


Abstract
Imprinted genes occur in discrete clusters that are coordinately regulated by shared DNA elements called Imprinting Control Regions. H19 and Igf2 are linked imprinted genes that play critical roles in development. Loss of imprinting (LOI) at the IGF2/H19 locus on the maternal chromosome is associated with the developmental disorder Beckwith Wiedemann Syndrome (BWS) and with several cancers. Here we use comprehensive genetic and genomic analyses to follow muscle development in a mouse model of BWS to dissect the separate and shared roles for misexpression of Igf2 and H19 in the disease phenotype. We show that LOI results in defects in muscle differentiation and hypertrophy and identify primary downstream targets: Igf2 overexpression results in over-activation of MAPK signaling while loss of H19 lncRNA prevents normal down regulation of p53 activity and therefore results in reduced AKT/mTOR signaling. Moreover, we demonstrate instances where H19 and Igf2 misexpression work separately, cooperatively, and antagonistically to establish the developmental phenotype. This study thus identifies new biochemical roles for the H19 lncRNA and underscores that LOI phenotypes are multigenic so that complex interactions will contribute to disease outcomes.
INTRODUCTION
Imprinting is a form of gene regulation where expression of an allele is dependent upon its parental origin. There are ∼100–200 imprinted genes in the human genome and imprinting is conserved across mammalian species. Imprinted genes are not randomly scattered but are organized into discrete clusters where monoallelic expression is dependent upon a shared DNA regulatory element called the Imprinting Control Region or ICR (1).
One imprinted gene cluster is the IGF2/H19 locus on human chromosome 11p15.5 or mouse distal chromosome 7. Imprinting in this 110 kb region is determined by the H19ICR which is located just upstream of the H19 promoter (2,3). As described in Figure 1A, the H19ICR organizes the locus so that transcription of the Igf2 and H19 genes is restricted to the paternal and maternal chromosomes, respectively.
Figure 1.
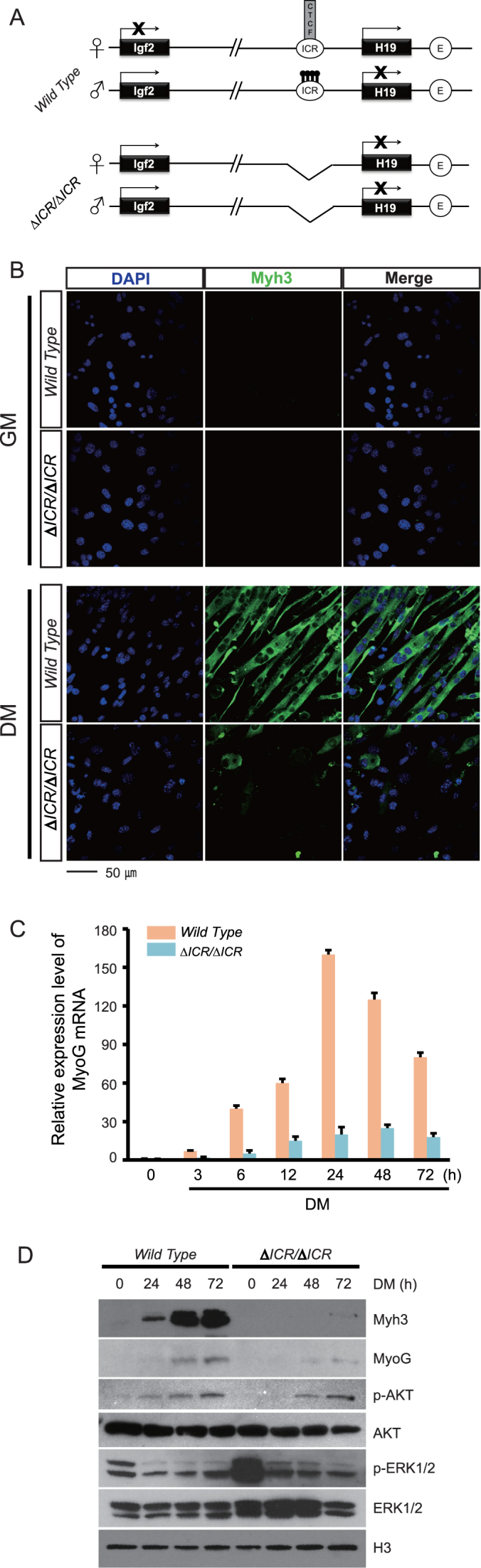
Differentiation defects in LOI myoblasts. (A) Cartoon depiction of the gene architecture and expression patterns in wild type and in ΔICR/ΔICR cells. Igf2 and H19 are about 80 kb apart on distal mouse chromosome 7. Parent-of-origin transcription depends upon the 2.4 kb H19ICR located just upstream of the H19 promoter. On the maternal chromosome, the H19ICR binds the CTCF genome organizing protein which positions the region into loop structures that prevent interaction of the Igf2 promoter with the shared muscle specific enhancer (E, unfilled circle) located 26 kb upstream of the H19 transcriptional start site. At the same time, these loop structures facilitate H19 promoter-enhancer interactions. Upon paternal inheritance, methylation of ICR CpGs prevents CTCF binding, thus enabling alternative loop structures that promote interactions between the paternal Igf2 promoter and the shared enhancers (60,61). In addition, developmentally programmed ‘spread’ of heterochromatin from the H19ICR to the adjacent H19 promoter prevents H19 transcription (62). (B) Primary myoblasts derived from wild type or ΔICR/ΔICR neonates were cultured in growth medium (GM) or in serum-depleted differentiation medium (DM) for 72 h. DAPI staining (blue) identifies nuclei and staining for Myh3 (green) identifies differentiating cells. The ΔICR/ΔICR cells in DM show highly aberrant morphology, indicating a severe differentiation defect. (C) Quantitation of Myogenin RNA by qRT-PCR (n = 3). Expression relative to GAPDH is reported. (D) Immunoblot analyses of cell extracts prepared from myoblasts (0 timepoint) and from cells grown in differentiating media for 24, 48 or 72 h. Differentiation markers, Myh3 and Myogenin, are significantly downregulated in LOI cells. Phosphorylated forms of Erk1/Erk2 peptides (also commonly referred to as MAPK3/1 or p44/p42) are also reduced in these cells, although total protein levels are unchanged. H3 is the loading control.
IGF2 (Insulin-like growth factor 2) encodes a peptide hormone with a wide array of functions promoting cell growth and proliferation. Its function in the context of muscle development has been studied extensively using both in vitro differentiation and in vivo loss of function mouse models (4–7). IGF2 peptide plays an important role in prenatal skeletal muscle growth and in muscle regeneration after injury in the adult. It has also been shown that IGF2 stimulates phosphoinositol-3 kinase and AKT signaling through activation of the InsR and Igf1R receptor kinases (7,8). The biochemical defects associated with IGF2 overdose, as caused by loss of imprinting mutations, have been less well characterized.
H19 does not encode any known peptide. Instead the gene's functional product is a 2.3 kb long non-coding RNA (lncRNA) whose biochemical activities have only recently begun to be elucidated (9). One role for the lncRNA is that it is the substrate used to generate two microRNAs (miRNAs), miR-675-3p and miR-675-5p (10). Genetic studies support a role for these miRNAs in placenta development (11) and in skeletal muscle differentiation and regeneration (12). The H19 lncRNA can also bind miRNAs. Gao and colleagues showed that H19 binding to let-7 miRNA can reduce let-7 bioavailability and thus regulate glucose metabolism in C2C12 cells (13). A third proposed biochemical function is that H19 lncRNA interacts directly with transcription factor proteins, including p53, to reduce their bioactivity (14–16). Mutant H19 alleles that specifically impair a single biochemical function would be of high value in sorting out the relative importance of these distinct mechanisms.
In different cancer models, H19 can have tumor suppressor or oncogenic properties (17–19). For example, restoration of H19 expression by plasmid transfection blocks tumorigenicity in rhabdosarcoma (20) but other studies support H19 oncogenic function in breast cancer (21). These apparent contradictions have led to proposals that H19 has fundamentally distinct roles dependent upon developmental stage (22).
