


Abstract
Long non-coding RNAs (lncRNAs) make up a significant portion of non-coding RNAs and are involved in a variety of biological processes. Accurate identification/annotation of lncRNAs is the primary step for gaining deeper insights into their functions. In this study, we report a novel tool, PLncPRO, for prediction of lncRNAs in plants using transcriptome data. PLncPRO is based on machine learning and uses random forest algorithm to classify coding and long non-coding transcripts. PLncPRO has better prediction accuracy as compared to other existing tools and is particularly well-suited for plants. We developed consensus models for dicots and monocots to facilitate prediction of lncRNAs in non-model/orphan plants. The performance of PLncPRO was quite better with vertebrate transcriptome data as well. Using PLncPRO, we discovered 3714 and 3457 high-confidence lncRNAs in rice and chickpea, respectively, under drought or salinity stress conditions. We investigated different characteristics and differential expression under drought/salinity stress conditions, and validated lncRNAs via RT-qPCR. Overall, we developed a new tool for the prediction of lncRNAs in plants and showed its utility via identification of lncRNAs in rice and chickpea.
INTRODUCTION
Recent studies have shown that transcription is pervasive and non-coding transcripts make up a significant portion of an organism's transcriptome (1,2). Even though non-coding RNAs (ncRNAs), are not translated into proteins, they play an important role in regulating expression of other coding transcripts (3,4). Long non-coding RNAs (lncRNAs) do not code for proteins and have minimum transcript length of 200 bp. Although many lncRNAs in plants and animals have been identified, their exact role in biological processes remains largely unknown (5,6). Some of recently discovered lncRNAs have been shown to perform regulatory roles in crucial biological processes via association with chromatin modifying complex, histone modifications and target mimicry (4,7,8).
The accurate prediction of lncRNAs remains one of the major problems in plants. Therefore, we need accurate and efficient computational methods to predict lncRNAs in plants to further investigate their roles. Next generation RNA-sequencing (RNA-seq) methods have given an opportunity to study the whole transcriptome of any organism and these data can be used to identify potential lncRNAs. Efficient methods/tools are needed to analyse the high-throughput data for discovery of lncRNAs. There are quite a few already existing tools, which can analyse the transcript sequences to assess their coding potential, such as coding potential calculator (CPC), coding-non-coding index (CNCI), coding-potential assessment tool (CPAT), lncRScan-SVM and predictor of lncRNAs and messenger RNAs based on an improved k-mer scheme (PLEK) (9–13). CPC, CNCI, lncRScan-SVM and PLEK use support vector machine (SVM) model to calculate the coding potential of transcripts (9,11–13), whereas CPAT uses linear regression model to discriminate coding and non-coding transcripts (10). These methods use different features of transcript sequences to build a classification model and have been shown to work well in animal datasets. However, to the best of our knowledge, there is no dedicated tool available, which can predict the lncRNAs with high accuracy in plants. It has been shown that models built with human data to predict ncRNAs or lncRNAs may work well with other closely related vertebrate species, but perform poorly with plant data (13). Thus, we need to build specific models for classification of coding and long non-coding transcripts in plants.
Here, we have developed a new tool, PLncPRO (Plant Long Non-Coding RNA Prediction by Random fOrest), to discover lncRNAs in plants via classifying coding and long non-coding transcripts. We have built classification models for different plant species to predict lncRNAs and tested our tool on various available datasets. We benchmarked the accuracy of PLncPRO vis-a-vis other existing programs using known set of lncRNAs in different plants. Furthermore, we developed dicot and monocot specific models to facilitate more robust discovery of lncRNAs in plants. We assessed the lncRNA prediction accuracy of PLncPRO in human and mouse also. In addition, we demonstrated the application of our tool via prediction of novel lncRNAs in two crop plants, chickpea and rice, under abiotic stress conditions. The availability of plant-specific lncRNA prediction tool and identification of stress-responsive lncRNAs will provide a useful resource for understanding lncRNA biology in plants.
