE, Huang H, Zhao M, Zhang X, Zhang A, Semonov MV et al. (2013). Wnt stabilization of beta‐catenin reveals principles for morphogen receptor‐scaffold assemblies. Science 340: 867–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman D, Xu W (2006). Beta‐catenin destruction complex: insights and questions from a structural perspective. Oncogene 25: 7482–7491. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Vogelstein B (1996). Lessons from hereditary colorectal cancer. Cell 87: 159–170. [DOI] [PubMed] [Google Scholar]
- Kirby CA, Cheung A, Fazal A, Shultz MD, Stams T (2012). Structure of human tankyrase 1 in complex with small‐molecule inhibitors PJ34 and XAV939. Acta Crystallogr Sect F Struct Biol Cryst Commun 68: 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida S, Yamamoto H, Hino S, Ikeda S, Kishida M, Kikuchi A (1999). DIX domains of Dvl and axin are necessary for protein interactions and their ability to regulate beta‐catenin stability. Mol Cell Biol 19: 4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S et al. (1998). Axin, a negative regulator of the Wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta‐catenin. J Biol Chem 273: 10823–10826. [DOI] [PubMed] [Google Scholar]
- Kohler EM, Chandra SH, Behrens J, Schneikert J (2009). Beta‐catenin degradation mediated by the CID domain of APC provides a model for the selection of APC mutations in colorectal, desmoid and duodenal tumours. Hum Mol Genet 18: 213–226. [DOI] [PubMed] [Google Scholar]
- Kohler EM, Derungs A, Daum G, Behrens J, Schneikert J (2008). Functional definition of the mutation cluster region of adenomatous polyposis coli in colorectal tumours. Hum Mol Genet 17: 1978–1987. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW et al. (1997). Constitutive transcriptional activation by a beta‐catenin‐Tcf complex in APC−/− colon carcinoma. Science 275: 1784–1787. [DOI] [PubMed] [Google Scholar]
- Kunttas‐Tatli E, Roberts DM, McCartney BM (2014). Self‐association of the APC tumor suppressor is required for the assembly, stability, and activity of the Wnt signaling destruction complex. Mol Biol Cell 25: 3424–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunttas‐Tatli E, Von Kleeck RA, Greaves BD, Vinson D, Roberts DM, McCartney BM (2015). The two SAMP repeats and their phosphorylation state in Drosophila adenomatous polyposis coli‐2 play mechanistically distinct roles in negatively regulating Wnt signaling. Mol Biol Cell 26: 4503–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Salic A, Kruger R, Heinrich R, Kirschner MW (2003). The roles of APC and axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol 1: E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Wang C, Liu X, Hua S, Liu X (2015). The roles of AXIN2 in tumorigenesis and epigenetic regulation. Fam Cancer 14: 325–331. [DOI] [PubMed] [Google Scholar]
- Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP et al. (2012). Wnt signaling through inhibition of beta‐catenin degradation in an intact axin1 complex. Cell 149: 1245–1256. [DOI] [PubMed] [Google Scholar]
- Li Z, Kroboth K, Newton IP, Nathke IS (2008). Novel self‐association of the APC molecule affects APC clusters and cell migration. J Cell Sci 121: 1916–1925. [DOI] [PubMed] [Google Scholar]
- Li Z, Nathke IS (2005). Tumor‐associated NH2‐terminal fragments are the most stable part of the adenomatous polyposis coli protein and can be regulated by interactions with COOH‐terminal domains. Cancer Res 65: 5195–5204. [DOI] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y et al. (2002). Control of beta‐catenin phosphorylation/degradation by a dual‐kinase mechanism. Cell 108: 837–847. [DOI] [PubMed] [Google Scholar]
- Liu D, Li L, Yang Y, Liu W, Wu J (2014). The axin2 rs2240308 polymorphism and susceptibility to lung cancer in a Chinese population. Tumour Biol 35: 10987–10991. [DOI] [PubMed] [Google Scholar]
- Liu J, Xing Y, Hinds TR, Zheng J, Xu W (2006). The third 20 amino acid repeat is the tightest binding site of APC for beta‐catenin. J Mol Biol 360: 133–144. [DOI] [PubMed] [Google Scholar]