The critical importance of precise regulation of IGF2 and H19 gene expression is underscored by disease phenotypes associated with loss of imprinting at the locus (23). Russell-Silver syndrome is associated with mutations that inhibit methylation of the ICR and thus promote binding of the CTCF protein to the paternal H19ICR. This results in decreased IGF2 and increased H19 from the paternal chromosome. Beckwith-Wiedemann syndrome (BWS) is associated with mutations that prevent CTCF binding to the maternal chromosome and that therefore result in biallelic or 2× dosage of IGF2 and decreased H19 RNA. Several mouse models have been developed that mimic parts of the BWS phenotype by altering the expression of H19 or of Igf2 (24–26). However, the combined effect of the lack of H19 and the overexpression of Igf2 has not been well characterized. Especially, the genetic interactions and interplay between H19 and Igf2 remain underexplored.
To address this question, we have modeled BWS by generating mice that carry a deletion of the H19ICR on the maternally inherited chromosome (27). These mice demonstrate molecular symptoms characteristic of BWS: low expression of H19 and biallelic Igf2 expression. Thus, it is a useful model system to study the molecular mechanisms of development in the context of BWS. In this study we use molecular, cellular, pharmacological, and transcriptome analyses to characterize differentiation of primary myoblast cell lines derived from wild type and from a panel of mutant and transgenic mice to study the relative significance of extra Igf2, of decreased H19, and the synergistic effects of the two changes. We chose this model system because of the powerful tools available to follow differentiation and development in vitro and to therefore identify downstream targets affected by Igf2/H19 misexpression. Also, our results might be directly relevant for understanding the link between LOI and rhabdosarcoma. We see that some key effects of the two genes are largely independent in regard to cell differentiation. Extra Igf2 prevents normal differentiation by over activation of the Erk1/2 kinases in the MAPK pathway. Decreased H19 expression prevents the downregulation of p53 activity that normally occurs during muscle cell differentiation. The persistent high levels of p53 in turn result in decreased pAKT/mTOR activation so that H19-deficient myotubes do not undergo normal hypertrophy. However, we also demonstrate situations where the simultaneous loss of H19/increased Igf2 increases phenotype severity and other cases where the two gene expression changes interact to ameliorate the mutant phenotype. Thus the final LOI phenotype is a complex and interesting interplay of the two genes.
MATERIALS AND METHODS
Animal studies
All mice were bred and housed in accordance with National Institutes of Health and United States Public Health Service policy. Animal research was approved through the Eunice Kennedy Shriver National Institute of Child Health and Human Development Animal Care and Use Committee. H19ΔICR (27), H19ΔEx1 (28), H19Δ13 (29), Igf2-(30) and H19 BAC transgenic (2) strains were generated as described.
Cell culture
Hind limb skeletal muscle was isolated from p2 to p3 pups. Myoblasts were isolated as described (31) and grown at 37°C in 10% CO2 in either F10 supplemented with 10% calf serum and 2.5 μg/ml fibroblast growth factor (GM, or Growth Media) or in DMEM supplemented with 1% horse serum (DM, or Differentiation Medium). The following supplements were used as described in the text: recombinant IGF2 (Peprotech, 100-12), BMS-754807 (Active Biochemicals, A-1013), PD 98059 (Cell Signaling, 9900), cAMP (Sigma, A6885). Cell fusion index was quantitated as described (32) (n = 100 for each of three independent cultures) and statistical significance was analyzed using the Student's t-test. Results are reported as mean ± standard error.
Immunofluorescence
At least three independent cultures were analyzed for each experiment. After growth times indicated in the text, cells were fixed with 4% paraformaldehyde (10 min at room temperature, RT), permeabilized with 0.5% Triton (10 min at RT), and blocked with 10% goat serum for 30 min at RT before incubation with antibodies. For MYH3 staining, primary antibodies were purchased from Santa Cruz Biotechnology, Inc., diluted 1:100 in blocking buffer, incubated at 4°C for 24 h, and visualized with Alexa-488 secondary antibody from Invitrogen diluted 1:500. For Myogenin staining, primary antibodies were purchased from Thermo Fisher, Inc., diluted 1:1000, incubated at 4°C for 24 h, and visualized with Alexa-594 secondary antibody, diluted 1:1000. Nuclear staining was with DAPI (Molecular Probe) diluted 1:1000 and incubated at RT for 30 min. Cells were imaged with Carl Zeiss 510 using 40× oil immersion objective. Images were composed and edited in LSM image software or Photoshop 7.0 (Adobe).
ELISA
IGF2 secreted peptide was measured with the Mouse IGF2 ELISA kit (Abcam, ab100696) on at least three independent cultures. Statistical significance was analyzed using the Student's t-test.
Quantitative RT-PCR
For each experiment, we used at least three independent cell cultures for RNA preparation using the RNeasy Plus Mini kit (Qiagen). RNAs were analyzed using ThermoFisher NANODROP 2000c to evaluate purity and yield and then stored at -70C. cDNA samples were prepared with and without reverse transcriptase according to the manufacturer's instructions (Transcriptor First Strand cDNA Synthesis Kit, Roche 04 897 030 001) using oligo-dT primers. cDNAs were analyzed using SYBR Green (Roche 04 887 352 001) on the Roche Cycler 480 (45 cycles with annealing at 60C) using primers described in Supplemental Table S3. For microRNA analyses we used mirVanaTM miRNA Isolation Kit and TaqMan MicroRNA Assays (ThermoFisher Scientific 4427975, Assay ID 001973 (U6), 001940 (miR-675-5p), 001941 (miR-675-3p), 002406 (let-7e), 002282 (let-7g) and 002221 (let-7i)). For each primer pair, we established standard curves to evaluate slope, y-intercepts, and PCR efficiency and to determine the dynamic range of the assay. Specificity of the reactions was confirmed by melting point analyses and by gel electrophoresis. The number of biological replicates is described in the relevant figures and each assay was run at least twice. Statistical significance was evaluated using a two-tailed, type 2 Student's t-test (Microsoft Excel). Results in all figures show mean ± standard error.
Immunoblotting
Cells were washed with ice-cold PBS, scraped, and pelleted by centrifugation. Whole-cell extracts were prepared using M-PER protein extraction reagents (Thermo Fisher). Protein concentrations of the lysates were assayed using a BCA protein assay kit (Pierce). Protein samples were fractionated by electrophoresis on 12% or 4–20% SDS-PAGE gels and then transferred to nitrocellulose. All antibodies were diluted in antibody enhancer buffer (HIKARI) and are described in Supplementary Table S2.
RNA sequencing
RNA was isolated from XX primary myoblast and 24-h-differentiated myotube cells using Tripure (Roche) followed by the RNeasy Micro kit (Qiagen). Samples with RNA Integrity numbers of ≥9 were used for sequencing. RNA-Seq was performed in two batches: (A) two wild type myoblasts, three wild type myotubes and two ΔICR/ΔICR myotube samples were sequenced at the NIH Intramural Sequencing Center using Illumina HiSeq2000 sequencers; and (B) two wild type myoblasts, two wild type myotubes and two ΔICR/ΔICR myoblasts were sequenced at the NICHD Molecular Genomics Core using an ABI SOLiD 5500xl sequencer. Ribosomal RNA was depleted from 1 to 5 μg total RNA using the RiboZero Gold kit (Illumina). Library preparation for the Illumina HiSeq2000 was performed using the TruSeq RNA Sample Prep V2 kit (Illumina). Pooled barcoded libraries were sequenced to generate paired-end 101 bp reads and raw data was aligned to mouse genome version mm9 using Tophat2 (33). For the ABI SOLiD sequencing, libraries were prepared using the SOLiD Whole Transcriptome Analysis kit (Thermo Fisher Scientific). Pooled barcoded libraries were sequenced to generate paired-end 35 × 75 bp reads and colorspace XSQ data was aligned to mouse genome version mm9 using Lifescope software (Thermo Fisher Scientific).