MATERIALS AND METHODS
Data description
We defined prediction of coding RNAs and lncRNAs as a binary classification problem and labelled coding sequences as positive and long non-coding sequences as negative. The lncRNA sequences for ten plant species (Amborella trichopoda, Arabidopsis thaliana, Chlamydomonas reinhardtii, Glycine max, Oryza sativa, Physcomitrella patens, Selaginella moellendorffii, Solanum tuberosum, Vitis vinifera and Zea mays) were downloaded from CANTATAdb as negative examples. CANTATAdb is an online repository for computationally identified plant lncRNAs, predicted from RNA-seq libraries (14). The positive examples, i.e. protein coding transcripts (pct) were downloaded from Phytozome v11 (15) for all the plant species. We split the data randomly into disjoint train and test sets for each species (Supplementary Table S1). The validation of constructed training models of dicots and monocots was performed using published lncRNA datasets for different plant species (Supplementary Table S2). We downloaded protein coding and lncRNA sequences from GENCODE for human (v24), and mouse (vM9) to test our tool on vertebrates (16). One training and two test datasets were generated for human and mouse by randomly splitting the data (Supplementary Table S3).
Feature extraction
To construct a random forest model, we extracted a 71 dimensional feature vector for each sequence in a given labelled (positive and negative) dataset. These features were selected based on the previous knowledge about coding and non-coding transcripts (17,18). We used two programs, Framefinder (19) and BLASTX (20) to extract some of the features.
To estimate quality of an open reading frame (ORF) present in a transcript, we used Framefinder software and extracted ORF score and coverage, designated as FF-score and ORF coverage, respectively.
Next, we used BLASTX program to find if the transcripts have a significant similarity to any known protein coding sequence in SWISS-PROT database (21). We extracted four relevant features by parsing the BLASTX output.
-
Number of hits, N: For a true protein coding sequence, a higher number of hits are expected. Thus, N is a good feature to distinguish between true coding and non-coding transcripts.
Many query sequences, however, can show random insignificant matches to a blast database. To handle this problem, we defined three more features, which can be helpful in discriminating between a true protein coding and a non-coding transcript sequence.
-
Significance Score, S: For a good quality hit, we expect a lower e-value. Thus, we defined S for a given query sequence as,
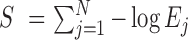 , where Ej is the e-value for jth match for the given query. If a given query has lower value of S, it is more likely to be a true protein coding sequence.
, where Ej is the e-value for jth match for the given query. If a given query has lower value of S, it is more likely to be a true protein coding sequence. -
Total bit score, B: The bit score in BLAST is a normalized measure derived from raw alignment score, which indicates the quality of alignment. We defined total bit score B for a given query sequence as,
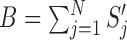 , where
, where 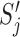 is the bit score for jth match for the given query. For a good quality hit, higher B is expected.
is the bit score for jth match for the given query. For a good quality hit, higher B is expected. -
Frame entropy, F: It captures information about how the hits are distributed in different reading frames. A hit just by chance is likely to be distributed among all possible frames. Frame entropy is based on Shannon's entropy function (22) and is defined as,
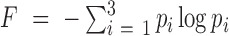 , where pi is the probability that hits are in the ith frame, calculated as
, where pi is the probability that hits are in the ith frame, calculated as 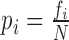 , where fi is the frequency of hits in ith frame and N is the total number of hits. When a query shows hit in one frame only, frame entropy is minimum (zero). If hits are equally distributed in all the three frames, frame entropy is maximum (1.5849).
, where fi is the frequency of hits in ith frame and N is the total number of hits. When a query shows hit in one frame only, frame entropy is minimum (zero). If hits are equally distributed in all the three frames, frame entropy is maximum (1.5849).
In addition, we extracted frequencies of each of 64 possible trimers of the four nucleotides from the input sequences to capture codon usage bias and length of the transcript.