- Liu YT, Dan QJ, Wang J, Feng Y, Chen L, Liang J et al. (2011). Molecular basis of Wnt activation via the DIX domain protein Ccd1. J Biol Chem 286: 8597–8608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchtenborg M, Weijenberg MP, Wark PA, Saritas AM, Roemen GM, van Muijen GN et al. (2005). Mutations in APC, CTNNB1 and K‐ras genes and expression of hMLH1 in sporadic colorectal carcinomas from the Netherlands Cohort Study. BMC Cancer 5: 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Peterson A, Garcia BA, Coombs G, Kofahl B, Heinrich R et al. (2007). Protein phosphatase 1 regulates assembly and function of the beta‐catenin degradation complex. EMBO J 26: 1511–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U et al. (2002). Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol 22: 1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, He X (2012). Frizzled and LRP5/6 receptors for Wnt/beta‐catenin signaling. Cold Spring Harb Perspect Biol 4: a007880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X (2009). Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Yokota C, Tamai K, Zeng X, He X (2008). Wnt signal amplification via activity, cooperativity, and regulation of multiple intracellular PPPSP motifs in the Wnt co‐receptor LRP6. J Biol Chem 283: 16115–16123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Wang J, Liu B, Pan W, Farr GH 3rd, Flynn C et al. (2001). Low‐density lipoprotein receptor‐related protein‐5 binds to axin and regulates the canonical Wnt signaling pathway. Mol Cell 7: 801–809. [DOI] [PubMed] [Google Scholar]
- Marin O, Bustos VH, Cesaro L, Meggio F, Pagano MA, Antonelli M et al. (2003). A noncanonical sequence phosphorylated by casein kinase 1 in beta‐catenin may play a role in casein kinase 1 targeting of important signaling proteins. Proc Natl Acad Sci U S A 100: 10193–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti L, Templeton CM, Ranes M, Paracuellos P, Cronin N, Beuron F et al. (2016). Tankyrase requires SAM domain‐dependent polymerization to support Wnt‐beta‐catenin signaling. Mol Cell 63: 498–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino‐Echarri E, Brocardo MG, Mills KM, Henderson BR (2016). Tankyrase inhibitors stimulate the ability of tankyrases to bind axin and drive assembly of beta‐catenin degradation‐competent axin puncta. PLoS One 11: e0150484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y et al. (2015). Modeling colorectal cancer using CRISPR‐Cas9‐mediated engineering of human intestinal organoids. Nat Med 21: 256–262. [DOI] [PubMed] [Google Scholar]
- Mazzoni SM, Fearon ER (2014). AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett 355: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney BM, Nathke IS (2008). Cell regulation by the Apc protein Apc as master regulator of epithelia. Curr Opin Cell Biol 20: 186–193. [DOI] [PubMed] [Google Scholar]
- McCartney BM, Price MH, Webb RL, Hayden MA, Holot LM, Zhou M et al. (2006). Testing hypotheses for the functions of APC family proteins using null and truncation alleles in Drosophila. Development 133: 2407–2418. [DOI] [PubMed] [Google Scholar]
- Melo FS, Kurtova AV, Harnoss JM, Kljavin N, Hoeck JD, Hung J et al. (2017). A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 543: 676–680. [DOI] [PubMed] [Google Scholar]
- Metcalfe C, Bienz M (2011). Inhibition of GSK3 by Wnt signalling – two contrasting models. J Cell Sci 124: 3537–3544. [DOI] [PubMed] [Google Scholar]
- Minde DP, Radli M, Forneris F, Maurice MM, Rudiger SG (2013). Large extent of disorder in adenomatous polyposis coli offers a strategy to guard Wnt signalling against point mutations. PLoS One 8: e77257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson‐Maduro J, Godsave S, Korinek V et al. (1996). XTcf‐3 transcription factor mediates beta‐catenin‐induced axis formation in Xenopus embryos. Cell 86: 391–399. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B et al. (1997). Activation of beta‐catenin‐Tcf signaling in colon cancer by mutations in beta‐catenin or APC. Science 275: 1787–1790. [DOI] [PubMed] [Google Scholar]
- Morrone S, Cheng Z, Moon RT, Cong F, Xu W (2012). Crystal structure of a tankyrase‐axin complex and its implications for axin turnover and tankyrase substrate recruitment. Proc Natl Acad Sci U S A 109: 1500–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P (1995). Regulation of intracellular beta‐catenin levels by the adenomatous polyposis coli (APC) tumor‐suppressor protein. Proc Natl Acad Sci U S A 92: 3046–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathke I (2006). Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nat Rev Cancer 6: 967–974. [DOI] [PubMed] [Google Scholar]