RNA-Seq analysis
Gene-wise read counts were analyzed for differential expression using DESeq2 (34). When comparing wild type myoblasts and myotubes, we used a linear model with a blocking factor to account for possible batch effects from sequencing. Contrasts were used to calculate the effects of genotype and sequencing block. We used a false discovery rate of 0.01 and a minimum of 2-fold difference to define differential expression. Finally, genes that were differentially expressed only due to the genotype effect (2268 genes) were used for subsequent analysis (Supplementary File 1). Since, a complete randomized design was not possible for ΔICR/ΔICR samples, we assumed that the same genes were affected by the sequencing block as in wild type. These 1003 genes were then removed from the analysis to yield 1193 differentially expressed genes in the LOI cells (Supplementary File 1).
When directly comparing wild type and ΔICR/ΔICR cells at each developmental stage, we avoided effects of sequencing block by conducting comparisons between samples sequenced in the same batch. However, since there were relatively few genes differentially expressed in myotubes using the above criteria, we used a less stringent FDR threshold of 0.1 for this comparison. For myoblasts, we used an FDR threshold of 0.01. Also, we did not enforce a minimum fold-change requirement for both myoblast and myotube cells. Differentially expressed genes were analyzed for functional enrichment with DAVID using default settings (35,36). Since many genes were differentially expressed in the LOI myoblasts compared to the wild type, we separately analyzed upregulated (1558) and downregulated (1403) genes for functional enrichment (Supplementary File 1). The log2 fold-changes calculated by DESeq2 for differentially expressed genes associated with the MAP kinase signaling pathway were plotted as a heatmap using the ggplot2 package in R.
Data access
RNA sequencing data were deposited in the NCBI Gene Expression Omnibus (GEO) under Series accession number GSE89136.
RESULTS
Loss of imprinting at the Igf2/H19 locus disrupts in vitro differentiation of primary myoblasts.
The H19ΔICR mutation is a deletion that removes the entire 2.4 kb H19ICR (Figure 1A). In skeletal muscle tissue and in isolated primary myoblasts, maternal inheritance of this deletion results in expression of the normally repressed maternal Igf2 allele and dramatically reduces expression of maternal H19 (27,37). Thus maternal inheritance of the deletion recapitulates the loss of imprinting (LOI) associated with BWS. Paternal inheritance does not alter H19 or Igf2 gene expression patterns. Thus ΔICR/ΔICR and ΔICR/+ muscle tissue and primary cells have equivalent phenotypes in regards to expression of H19 and Igf2. Accordingly, LOI phenotypes described below are likewise equivalent in ΔICR/ΔICR and ΔICR/+ cell lines.
To understand the downstream cellular consequences of LOI, we characterized primary myoblast cells isolated from wild type and mutant neonatal littermates. Upon replacement with serum-depleted media, wild type myoblasts efficiently differentiate into myotubes so that within 72 h, >90% of cells elongate, fuse, and express myotube-specific markers such as Myh3 and MyoG (Myogenin). ΔICR/ΔICR myoblasts look and behave much like wild type cells when cultured in serum replete medium (Figure 1B, see ‘GM’ panels); however, upon serum depletion, they show dramatic defects in their ability to differentiate into myotubes. In addition to their aberrant morphology (Figure 1B, see ‘DM’ panels), these deficiencies can be quantitated as 6-fold reductions in cell fusion (wild type = 90±4%, LOI = 15 ± 2%, n = 3, P < 0.001) and in limited expression of myotube specific RNA (Figure 1C, Supplemental Figure S1) and proteins (Myh3 and MyoG) (Figure 1D).
We note here that our western blots (Figure 1D) show that LOI cells have significantly higher levels of phosphorylated Erk1/2 peptides but this will be discussed in detail below.
Extra IGF2 peptide accounts for many of the differentiation defects in LOI cells
Western blot analyses of myoblast cell extracts and ELISA-based quantitation of secreted IGF2 show a 1.8-fold increase in IGF2 peptide in ΔICR/ΔICR cells (Figure 2A) (n = 5, P = 0.057), consistent with the ΔICR mutation's effect on Igf2 imprinting. However, quantitation of secreted peptide after initiation of in vitro differentiation showed that the differences between wild type and mutant myotubes increase to 3.5-fold (to about 140 ng/ml) suggesting that impairment in the differentiation process induces additional IGF2 synthesis or prevents its uptake and/or degradation in LOI cells (Figure 2B).
Figure 2.

High levels of Igf2 are responsible for the differentiation defects in LOI myoblasts. (A) ELISA analysis of secreted IGF2 in myoblast cell cultures show an increase of 1.8-fold relative to wild type cells (n = 5, P = 0.057). (B) ELISA analyses of secreted IGF2 in cells grown in Differentiation Medium (DM) for 24, 48 and 72 h show progressive accumulation of IGF2 in ΔICR/ΔICR cells at significantly higher levels compared to wild type. (C–E) Added IGF2 peptide prevents normal differentiation in wild type cells. The normal myotube morphology is disrupted in a dose-dependent manner with added IGF2, recapitulating the LOI differentiation phenotype. Myh3 and Myogenin are also downregulated and cell fusion is significantly reduced. (F–H) IGF2 inhibition rescues differentiation in ΔICR/ΔICR cells. BMS-754807 inhibits IGF1 and InsR receptor kinase activity. When treated with BMS-754807, ΔICR/ΔICR cells show improved myotube morphology, increased Myh3 and Myogenin expression and significantly higher fusion rates. (C, F) DAPI (blue) and Myh3 (green) staining (D, G) Immunoblot analyses of cell extracts quantitates expression of differentiation markers, Myh3 and Myogenin (MyoG). H3 is a loading control. (E, H) Quantitation of cell fusion (n = 3). For C–H, cells were grown 72 h in DM. *P < 0.05; **P < 0.01; ***P < 0.001.
To understand the functional importance of this extra IGF2 in the differentiation defect of LOI myotubes, we added IGF2 peptide to wild type primary cell cultures and recapitulated the mutant phenotype. Exogenous IGF2 peptide prevents efficient differentiation of wild type myoblasts into elongated, fused myotubes (Figure 2C–E). Moreover, we rescued much of the mutant cell phenotype by culturing differentiating cells with BMS-754807, a small molecule inhibitor of Igf1R and InsR receptor kinases. (38). That is, in the presence of the antagonist, LOI myoblasts obtain an elongated morphology (Figure 2F), express myotube markers (Figure 2F and G), and fuse with almost normal efficiency (Figure 2H).
To address concerns that inappropriate doses or non-specific effects resulted in mis-interpretation of these drug studies, we used a genetic approach to rescue IGF2 levels in our LOI model by comparing primary cells from ΔICR/Igf2+ and ΔICR/Igf2- littermates (Figure 3A). Igf2- is an allele that removes all Igf2 peptide coding sequences while leaving imprinting of the locus intact (30). In ΔICR/Igf2- pups, the ICR deletion on the maternal chromosome results in the expected LOI phenotype (activation of maternal Igf2 concomitant with reduced expression of H19), however, the mutation of the paternal Igf2 allele prevents synthesis of functional Igf2 mRNA from that chromosome. Thus the net effect of the combined mutations is to return the animals to 1× dosage levels of IGF2 peptide (Figure 3B and C) without restoring H19 RNA to wild type levels (Figure 3D). In ΔICR/Igf2- cells, key aspects of myotube differentiation are restored toward wild type including an elongated morphology (Figure 3E), expression of myotube markers (Figure 3B and E), and normal cell fusion (Figure 3F). Thus altogether, we conclude that key differentiation defects in LOI myoblasts depend on the 2-fold increase in Igf2 expression associated with LOI.
Figure 3.