Model construction
For a given training set, we constructed a random forest model with 1000 trees after extracting the above mentioned features from the input sequences. Random forest is an ensemble learning method for classification and regression, which constructs a number of decision trees from the training data and reports the output after computing results from all the trees in the forest (22). For classification, it outputs the mode of the classes produced by individual decision tree. Random forests can efficiently handle large datasets with many variables and also provide relative feature ranking, which can be helpful in interpreting the model and data. In addition, random forest algorithm does not require a separate cross-validation set, as it reports out-of-bag (OOB) score (an estimate of generalization error) which is equivalent to cross-validation error. This is particularly useful, when enough data is not available to make a separate cross-validation set. Our program builds five random forest models and chooses the one with the highest OOB score. The model built using the training data is then used for classification of unlabelled data. To measure the performance of classification, we used following metrics:
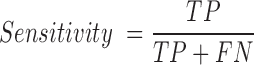 |
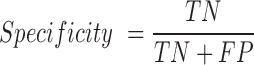 |
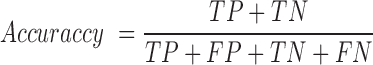 |
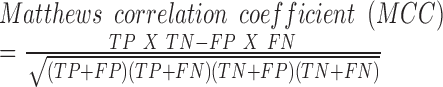 |
where
TP: Number of true positives (coding transcripts correctly classified as coding)
TN: Number of true negatives (long non-coding transcripts classified as lncRNA)
FP: Number of false positives (long non-coding transcripts incorrectly classified as coding RNA)
FN: Number of false negatives (coding transcripts incorrectly classified as lncRNA)
Program description
PLncPRO takes a set of training transcript sequences as input, builds a random forest classifier and outputs the random forest model in a file. Once a model has been trained, it can be used to classify unknown transcript sequences and label them as potential coding or long non-coding sequence (1 or 0, respectively). Figure 1 shows the schematic workflow of PLncPRO algorithm. PLncPRO is multi-threaded and memory-efficient standalone program, which can run on any environment with python. We have empirically set some default computing parameters, such as number of trees and minimum length of lncRNA for PLncPRO, which can be changed by the user as per requirement.
Figure 1.

Schematic workflow of PLncPRO. The top panel shows building of the model using training data. Two files (training data) containing protein coding (mRNA) and long non-coding transcripts (lncRNA) are given as input. PLncPRO extracts features and build a random forest model using these features. The bottom panel shows classification of transcripts by PLncPRO. Test data containing transcripts are given as input and PLncPRO classifies transcripts using the training model with probability score. The transcripts with non-coding probability score ≥0.5 (default parameter) are identified as the lncRNAs.
Prediction of lncRNAs in rice and chickpea and differential expression analysis
We used transcriptome data of rice (O. sativa) and chickpea (Cicer arietinum) under abiotic stress conditions (drought and salinity) from previous reports (24,25) to identify lncRNAs. Rice transcriptome represented assembly generated using RNA-seq data from control, desiccation and salinity stress conditions (nine samples) for three rice cultivars, IR64 (stress-sensitive), Nagina 22 (N22, drought-tolerant) and Pokkali (salinity-tolerant) at the seedling stage (24). Chickpea transcriptome represented assembly generated using RNA-seq data of 30 samples representing control, drought/salinity stress for four chickpea genotypes ICC 1882 (drought-sensitive), ICC 4958 (drought-tolerant), ICCV 2 (salinity-sensitive) and JG 62 (salinity-tolerant) at vegetative/early reproductive and late reproductive stages (25). The description of samples has been summarized in Supplementary Table S4. A total of 42 064 and 33 179 transcripts from rice and chickpea, respectively, representing the longest isoform of each transcript were used for prediction of lncRNAs. LncRNAs in chickpea and rice were identified using two different probability cut-offs (≥0.5 and ≥0.8) with other parameters set at default. The novel lncRNAs in rice and chickpea were identified via comparing the lncRNAs identified in this study with already published reports (8,14,26–29) using CD-Hit tool with c ≥ 0.8 (30). Various characteristics of lncRNAs and mRNAs were determined using custom scripts. The statistical significance of differences in various characteristics between lncRNAs and mRNAs was calculated using Wilcoxon rank sum test in R (31).
The high-confidence lncRNAs (probability cut-off ≥ 0.8) identified in rice and chickpea were analyzed for differential expression via Cuffdiff. The lncRNAs were considered as differentially expressed, if the absolute value of log2 fold change between the two given samples was ≥1 with P-value ≤0.05.