- Ng SS, Mahmoudi T, Danenberg E, Bejaoui I, de Lau W, Korswagen HC et al. (2009). Phosphatidylinositol 3‐kinase signaling does not activate the Wnt cascade. J Biol Chem 284: 35308–35313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noutsou M, Duarte AM, Anvarian Z, Didenko T, Minde DP, Kuper I et al. (2011). Critical scaffolding regions of the tumor suppressor axin1 are natively unfolded. J Mol Biol 405: 773–786. [DOI] [PubMed] [Google Scholar]
- Okada K, Bartolini F, Deaconescu AM, Moseley JB, Dogic Z, Grigorieff N et al. (2010). Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J Cell Biol 189: 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterveen T, Coudreuse DY, Yang PT, Fraser E, Bergsma J, Dale TC et al. (2007). Two functionally distinct axin‐like proteins regulate canonical Wnt signaling in C. elegans . Dev Biol 308: 438–448. [DOI] [PubMed] [Google Scholar]
- Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW (1997). Serine phosphorylation‐regulated ubiquitination and degradation of beta‐catenin. J Biol Chem 272: 24735–24738. [DOI] [PubMed] [Google Scholar]
- Peterson‐Nedry W, Erdeniz N, Kremer S, Yu J, Baig‐Lewis S, Wehrli M (2008). Unexpectedly robust assembly of the axin destruction complex regulates Wnt/Wg signaling in Drosophila as revealed by analysis in vivo. Dev Biol 320: 226–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piao S, Lee SH, Kim H, Yum S, Stamos JL, Xu Y et al. (2008). Direct inhibition of GSK3beta by the phosphorylated cytoplasmic domain of LRP6 in Wnt/beta‐catenin signaling. PLoS One 3: e4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P (2007). The many ways of Wnt in cancer. Curr Opin Genet Dev 17: 45–51. [DOI] [PubMed] [Google Scholar]
- Polakis P (2012). Wnt signaling in cancer. Cold Spring Harb Perspect Biol 4: a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronobis MI, Deuitch N, Posham V, Mimori‐Kiyosue Y, Peifer M (2017). Reconstituting regulation of the canonical Wnt pathway by engineering a minimal beta‐catenin destruction machine. Mol Biol Cell 28: 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronobis MI, Rusan NM, Peifer M (2015). A novel GSK3‐regulated APC:axin interaction regulates Wnt signaling by driving a catalytic cycle of efficient betacatenin destruction. Elife 4: e08022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe MJ, Itoh K, Sokol SY (2000). A positive role for the PP2A catalytic subunit in Wnt signal transduction. J Biol Chem 275: 35680–35683. [DOI] [PubMed] [Google Scholar]
- Roberts DM, Pronobis MI, Poulton JS, Waldmann JD, Stephenson EM, Hanna S et al. (2011). Deconstructing the sscatenin destruction complex: mechanistic roles for the tumor suppressor APC in regulating Wnt signaling. Mol Biol Cell 22: 1845–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M, Ibrahim AE, Mieszczanek J, Bienz M (2014). LEF1 and B9L shield beta‐catenin from inactivation by axin, desensitizing colorectal cancer cells to tankyrase inhibitors. Cancer Res 74: 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosales‐Reynoso MA, Arredondo‐Valdez AR, Wence‐Chavez LI, Barros‐Nunez P, Gallegos‐Arreola MP, Flores‐Martinez SE et al. (2016). AXIN2 polymorphisms and their association with colorectal cancer in Mexican patients. Genet Test Mol Biomarkers 20: 438–444. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P (1996). Binding of GSK3beta to the APC‐beta‐catenin complex and regulation of complex assembly. Science 272: 1023–1026. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Munemitsu S, Polakis P (1997a). Loss of beta‐catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res 57: 4624–4630. [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El‐Gamil M, Albert I, Porfiri E, Polakis P (1997b). Stabilization of beta‐catenin by genetic defects in melanoma cell lines. Science 275: 1790–1792. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Tice DA, Polakis P (2001). Axin‐dependent phosphorylation of the adenomatous polyposis coli protein mediated by casein kinase 1epsilon. J Biol Chem 276: 39037–39045. [DOI] [PubMed] [Google Scholar]