Restoring monoallelic expression of Igf2 in LOI cells rescues their differentiation defects. (A) Genetic scheme to restore monoallelic expression of Igf2 in LOI cells. In mouse genetic nomenclature, maternal alleles are indicated first. For example, ΔICR/Igf2- means that the maternal chromosome carries the ICR deletion and the paternal chromosome carries the Igf2 deletion described in the text. Here we crossed ΔICR/ΔICR mice with Igf2+/- mice to obtain two genotypes of interest: ΔICR/Igf2+, which phenocopies ΔICR/ΔICR (extra Igf2, low H19) and ΔICR/Igf2- which genetically rescues Igf2 expression while maintaining LOI levels of H19. (B, C) Ablation of the paternal Igf2 allele in LOI cells restores expression of IGF2 to wild type levels. (B) Immunoblot analyses. (C) ELISA analyses. (D) H19 expression relative to GAPDH was determined by qRT-PCR. Expression in wild type cells is set at 1. The ΔICR/Igf2- and ΔICR/Igf2+ cells have significantly downregulated H19 levels, similar to ΔICR/ΔICR. (E, F) Ablation of paternal Igf2 in LOI cells rescues differentiation defects. ΔICR/Igf2- myotubes have improved morphology with near-normal fusion, while ΔICR/Igf2+ cells have aberrant myotube morphology similar to LOI cells. (E) DAPI (blue) and Myh3 (green) staining. (F) Quantitation of cell fusion. In all experiments, cells were grown for 72 h in differentiation media and at least 3 independent cultures were analyzed. Statistical significance was evaluated by t-test, comparing to wild type controls. **P < 0.01; ***P < 0.001.
Next we wanted to determine if H19-deficiency is a necessary pre-requisite for observing the effects of increased IGF2. Therefore we generated primary cell lines where Igf2 and H19 expression from the endogenous locus were altered by the ICR deletion but where H19 mRNA levels were restored by expression from an unlinked single copy Bacterial Artificial Chromosome (BAC) transgene (Figure 4A and B). In these experiments, LOI was achieved with a deletion mutation called H19Δ13 (29). Like ΔICR, the Δ13 allele deletes the entire ICR but it also deletes the H19 promoter and RNA coding sequences. Maternal inheritance results in activation of maternal Igf2 allele while the paternal inheritance has no detectable effect on transcription. Thus maternal inheritance of either ΔICR or Δ13 deletions phenocopies molecular LOI defects associated with BWS.
Figure 4.

Restoration of H19 RNA in LOI cells does not rescue differentiation defects. (A) Genetic scheme to restore H19 in LOI cells. The Δ13 allele carries a deletion of the ICR and the H19 promoter and coding region and therefore causes LOI equivalent to the effect of ΔICR. (B) A Bacterial Artificial Chromosome (BAC) transgene provides wild type levels of H19 RNA, rescuing it from near-zero in the Δ13/Δ13 cells. H19 expression relative to GAPDH was determined by qRT-PCR and normalized to expression in wild type cells. (C–E) Analysis of differentiation defects after restoration of H19. (C) DAPI (blue) and Myh3 (green) staining. (D) Myh3 RNA was quantitated as described in B and normalized to H19 expression in wild type myoblasts. (E) Quantitation of cell fusion. All analyses were on cells grown in DM for 72 h, n = 3. Note that Δ13/Δ13 + H19 BAC cells show major differentiation defects relative to wild type cells for myogenin expression (P < 0.01) and cell fusion (P < 0.01). However, compared to Δ13/Δ13 cells without the BAC transgene, we also see modest improvements in myogenin expression (P = 0.088) and cell fusion (P = 0.078). **P < 0.01.
In these cell lines, the fundamental problems with differentiation are still apparent (Figure 4C–E). Altogether, we conclude that biallelic Igf2 is enough to prevent normal differentiation even when H19 is expressed at wild type levels. However, as we will address in the Discussion, LOI cells with the H19 transgene do not behave identically to cells without: restoration of H19 does result in some improvements in myogenin expression (P = 0.088) and in cell fusion (P = 0.078) (Figure 4D and E).
The MAPK pathway is dysregulated in LOI cells
To understand how the process of differentiation is altered in LOI cells, we performed RNA-sequencing to determine the transcriptional status of ΔICR/ΔICR and wild type myoblasts and of myotubes early in the differentiation process (24 h after serum removal).
First, we compared myoblasts to differentiating myotubes to identify genes whose expression changes during the 24 h in differentiation medium. At a false discovery rate (FDR) of 1% we found that the expression of 2268 genes was changed at least two-fold during the myoblast-myotube transition in wild type cells (Supplementary File 1). However, only 555 of these genes (24.4%) changed in expression in ΔICR/ΔICR cells upon removal of serum. That is, >75% of the genes that are changed during normal myoblast differentiation are misregulated in LOI cells suggesting global defects in establishing the new gene expression patterns necessary for myotube formation (Figure 5A).
Figure 5.

MAP kinase signaling is altered in ΔICR/ΔICR myoblasts and myotubes. (A and B) Comparison of gene expression changes that occur during myoblast differentiation in wild type and in ΔICR/ΔICR cells. We used RNA sequencing to identify genes where expression changed by >2-fold (FDR < 0.01) after 24 h in differentiation medium. (A) Venn diagram shows that more than 75% (1713 out of 2268) genes changed during normal myoblast differentiation are misregulated in LOI cells. (B) Top five pathways altered in wild type, but not ΔICR/ΔICR cells, during differentiation. Cell cycle and DNA replication pathways are highly over-represented among these genes. (C and D) Direct comparison of wild type and LOI cells at each developmental stage. (C) Selected pathways enriched in genes differentially expressed in LOI cells compared to wild type at the myoblast and myotube stages. Cell cycle, p53 signaling and the upstream MAP kinase signaling pathways are affected in both myoblast and myotube cells. (D) Heatmap of genes in the MAP kinase signaling pathway that are differentially expressed in LOI vs. wild type cells at the myoblast or myotube stage. Differentially expressed genes in this pathway show the same effect of the LOI mutation in both cell types, indicating early developmental defects.
Pathway analyses showed that these 1713 mis-regulated genes were highly enriched for cell cycle and other pathways associated with the cell cycle, such as DNA replication and p53 signaling (Figure 5B). Looking at the cell cycle pathway in detail, we noted that a large proportion of associated genes were aberrantly expressed in LOI cells: most affected genes are down regulated during normal differentiation in wild type, but remain unchanged during differentiation of LOI cells (Supplemental Figure S2). The p53 signaling pathway also shows similar patterns (Supplemental Figure S3): during normal differentiation expression of cyclin genes is turned down but in ΔICR/ΔICR cells these changes do not occur. Thus, our RNA-Seq data confirms that the LOI cells have global problems and suggests that these defects likely stem from an aberrant cell cycle. In fact, the differences in cell cycle are so widespread in mutant cells that it suggested that upstream pathways might be altered.
To pinpoint these fundamental upstream defects we compared wild type cells to ΔICR/ΔICR cells at each stage of development. That is, we directly compared wild type myoblasts to ΔICR/ΔICR myoblasts to examine the differences caused by the LOI mutation at this pre-differentiation stage (Supplementary File 1). Then we repeated this analysis in the differentiating cells. As before, we found that the cell cycle pathway was highly enriched among genes that were up regulated in LOI cells with respect to wild type. But through this analysis we also identified genes in Mitogen-activated protein kinase (MAPK) signaling pathways as aberrantly expressed in LOI myoblasts and in early myotubes (Figure 5C, Supplemental Figure S4A). Detailed examination of individual genes of the MAPK pathway showed that most of the associated genes had similar fold-differences in both myoblasts and myotubes (Figure 5D), suggesting the presence of intrinsic defects in this pathway even before differentiation began. Key results were validated by qRT-PCR using RNAs generated from independently derived cell lines (Supplemental Figure S4B).
MAPKs are protein Ser/Thr kinases that convert extracellular stimuli into a plethora of cellular responses (39). At least 14 MAPKs have been characterized in mammals and the three most extensively characterized subgroups include ERK1/ERK2, p38, and JNK kinases. p38 and JNK are most often activated by environmental stresses. During myotube differentiation, both pathways are activated and this activation is already known to be essential for muscle development (40,41). In contrast, ERK1/2 kinase activity is activated by several growth factor receptors including the insulin receptors that mediate IGF2 stimulation (42). Furthermore, ERK1/2 kinases are known to play essential roles in promoting cell proliferation in many kinds of cells. Finally, consistent with a role in muscle cells, during normal myotube differentiation, ERK1/2 activity is downregulated as myoblasts differentiate into myotubes (43,44). Altogether, these facts suggested that hyperactivation of Erk1/2 might explain the LOI phenotype. (Note that Erk1/2 are also widely known as MAPK3/1 or peptides 44/42.)