Reverse transcriptase-quantitative polymerase chain reaction (RT-qPCR) validation
To validate the expression profiles, 13 and 21, differentially expressed lncRNAs under different stress conditions/genotype/developmental stage, from chickpea and rice, respectively, were selected. Primers were designed using Primer Express (version 3.0) software (Applied Biosystems, Foster City, CA, USA). The details of the primer sequences used in this study are provided in Supplementary Table S5. RT-qPCR analysis of lncRNAs was performed using total RNA isolated from different samples as described previously (24,25). For each sample, at least two biological replicates and three technical replicates for each biological replicate were used. Most suitable house-keeping genes, Elongation factor 1 alpha (EF1α) and Ubiquitin (UBQ5), were used to normalize the transcript level for chickpea and rice, respectively (32,33). Fold change in the transcript level was determined using the standard 2−ΔΔCT method. Log2 values of average fold change (average of two biological replicates and three technical replicates of each biological replicate) for each lncRNA were used to determine correlation between RT-qPCR and RNA-seq data.
RESULTS
Development of models for prediction of lncRNAs in plants
We developed PLncPRO tool for prediction of lncRNAs in plants based on various sequence features extracted from the training data using random forest algorithm. The features [FF score, ORF coverage, BLASTX results (N, S, B and F), trimer frequency and transcript length] used in PLncPRO represented a combination of the features used in other available lncRNA prediction tools (Supplementary Table S6). We constructed training models for different plant species using a set of known lncRNAs and coding RNAs (Supplementary Table S1). The OOB score of the models for all the plant species generated by PLncPRO was high (≥0.90 except for S. tuberosum; Supplementary Table S7), indicating high cross-validation accuracy. The species-specific models trained on data from individual plant species were used for prediction of lncRNAs on the test datasets of all the plant species. In terms of accuracy, the species-specific models performed best for the same species (Table 1). The accuracy of prediction ranged from 83% (S. tuberosum) to 97.9% (A. trichopoda) with high sensitivity (81–98%) and specificity (85 – 98%) (Supplementary Table S7). Next, we analyzed the Mathew's correlation coefficient (MCC) and area under ROC curve to assess the quality of prediction. Both MCC and area under ROC curve were also high for all the plant models analysed (Supplementary Table S7). However, we found that the prediction accuracy with S. tuberosum model was in the range of 50–83% (Table 1). We suspect that the data (known lncRNAs and/or coding RNAs) for S. tuberosum were not good, which resulted in low performance of S. tuberosum model. The performance of PLncPRO on test dataset of C. reinhardtii was poor using other plant models. However, using C. reinhardtii specific model, PLncPRO achieved an accuracy of 97%. This may be because of significant difference between the sequence makeup of C. reinhardtii (single-celled algae) and other plants (higher plants).
Table 1. Performance (percentage accuracy) of PLncPRO on different plant test data sets.
| Model used | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Plant species | At | Gm | Vv | St | Os | Zm | Amt | Pp | Sm | Cr |
| A. thaliana (At) | 95.70 | 94.54 | 93.72 | 91.36 | 89.72 | 93.72 | 62.61 | 92.03 | 84.69 | 54.05 |
| G. max (Gm) | 91.52 | 92.30 | 91.22 | 86.44 | 83.75 | 92.02 | 61.55 | 89.88 | 82.36 | 53.58 |
| V. vinifera (Vv) | 93.03 | 92.62 | 93.64 | 86.34 | 87.15 | 92.35 | 66.94 | 90.79 | 85.93 | 56.89 |
| S. tuberosum (St) | 73.98 | 74.07 | 74.13 | 83.05 | 74.51 | 74.69 | 67.48 | 72.13 | 72.03 | 50.48 |
| O. sativa (Os) | 87.41 | 88.79 | 90.09 | 86.07 | 93.93 | 88.45 | 81.30 | 84.32 | 83.36 | 71.05 |
| Z. mays (Zm) | 86.09 | 88.62 | 86.61 | 85.24 | 88.49 | 91.22 | 80.95 | 87.06 | 76.66 | 66.55 |
| A. trichopoda (Amt) | 70.20 | 75.43 | 75.82 | 89.11 | 88.62 | 77.81 | 97.92 | 73.71 | 75.56 | 64.48 |
| P. patens (Pp) | 87.26 | 86.55 | 87.84 | 83.03 | 79.76 | 87.53 | 62.58 | 90.74 | 74.86 | 60.73 |
| S. moellendorffii (Sm) | 85.22 | 87.63 | 86.53 | 72.91 | 88.57 | 82.52 | 74.70 | 74.04 | 93.70 | 74.70 |
| C. reinhardtii (Cr) | 75.49 | 74.42 | 75.37 | 56.21 | 73.76 | 67.66 | 54.28 | 74.25 | 80.31 | 96.78 |
Prediction accuracy using the model of same species is highlighted in bold.