- Salahshor S, Woodgett JR (2005). The links between axin and carcinogenesis. J Clin Pathol 58: 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz‐Romond T, Fiedler M, Shibata N, Butler PJ, Kikuchi A, Higuchi Y et al. (2007a). The DIX domain of dishevelled confers Wnt signaling by dynamic polymerization. Nat Struct Mol Biol 14: 484–492. [DOI] [PubMed] [Google Scholar]
- Schwarz‐Romond T, Metcalfe C, Bienz M (2007b). Dynamic recruitment of axin by dishevelled protein assemblies. J Cell Sci 120: 2402–2412. [DOI] [PubMed] [Google Scholar]
- Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM (1999). Regulation of beta‐catenin signaling by the B56 subunit of protein phosphatase 2A. Science 283: 2089–2091. [DOI] [PubMed] [Google Scholar]
- Shiomi K, Uchida H, Keino‐Masu K, Masu M (2003). Ccd1, a novel protein with a DIX domain, is a positive regulator in the Wnt signaling during zebrafish neural patterning. Curr Biol 13: 73–77. [DOI] [PubMed] [Google Scholar]
- Smith S, Giriat I, Schmitt A, de Lange T (1998). Tankyrase, a poly(ADP‐ribose) polymerase at human telomeres. Science 282: 1484–1487. [DOI] [PubMed] [Google Scholar]
- Smits R, Kielman MF, Breukel C, Zurcher C, Neufeld K, Jagmohan‐Changur S et al. (1999). Apc1638T: a mouse model delineating critical domains of the adenomatous polyposis coli protein involved in tumorigenesis and development. Genes Dev 13: 1309–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobrado P, Jedlicki A, Bustos VH, Allende CC, Allende JE (2005). Basic region of residues 228‐231 of protein kinase CK1alpha is involved in its interaction with axin: binding to axin does not affect the kinase activity. J Cell Biochem 94: 217–224. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spink KE, Polakis P, Weis WI (2000). Structural basis of the axin‐adenomatous polyposis coli interaction. EMBO J 19: 2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos JL, Chu ML, Enos MD, Shah N, Weis WI (2014). Structural basis of GSK‐3 inhibition by N‐terminal phosphorylation and by the Wnt receptor LRP6. Elife 3: e01998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamos JL, Weis WI (2013). The beta‐catenin destruction complex. Cold Spring Harb Perspect Biol 5: a007898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Fu C, Ishikawa S, Stella A, Kojima M, Shitoh K et al. (2008). APC is essential for targeting phosphorylated beta‐catenin to the SCFbeta‐TrCP ubiquitin ligase. Mol Cell 32: 652–661. [DOI] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I et al. (2010). Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell 143: 1136–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamai K, Zeng X, Liu C, Zhang X, Harada Y, Chang Z et al. (2004). A mechanism for Wnt coreceptor activation. Mol Cell 13: 149–156. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Mashima T, Mizutani A, Sato A, Aoyama A, Gong B et al. (2017). APC mutations as a potential biomarker for sensitivity to tankyrase inhibitors in colorectal cancer. Mol Cancer Ther 16: 752–762. [DOI] [PubMed] [Google Scholar]
- Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS et al. (2010). Small‐molecule inhibition of Wnt signaling through activation of casein kinase 1alpha. Nat Chem Biol 6: 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorstensen L, Lind GE, Lovig T, Diep CB, Meling GI, Rognum TO et al. (2005). Genetic and epigenetic changes of components affecting the WNT pathway in colorectal carcinomas stratified by microsatellite instability. Neoplasia 7: 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsen TE, Pedersen NM, Wenzel EM, Schultz SW, Brech A, Liestol K et al. (2015). Structure, dynamics, and functionality of tankyrase inhibitor‐induced degradasomes. Mol Cancer Res 13: 1487–1501. [DOI] [PubMed] [Google Scholar]
- Thorvaldsen TE, Pedersen NM, Wenzel EM, Stenmark H (2017). Differential roles of AXIN1 and AXIN2 in tankyrase inhibitor‐induced formation of degradasomes and beta‐catenin degradation. PLoS One 12: e0170508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolwinski NS, Wehrli M, Rives A, Erdeniz N, DiNardo S, Wieschaus E (2003). Wg/Wnt signal can be transmitted through arrow/LRP5,6 and axin independently of Zw3/Gsk3beta activity. Dev Cell 4: 407–418. [DOI] [PubMed] [Google Scholar]