Erk2 itself is one of the RNAs in the MAPK pathway that are increased in abundance in ΔICR/ΔICR myoblasts (30% increase, FDR = 0.005) and consistent with this observation, mutant myoblasts show a modest increase in total Erk2 peptide (Figure 1D). Much more strikingly, LOI myoblasts show a large increase in the phosphorylated or activated forms of Erk2 and of Erk1 (Figure 1D). Just as in wild type cells, phosphorylated Erk1/2 levels decrease as LOI myoblasts differentiate into myotubes but since LOI myoblasts begin with elevated levels of active enzyme, they are slower in returning to the low levels typical of myotubes so that early stages of differentiation are also marked with hyperactivation of these kinases.
To test directly for functional importance of ERK1/2 kinase activation in the LOI phenotype, we treated ΔICR/ΔICR cells with PD98059, an inhibitor of the MEK1/2 kinases that activate ERK1/2 (45,46). At our doses, this treatment did not prevent differentiation of wild type myoblasts but did restore normal differentiation into myotubes in LOI cells. That is, treated ΔICR/ΔICR myoblasts differentiate into myotubes with normal morphology (Figure 6A), express myotube markers (Figure 6B), and show near wild type levels of fusion (Figure 6C). Altogether these data indicate that the extra Igf2 RNA/H19 lncRNA depletion associated with LOI results in hyperactivation of ERK1 and ERK2 which in turn prevents myotube differentiation.
Figure 6.

Inhibition of MAPK activity in LOI cells rescues defective differentiation. Wild type and ΔICR/ΔICR cells were grown in differentiation media with PD 98059 (0.1 μM) to inhibit activation of the Erk1/2 kinases. ΔICR/ΔICR myotubes show normal morphology upon treatment with PD 98059. This rescue is reflected in Myh3 and Myogenin expression and increased fusion levels. (A) DAPI (blue) and Myh3 (green) staining. (B) Immunoblot analysis of Myh3 and Myogenin proteins. H3 is a loading control. (C) Quantitation of cell fusion. ***P < 0.001.
Consistent with the idea that disruption of ERK1/2 pathways through the insulin receptor is singularly important in the LOI phenotype, we did not detect any differences in levels of either total or activated p38 between wild type and LOI cells (Supplemental Figure S5A). We were also unable to rescue myotube differentiation of ΔICR/ΔICR myoblasts with SB202190, a direct inhibitor of p38 kinase activity, or with inhibitors to Epidermal Growth Factor, Tumor Growth Factor-β, or Bone Morphogenic Protein pathways (Supplemental Figure S5B, C and data not shown).
H19 lncRNA regulates p53 levels and is required for normal myotube hypertrophy
In Figure 3E and F, we noted that reduction of Igf2 to a 1× gene dosage was highly successful in rescuing the differentiation defects in LOI myoblasts even while H19 RNA levels remained low and we therefore concluded that differentiation defects were primarily associated with extra Igf2. However, even a cursory examination of the morphology of ΔICR/Igf2- myotubes indicates that it is not equivalent to that of wild type cells: ΔICR/Igf2- cells are significantly smaller in diameter (Figure 3E) (WT = 12.2± 5.4 μm, ΔICR/Igf2- = 6.2±5.0 μm, n = 300, P < 0.001). These results indicate that H19 RNA is essential for normal muscle cell hypertrophy.
Muscle hypertrophy is an essential process that occurs during normal development and in response to muscle injury or stress. It is now established that hypertrophy is directly dependent upon activation of the AKT/mTOR pathway (47–50).
To elucidate the role of the H19 gene in hypertrophy, we analyzed myotube formation in H19ΔEx1/H19+ primary cells. H19ΔEx1is an allele of H19 that carries a 1 kb internal deletion within H19 exon 1. This allele is transcribed at high levels but the truncated RNA is of such decreased stability its steady state levels are reduced >200-fold (28). Most relevant to this study, DNA sequences encoding H19 derived miRNA-675 isoforms are not deleted and mi675-3p and mi675-5p remain expressed (Figure 7A). Thus the persistence of hypertrophy defects in H19ΔEx1/H19+ myotubes (Figure 7B, top panels) (cell diameters: WT = 12.2 ± 5.0 μm, H19ΔEx1/H19+ = 5.8 ± 3.1 μm, P < 0.001) indicates it is the H19 lncRNA that is critical.
Figure 7.

H19 regulates p53 peptide levels and activity and is required for activation of the AKT/mTOR pathway. (A) Expression of H19 lncRNA and of miRNAs 675-3p and 675-5p in H19ΔEx1/H19+ myoblasts. RNAs were assayed as described in Methods and are reported relative to their expression in wild type cells. The >200-fold change in H19lncRNA expression in LOI cells dwarfs the change in miR-675–3p and -5p expression. N = 3. (B) Maternal inheritance of H19ΔEx1 results in reduced hypertrophy that can be rescued by treatment with cAMP. Cells were cultured for 72 h in differentiation medium with 0 or with 50 μM cAMP and costained for DAPI (blue) and Myh3 (Green). Upon cAMP treatment, the H19Δex1/+ cells display hypertrophy similar to wild type cells. (C) Expression of let-7 miRNAs and of let7 and of p53 mRNA biomarkers in H19-deficient myotubes. Cells were cultured 48 h in Differentiation Medium (DM) and analyzed as described in A. N = 3. (D) Immunoblot analyses of wild type (W.T.) and of H19ΔEx1/H19+ myoblasts (DM–) and myotubes (DM+). AKT/p-AKT, p70/p-p70, and rpS6/p-rpS6 were analyzed to monitor activation of AKT/mTOR pathways. β-tubulin is included as a loading control. p53 expression stays high in H19ΔEx1/+ cells in DM, while Myogenin is downregulated. p-AKT, p-p70 and p-rpS6 are also reduced, indicating reduced activation of AKT/mTOR pathways. (E) cAMP treatment causes rapid activation of pAKT/mTOR pathways in both wild type and H19ΔEx1/H19+ myotubes. 50 μM cAMP was added for the times indicated when cells were moved to differentiation medium. (F) Cartoon summary of H19 lncRNA function in myotube hypertrophy in wild type and H19-deficient cells. p53 controls InsR/Igf1R expression, which in turn activates AKT/mTOR signaling and induces muscle hypertrophy. H19 lncRNA adds another layer of control in wild type cells, negatively regulating p53 bioavailability. In H19ΔEx1/+ cells, the lack of H19 expression results in increased p53 and lower InsR/Igf1R levels, causing reduced AKT/mTOR signaling and less hypertrophy. The addition of cAMP to these cells bypasses the H19-p53-InsR/Igf1R control, directly activating AKT/mTOR signaling and inducing hypertrophy. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Recent studies using a C2C12 model showed that H19 lncRNA can function as a molecular sponge to reduce let7 miRNA bioavailability and thereby affect AKT/mTOR signaling (13). To test if this mechanism applied to our primary cell lines, we analyzed expression of let-7 miRNAs and more importantly, of genes know to be readouts of let7 repression. Although Igf1R and InsR expression are reduced about 2-fold in H19-deficient cells, other well established targets, such as LPL and HMGA2, are unaffected (Figure 7C). Because let7 target genes are not consistently misregulated and because alternative mechanisms can readily explain misregulation of Igf1R and InsR (see below), we concluded that reduced let7 bioactivity was not a good explanation for the hypertrophy defects in H19ΔEx1/H19+ cells.
As a sidebar, we note that H19 lncRNA levels are >1400-fold reduced in C2C12 myotubes relative to H19 lncRNA levels in primary cells (Supplemental Table S3). Given this difference, it is not surprising that H19 lncRNA is playing a distinct biochemical role in the two cell types.
Two recent studies using tumor cell models suggested that H19 lncRNA can regulate cell physiology by interacting directly with p53 protein to reduce its function (14–16). According to this mechanism, the lack of H19 lncRNA in H19ΔEx1/H19+ myotubes would prevent normal modulation of p53 function. Consistent with this potential mechanism, our RNA sequencing analyses showed that p53 pathway genes were not appropriately regulated when LOI myoblasts differentiated into myotubes (Supplemental Figure S2). As already described above, Igf1R and InsR, two known targets of p53 repression (51,52) show reduced expression in H19ΔEx1/H19+ myotubes (Figure 7C). Bax is a gene often used as a reporter to monitor p53 function and its expression is increased as predicted by p53 hyperactivity (Figure 7C). To directly address the effect of H19, we performed immunoblotting assays that confirmed that p53 peptide levels are not appropriately downregulated in H19ΔEx1/H19+ myotubes (Figure 7D, top panel). Since p53 RNA levels are unaffected in mutant cells (Figure 7C), these results demonstrate that H19 regulates p53 protein and thereby regulates p53 target genes.
As predicted by decreased p53 function (53,54) and the consequent decrease in Igf1R/InsR functions (51,52), mTOR/AKT pathway activation is decreased in H19-deficient myotubes. That is, phosphorylated but not total peptide levels for AKT, p70, and p-rpS6 are all reduced in H19ΔEx1/H19+ myotubes (Figure 7D). It is well established that cAMP treatment can circumvent Igf1R/InsR receptor kinases to directly stimulate mTOR/pAKT (55). Here we confirm that this applies to our primary cell models: cAMP rapidly activates mTOR/pAKT signaling pathways in both wild type and H19-deficient cells (Figure 7E). Most importantly, cAMP treatment restores normal hypertrophy in H19ΔEx1/H19+ myotubes (Figure 7B, bottom panels). Altogether, these results indicate that H19 lncRNA works through p53 to activate AKT/mTOR and thereby promote hypertrophy (Figure 7F).
DISCUSSION
Imprinted genes are not randomly scattered throughout the genome but are organized into discrete clusters where monoallelic expression is dependent upon shared regulatory regions called Imprinting Control Regions (ICRs). Disruptions in imprinting by genetic or epigenetic mutations at ICRs will therefore almost invariably result in disruption of normal expression patterns in multiple genes. Thus LOI diseases are likely to be multigenic and complex in etiology and defined model systems such as those described here that allow single gene effects to be evaluated in isolation and in combination will be required to elucidate the separate and the synergistic effects of gene misexpression.
LOI at the IGF2/H19 locus is associated with two distinct syndromes based on parental origin of the defects. Defects on the maternal chromosome can lead to ectopic expression of maternal IGF2 and reduced H19 mRNA and are associated with BWS and with several pediatric cancers including rhabdosarcoma and Wilms’ tumor (20,56). In this study, we analyze in vitro differentiation of wild type and LOI primary myoblasts to identify the downstream molecular effects of maternal LOI. We identify phenotypes that are distinct effects of Igf2 overexpression and of H19 depletion but also identify genetic interactions including both synergy and suppression (Figure 8).
Figure 8.

Interactions between H19 and Igf2 regulate muscle cell development. Imprinted expression of H19 and Igf2 genes is determined by the H19ICR. Disruptions in ICR function on the maternal chromosome result in biallelic expression of Igf2 and reduced expression of H19 while disruptions on the paternal chromosome result in reduced expression of Igf2 and biallelic expression of H19. H19 lncRNA directly represses Igf2 RNA levels (63). In addition, H19 is required for normal activation of the AKT/mTOR signaling pathways because H19 lncRNA modulates p53 levels and activity and therefore reduces p53-mediated repression of InsR and Igf1R. Igf2 mRNA encodes IGF2 peptide which activates MAPK signaling through the InsR and Igf1R receptor kinases. MAPK (or Erk1/2) signaling is normally downregulated as myoblasts differentiate into myotubes but excessive IGF2 in LOI cells results in hyperactivation of Erk1/2 and prevents normal differentiation. In addition, IGF2 activates AKT/mTOR signaling. In LOI myoblasts, the loss of H19 and the extra IGF seem to cancel out so that AKT signaling is relatively normal.
LOI myoblasts already have significantly altered transcriptomes which results in an inability to differentiate into myotubes (Figure 1). This differentiation defect is primarily associated with overexpression of Igf2 and can be reversed by a second-site genetic mutation to restore monoallelic expression of Igf2 or by pharmacologic inhibition of IGF2 receptor kinases but not by restoring H19 expression (Figures 2–4). Analyses of transcriptomes in wild type and mutant cells suggested a critical role for cell cycle regulation and specifically for the MAPK pathway (Figure 5). In fact, Erk1/2 kinases are hyperactivated in LOI myoblasts and differentiation is restored in mutant cells by inhibiting activity of the MEK1/2 kinases that phosphorylate the Erk1/2 peptides (Figure 6).
While restoration of H19 cannot restore normal differentiation to LOI myoblasts, our data indicate that H19 does contribute to differentiation. In Figure 4, we see that restoring H19 to LOI cells improves both expression of myotube markers and cell fusion efficiency. Likewise, we see a reduction in expression of myotube markers when H19 is mutated in cells with undisturbed imprinting of Igf2 (See MyoG blot in Figure 7D). These results are consistent with other studies on H19 function in muscle in vivo (4,12,57). However, it is important to note that in all these models, it is difficult to ascertain a function for H19 that is truly distinct from that of Igf2 because it is well established that H19 lncRNA directly represses Igf2 (9). In agreement with this previous report, in our models we also saw that both Igf2 RNA levels and IGF2 peptide levels are increased ∼20% upon mutation of H19 (data not shown). Since the 1.8-fold increase in Igf2 due to LOI completely blocks myoblast differentiation, it is reasonable to postulate that the 20% increase might result in partial blocks. Thus the differentiation defect is best viewed as a synergistic one where loss of imprinting at the Igf2 gene is the primary problem but one that is exacerbated by loss of H19 and the consequent further increase in cellular Igf2.
Although H19 plays some role in differentiation, in our model system its primary role is to promote myotube hypertrophy. This finding is consistent with and helps explain results in recent publications describing a critical role for H19 in muscle regeneration in vivo (12,57). Our results indicate that H19 lncRNA works to reduce p53 peptide. In wild type cells, differentiation into myotubes is associated with a 60-fold increase in H19 lncRNA (Supplemental Table S1). Our data show that this increased H19 lnc RNA results in reduced p53 peptide (Figure 7D) and p53 function (Figure 7C). In H19-deficient myotubes, p53 levels and activity remain aberrantly high. The consequent reduction in AKT/mTOR signaling is likely caused by reduced activation of Igf1R and InsR since these are directly repressed by p53 (51,52). Addition of cAMP to mutant cells rescues AKT/mTOR activation (55) (Figure 7E) and hypertrophy (Figure 7B) demonstrating that insufficient activation of AKT is the direct cause of abnormal hypertrophy in H19-deficient cells described in Figure 7F.
This novel mechanism for H19 lncRNA function in regulating AKT activation is different than one proposed based on a very powerful analysis of C2C12 cell differentiation performed by Gao and colleagues (13). That study showed that H19 lncRNA regulates muscle cell physiology by reducing let7 bioavailability. However, in primary cells, let7 biomarkers are not altered in a way that is consistent with a major role for let7 miRNA (Figure 7C). In accord with the idea that H19 lncRNA biochemical function in C2C12 and in primary cell is very different, total H19 lncRNA levels are reduced >1400-fold in C2C12 myotubes while p53 levels are 8-fold higher (Supplemental Table S1). The high levels of H19 (and Igf2) in primary cells might suggest that they are a more appropriate model for understanding the role of this locus in fetal and neonatal development while C2C12 might better model adult muscle cell development and differentiation. See Raveh et al. for a review of data suggesting that H19 functions in adult and embryonic cells are very different (22).
We used the ΔEx1 deletion mutation to rule out a significant role for H19-derived miRNAs 675-3p and -5p in promoting the hypertrophy phenotype of interest in this study (Figure 8). The minimal effect of the deletion on miRNA levels was initially a surprise but made sense upon reflection. Transcription studies show that transcription of H19 lncRNA remains high in this mutation even though steady state levels are drastically reduced (28). If the miRNA processing occurs using nascent transcripts, there is no expectation that miRNA levels should be reduced by the ΔEx1 mutation. In addition, it should be noted that miRNA 675 levels are very, very low relative to H19 lncRNA levels. Thus the overwhelming portion of H19 lncRNA is not processed. Furthermore, the levels of miRNA 675 in a cell type do not correlate well with levels of H19 lncRNA. Thus H19 lncRNA does not appear to be the limiting factor in determining miRNA 675 levels in many circumstances.