- Valvezan AJ, Zhang F, Diehl JA, Klein PS (2012). Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase‐3 (GSK‐3) activity. J Biol Chem 287: 3823–3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinyoles M, Del Valle‐Perez B, Curto J, Vinas‐Castells R, Alba‐Castellon L, Garcia de Herreros A et al. (2014). Multivesicular GSK3 sequestration upon Wnt signaling is controlled by p120‐catenin/cadherin interaction with LRP5/6. Mol Cell 53: 444–457. [DOI] [PubMed] [Google Scholar]
- Voloshanenko O, Erdmann G, Dubash TD, Augustin I, Metzig M, Moffa G et al. (2013). Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun 4: 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M et al. (2004). Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell 7: 871–883. [DOI] [PubMed] [Google Scholar]
- Woodgett JR (1990). Molecular cloning and expression of glycogen synthase kinase‐3/factor A. EMBO J 9: 2431–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Huang H, Garcia Abreu J, He X (2009). Inhibition of GSK3 phosphorylation of beta‐catenin via phosphorylated PPPSPXS motifs of Wnt coreceptor LRP6. PLoS One 4: e4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP (2003). Structure of a beta‐TrCP1‐Skp1‐beta‐catenin complex: destruction motif binding and lysine specificity of the SCF(beta‐TrCP1) ubiquitin ligase. Mol Cell 11: 1445–1456. [DOI] [PubMed] [Google Scholar]
- Xing Y, Clements WK, Kimelman D, Xu W (2003). Crystal structure of a beta‐catenin/axin complex suggests a mechanism for the beta‐catenin destruction complex. Genes Dev 17: 2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing Y, Clements WK, Le Trong I, Hinds TR, Stenkamp R, Kimelman D et al. (2004). Crystal structure of a beta‐catenin/APC complex reveals a critical role for APC phosphorylation in APC function. Mol Cell 15: 523–533. [DOI] [PubMed] [Google Scholar]
- Xu W, Kimelman D (2007). Mechanistic insights from structural studies of beta‐catenin and its binding partners. J Cell Sci 120: 3337–3344. [DOI] [PubMed] [Google Scholar]
- Xue B, Romero PR, Noutsou M, Maurice MM, Rudiger SG, William AM Jr et al. (2013). Stochastic machines as a colocalization mechanism for scaffold protein function. FEBS Lett 587: 1587–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Hinoi T, Michiue T, Fukui A, Usui H, Janssens V et al. (2001). Inhibition of the Wnt signaling pathway by the PR61 subunit of protein phosphatase 2A. J Biol Chem 276: 26875–26882. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A (1999). Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase‐3beta regulates its stability. J Biol Chem 274: 10681–10684. [DOI] [PubMed] [Google Scholar]
- Yang E, Tacchelly‐Benites O, Wang Z, Randall MP, Tian A, Benchabane H et al. (2016). Wnt pathway activation by ADP‐ribosylation. Nat Commun 7: 11430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N, Kakunda M, Pham V, Lill JR, Du P, Wongchenko M et al. (2015). HSP105 recruits protein phosphatase 2A to dephosphorylate beta‐catenin. Mol Cell Biol 35: 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Fagotto F, Zhang T, Hsu W, Vasicek TJ, Perry WL 3rd et al. (1997). The mouse fused locus encodes axin, an inhibitor of the Wnt signaling pathway that regulates embryonic axis formation. Cell 90: 181–192. [DOI] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R et al. (2005). A dual‐kinase mechanism for Wnt co‐receptor phosphorylation and activation. Nature 438: 873–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan T, Rindtorff N, Boutros M (2017). Wnt signaling in cancer. Oncogene 36: 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yang J, Liu Y, Chen X, Yu T, Jia J et al. (2009). PR55 alpha, a regulatory subunit of PP2A, specifically regulates PP2A‐mediated beta‐catenin dephosphorylation. J Biol Chem 284: 22649–22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lou Y, Zheng X, Wang H, Sun J, Dong Q et al. (2015). Wnt blockers inhibit the proliferation of lung cancer stem cells. Drug Des Devel Ther 9: 2399–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA et al. (2011). RNF146 is a poly(ADP‐ribose)‐directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol 13: 623–629. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Qiu WJ, Chan SC, Han J, He X, Lin SC (2002). Casein kinase I and casein kinase II differentially regulate axin function in Wnt and JNK pathways. J Biol Chem 277: 17706–17712. [DOI] [PubMed] [Google Scholar]