Our results do not contradict other studies that have suggested an important role of mi675. In fact, this allele should prove very useful to many investigators seeking to distinguish the separate roles of H19 miRNA and lncRNA.
The regulation of AKT/mTOR pathways is an example of mutual suppression by the two transcriptional defects associated with the LOI mutation. As described above, activation of AKT can occur through the Igf1R and InsR receptors and is therefore responsive to IGF2 peptide so that high levels of IGF2 lead to super activation of AKT (7,58). However, as just discussed above, loss of H19 results in decreased activation of AKT. In LOI cells, these two effects seem to balance out since phosphorylated AKT, rpS6, and p70 are all present at wild type levels (data not shown). Thus by affecting both genes in opposite ways, LOI eliminates the AKT/mTOR activation phenotypes associated with misexpression of Igf2 or of H19 alone (Figure 8).
LOI diseases are complicated in several ways and it has often been difficult to understand their broad phenotypes that can vary significantly from patient to patient. Recent work from the Bartolomei and Raj labs showed that within a single patient, LOI can be stochastic from cell to cell and this variability likely contributes to the wide spectrum of these disorders (59). Here, we highlight a second complexity, that LOI diseases are invariably multigenic and that altered expression of the affected genes will have individual and interactive effects that are likely to vary in importance from cell type to cell type. It is now clear that Igf2 and H19 each play important roles in muscle development and in muscle regeneration in response to injury. In this study, we use a loss of imprinting model to further elucidate the molecular mechanisms for function of these two genes and especially to understand unique defects associated with misregulation of these genes that play both opposing and supporting roles in regulating key signaling pathways including MAPK and AKT/mTOR (Figure 8). We specifically analyzed the implications of these disruptions in muscle cell development and differentiation but note that these pathways are of importance in key developmental events in other cell types. Finally, we characterize a novel allele of H19 that will be invaluable in distinguishing the phenotypes associated with the mi675 and with the 2.3 kb H19 lncRNA.
DATA AVAILABILITY
RNA sequencing data were deposited in the NCBI Gene Expression Omnibus (GEO) under Series accession number GSE89136.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the Molecular Genomics Core at the NICHD and the NIH Intramural Sequencing Center for assistance during our sequencing experiments. The authors have no financial conflicts of interest to report.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Division of Intramural Research of the Eunice Kennedy Shriver National Institute of Child Health and Human Development [ZIA HD001804]. Funding for open access charge: National Insitutes of Health [ZIA HD001804].
Conflict of interest statement. None declared.
REFERENCES
- 1. Barlow D., Bartolomei M.. Genomic imprinting in mammals. Cold Spring Harbor Perspect. Biol. 2014; 6:a018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaffer C.R., Srivastava M., Park K., Ives E., Hsieh S., Batlle J., Grinberg A., Huang S.P., Pfeifer K.. A transcriptional insulator at the imprinted H19/Igf2 Locus. Genes Dev. 2000; 14:1908–1919. [PMC free article] [PubMed] [Google Scholar]
- 3. Thorvaldsen J.L., Duran K.L., Bartolomei M.S.. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998; 12:3693–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borensztein M., Monnier P., Court F., Louault Y., Ripoche M., Tiret L., Yao Z., Tapscott S., Forne T., Montarras D. et al. . Myod and H19-Igf2 locus interactions are required for diaphragm formation in the mouse. Development. 2013; 140:1231–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rotwein P. Insulin-like growth factor action and skeletal muscle growth, an in vivo perspective. Growth Horm. IGF Res. 2003; 13:303–305. [DOI] [PubMed] [Google Scholar]
- 6. Wilson E., Hsieh M., Rotwein P.. Autocrine growth factor signaling by insulin-like growth factor-II mediates MyoD-stimulated myocyte maturation. J. Biol. Chem. 2003; 278:41109–41113. [DOI] [PubMed] [Google Scholar]
- 7. Wilson E., Rotwein P.. Control of MyoD function during initiation of muscle differentiation. J. Biol. Chem. 2006; 281:29962–29971. [DOI] [PubMed] [Google Scholar]
- 8. Woelfle J., Chia D., Massart-Schlesigner M., Moyano P., Rotwein P.. Molecular physiology, pathology, and regulation of the growth hormone/insulin-like growth factor-I system. Pediatr. Nephrol. 2005; 20:295–302. [DOI] [PubMed] [Google Scholar]
- 9. Gabory A., Jammes H., Dandolo L.. The H19 locus: role of an imprinted non-coding RNA in growth and development. BioEssays. 2010; 32:473–480. [DOI] [PubMed] [Google Scholar]
- 10. Cai X., Cullen B.. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA. 2007; 13:313–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Keniry A., Oxley D., Monnier P., Kyba M., Dandolo L., Smits G., Reik W.. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and IgfR1. Nat. Cell Biol. 2012; 14:659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dey B., Pfeifer K., Dutta A.. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5- to promote skeletal muscle differentiation and regeneration. Genes Dev. 2014; 28:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gao Y., Wu F., Zhou J., Yan L., Jurczak M., Hui-Young L., Yang L., Mueller M., Xiao-Bo Z., Dandolo L. et al. . The H19/let-7 double negative feedback loop contributes to glucose metabolism in muscle cells. Nucleic Acids Res. 2014; 42:13799–13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yang F., Bi J., Xue X., Zheng L., Zhi K., Hua J., Fang G.. Up-regulated long non-coding RNA H19 contributes to proliferationof gastric cancer cells. FEBS J. 2012; 279:3159–3165. [DOI] [PubMed] [Google Scholar]
- 15. Zhang L., Zhou Y., Huang T., Cheng A.S.I., Yu J., Kang W., To K.F.. The interplay of lncRNA-H19 and its binding partners in physiological process and gastric carcinogenesis. Int. J. Mol. Sci. 2017; 18:E450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hadji F., Boulanger M., Guay S., Gaudreault N., Amellah S., Mkanne G., Bouchareb R., Marchand J., Nsaibia M., Guauque-Olarte S. et al. . Altered methylation of long noncoding RNA H19 in calcific aortic valve disease promotes mineralization by silencing NOTCH1. Circulation. 2016; 134:1848–1862. [DOI] [PubMed] [Google Scholar]
- 17. Barsyte-Lovejoy D., Lau S., Boutros P., Khosravi F., Jurisica I., Andrulis I., Tsao M., Penn L.. The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 2006; 66:5330–5337. [DOI] [PubMed] [Google Scholar]
- 18. Matouk I., DeGroot N., Mezan S., Ayesh S., Abu-Iail R., Hochberg A., Galun E.. The H19 non-coding RNA is essential for human tumor growth. PLoS ONE. 2007; 2:e845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yoshimizu T., Miroglio A., Ripoche M., Gabory A., Vernucci M., Riccio A., Colnot S., Godard C., Terris B., Jammes H. et al. . The H19 locus acts in vivo as a tumour repressor. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:12417–12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hao Y., Crenshaw T., Moulton T., Newcomb E., Tycko B.. Tumour-suppressor activity of H19 RNA. Nature. 1993; 365:764–767. [DOI] [PubMed] [Google Scholar]
- 21. Berteaux N., Lottin S., Monte D., Pinte S., Quatannens B., Coll J., Hondermarck H., Curgy J., Dugimont T., Adriaenssens E.. H19 mRNA-like noncoding RNA promoter promotes breast cancer cell proliferation through positive contol by E2F1. J. Biol. Chem. 2005; 280:29625–29636. [DOI] [PubMed] [Google Scholar]
- 22. Raveh E., Matouk I., Gilon M., Hochberg A.. The H19 long non-coding RNA in cancer initiation, progression, and metastasis – a proposed unifying theory. Mol. Cancer. 2015; 14:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacob K., Robinson W., Lefebvre L.. Beckwith-Wiedemann and Silver-Russell syndromes: opposite developmental imbalances in imprinted regulators of placental function and embryonic growth. Clin. Genet. 2013; 84:326–334. [DOI] [PubMed] [Google Scholar]
- 24. Caspary T., Cleary M., CC B., Guan X.-J., Tilghman S.. Multiple mechanisms regulate imprinting of the distal chromosome 7 gene cluster. Mol. Cell Biol. 1998; 18:3466–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reik W., Brown K.W., Schneid H., Le Bouc Y., Bickmore W., Maher E.R.. Imprinting mutations in the Beckwith-Wiedemann syndrome suggested by an altered imprinting pattern in the IGF2-H19 domain. Hum. Mol. Genet. 1995; 4:2379–2385. [DOI] [PubMed] [Google Scholar]
- 26. Sun F.L., Dean W.L., Kelsey G., Allen N.D., Reik W.. Transactivation of Igf2 in a mouse model of Beckwith-Wiedemann syndrome. Nature. 1997; 389:809–815. [DOI] [PubMed] [Google Scholar]
- 27. Srivastava M., Hsieh S., Grinberg A., Williams-Simon L., Huang S.-P., Pfeifer K.. H19 and Igf2 monoallelic expression is regulated in two distinct ways by a shared cis acting element. Genes Dev. 2000; 14:1186–1195. [PMC free article] [PubMed] [Google Scholar]
- 28. Srivastava M., Frolova E., Rottinghaus B., Boe S., Grinberg A., Lee E., Lover P., Pfeifer K.. Imprint control element mediated secondary methylation imprints at the Igf2/H19 locus. JBC. 2003; 278:5977–5983. [DOI] [PubMed] [Google Scholar]
- 29. Leighton P.A., Ingram R.S., Eggenschwiler J., Efstratiadis A., Tilghman S.M.. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature. 1995; 375:34–39. [DOI] [PubMed] [Google Scholar]
- 30. DeChiara T.M., Robertson E.J., Efstratiadis A.. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991; 64:849–859. [DOI] [PubMed] [Google Scholar]
- 31. Bois P., Grosveld G.. FKHR (FOX01a) is required for myotube fusion of primary mouse myoblasts. EMBO J. 2003; 22:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. von Maltzahn J., Bentzinger C.F., Rudnicki M.A.. Wnt7a-Fzd7 signalling directly activates the Akt/mTOR anabolic growth pathway in skeletal muscle. Nat. Cell Biol. 2011; 14:186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Love M., Huber W., Anders S.. Moderated estimation of fold-change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Huang da W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 36. Huang da W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eun B., Sampley M., Good A., Gebert C., Pfeifer K.. Promoter cross-talk via a shared enhancer explains paternally biased expression of Nctc1 at the Igf2/H19/Nctc1 imprinted locus. Nucleic Acids Res. 2013; 42:817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carboni J., Wittman M., Yang Z., Lee F., Greer A., Hurlburt W., Hillerman S., Cao C., Cantor G., Dell-John J. et al. . BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009; 8:3341–3349. [DOI] [PubMed] [Google Scholar]
- 39. Cargnello M., Roux P.. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011; 75:50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Keren A., Tamir Y., Bengal E.. The p38 MAPK signaling pathway: a major regulator of skeletal muscle development. Mol. Cell Endocrinol. 2006; 252:224–230. [DOI] [PubMed] [Google Scholar]
- 41. Khurana A., Dey C.. Involvement of c-Jun N-terminal kinase activities in skeletal muscle differentiation. J. Muscle Res. Cell Motility. 2004; 25:645–655. [DOI] [PubMed] [Google Scholar]
- 42. Boulton T., Yancopoulos G., Gregory J., Slaughter C., Moomaw C., Hsu J., Cobb M.. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science. 1990; 249:64–67. [DOI] [PubMed] [Google Scholar]
- 43. Bennett A., Tonks N.. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Science. 1997; 278:1288–1291. [DOI] [PubMed] [Google Scholar]
- 44. Khurana A., Dey C.. Subtype specific roles of mitogen activated protein kinases in L6E9 skeletal muscle cell differentiation. Mol. Cell Biochem. 2002; 238:27–39. [DOI] [PubMed] [Google Scholar]
- 45. Alessi D., Cuenda A., Cohen P., Dudley D., Saltiel A.. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J. Biol. Chem. 1995; 270:274889–274894. [DOI] [PubMed] [Google Scholar]
- 46. Dudley D., Pang L., Decker S., Bridges A., Saltiel A.. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:7686–7689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bodine S., Stitt T., Gonzalez M., Kline W., Stover G., Bauerlein R., Zlotchenko E., Scrimgeour A., Lawrence J., Glass D. et al. . Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001; 3:1014–1019. [DOI] [PubMed] [Google Scholar]
- 48. Glass D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Biochem. Cell Biol. 2005; 37:1974–1984. [DOI] [PubMed] [Google Scholar]
- 49. Le Grand F., Jones A., Seale V., Scime A., Rudnicki M.A.. Wnt7a activates the planar cell polarity pathway to drive the symmetric expansion of satellite stem cells. Cell Stem Cell. 2009; 4:535–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rochat A., Fernandez A., Vandromme M., Moles J., Bouschet T., Carnac G., Lamb N.J.. Insulin and wnt1 pathways cooperate to induce reserve cell activation in differentiation and myotube hypertrophy. Mol. Biol. Cell. 2004; 15:4544–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Werner H., Karnieli E., Rauscher F., LeRoith D.. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:8318–8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Webster N., Resnik J., Reichart D., Strauss B., Haas M., Seely B.. Repression of the insulin receptor promoter by the tumor suppressor gene product p53: a possible mechanism for receptor overexpression in breast cancer. Cancer Res. 1996; 56:2781–2788. [PubMed] [Google Scholar]
- 53. Feng Z., Zhang H., Levine A., Jin S.. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Levine A., Feng Z., Mak T., You H., Jin S.. Coordination and communication between the p53 and IGF1-AKT-TOR signal transduction pathways. Genes Dev. 2006; 20:267–275. [DOI] [PubMed] [Google Scholar]
- 55. Berdeaux R., Stewart R.. cAMP signaling in skeletal muscle adaptation: hypertrophy, metabolism, and regeneration. Am. J. Physiol. Endocrinol. Metab. 2012; 2012:E1–E17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feinberg A., Tycko B.. The history of cancer epigenetics. Nat. Rev. Cancer. 2004; 4:143–153. [DOI] [PubMed] [Google Scholar]
- 57. Martinet C., Monnier P., Louault Y., Benard M., Gabory A., Dandolo L.. H19 controls reactivation of the imprinted gene network during muscle regeneration. Development. 2016; 143:962–971. [DOI] [PubMed] [Google Scholar]
- 58. Yoon M., Chen J.. PLD regulated myoblast differentiation through the mTOR-IGF2 pathway. J. Cell Sci. 2008; 121:282–289. [DOI] [PubMed] [Google Scholar]
- 59. Ginart P., Kalish J., Jiang C., Yu A., Bartolomei M., Raj A.. Visualizing allele-specific expression in single cells reveals epigenetic mosaicism in an H19 loss-of-imprinting mutant. Genes Dev. 2016; 30:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iberaabdullah F., Vigneau S., Bartolomei M.. Genomic imprinting mechanisms in mammals. Mutat. Res. 2008; 647:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Murrell A. Setting up and maintaining differential insulators and boundaries for genomic imprinting. Biochem. Cell Biol. 2011; 89:469–478. [DOI] [PubMed] [Google Scholar]
- 62. Gebert C., Rong Q., Jeong S., Iben J., Pfeifer K.. H19ICR mediated transcriptional silencing does not require target promoter methylation. Biochem. Biophys. Res.. 2016; 476:121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gabory A., Ripoche M.A., Le Digarcher A., Watrin F., Ziyyat A., Forné T., Jammes H., Ainscough J.F., Surani M.A., Journot L. et al. . H19 acts as a trans regulator of the imprinted gene network controlling growth in mice. Development. 2009; 136:3413–3421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data were deposited in the NCBI Gene Expression Omnibus (GEO) under Series accession number GSE89136.