While building the model, we assessed relative feature ranking for each plant as determined by the random forest algorithm. ORF coverage and FF-score were found to be the most important features for all the higher plants, whereas ORF coverage ranked lower in C. reinhardtii and FF-score ranked lower in A. trichopoda (Figure 2A and B; Supplementary Figure S1). The relative ranking of BLASTX features and trimer's frequency in different plant models showed clear distinction between monocot and dicot plant models. The trimers ranked higher in most of the monocot plants (A. trichopoda, O. sativa, S. tuberosum and Z. mays), whereas BLASTX and entropy based features ranked high in majority of dicot plants (A. thaliana, G. max, P. patens, and V. vinifera) (Figure 2A and B; Supplementary Figure S1).
Figure 2.

Feature ranking and prediction accuracy of PLncPRO using species-specific models. (A and B) Relative feature ranking in A. thaliana (A) and O. sativa (B) obtained using PLncPRO. The relative feature ranking for other plants is given in Supplementary Figure S1. (C) ROC curves for test sets of different plants using species-specific models. The accuracy of prediction for each model has been given.
Consensus models for dicots and monocots
Each species-specific model performed best on its own data and on closely related species (Table 1). For example, the prediction of lncRNAs in A. thaliana using its own model revealed an accuracy of 95.70%, while using G. max model an accuracy of 94.54% was achieved. However, it was quite less (89.72%) using O. sativa model (Table 1). Similarly, for O. sativa, using its own model, an accuracy of 93.93% was obtained, however, using A. thaliana and G. max models an accuracy of 87.41% and 88.79%, respectively, was achieved (Table 1). These observations support the fact that sequence make-up of coding and lncRNAs is more conserved in closely related species. With lack of proper annotation for many plant species, the model constructed from closely related model organisms can be used for lncRNA prediction. Therefore, we constructed consensus models for dicots and monocots for prediction of lncRNAs. We constructed a single model for monocots (using training data of O. sativa and Z. mays) and dicots (using training data of A. thaliana, G. max and V. vinifera). We did not include S. tuberosum in dicot model, as its accuracy within the dicot group was relatively poor. The OOB score of the consensus dicot and monocot models was 0.94 and 0.92, respectively, which indicated high cross-validation accuracy. The relative feature rankings obtained using dicot and monocot models showed some differences. For example, trimers ranked lower in dicot model (Figure 3A) as compared to the monocot model (Figure 3B). However, FF-score and ORF coverage ranked top in both the models. We used dicot and monocot models on the test data of all the plant species and found that accuracy of dicot and monocot models was sufficiently high for lncRNA prediction in dicot and monocot plants, respectively (Figure 3C and D, Supplementary Table S8). The performance of dicot and monocot models was comparable to the species-specific models on dicot and monocot plants, respectively (Table 1; Supplementary Table S8). For example, in A. thaliana, plant specific model gave an accuracy of 95.7%, while consensus dicot model achieved an accuracy of 95.56%. Likewise, the plant specific model showed an accuracy of 93.93% in O. sativa, while consensus monocot model gave an accuracy of 93.75% (Table 1; Supplementary Table S8).
Figure 3.

Feature ranking and prediction accuracy of dicot and monocot specific models using PLncPRO. (A and B) Relative feature ranking using dicot (A), and monocot model (B). (C and D) The ROC curves of all plant test datasets using dicot model (C) and monocot model (D). The accuracy of prediction for each model has also been given.
Further, we tested prediction accuracy of consensus dicot and monocot models on published lncRNA datasets of A. thaliana, S. lycopersicum, P. trichocarpa, Gossypium spp., Cucumis sativus, Medicago truncatula, Z. mays and O. sativa (Figure 4). These lncRNAs have been identified using different methods/pipelines (8,28,34–40). The results showed that the consensus monocot and dicot models exhibited reasonably high accuracy of lncRNAs prediction in these plants ranging from 83% to 93.5% for monocots, and 91.6% to 99.5% for dicots (Figure 4). The higher accuracy achieved with these datasets demonstrated that PLncPRO can predict lncRNAs in previously unseen data of different plant species with high accuracy and the models generated did not overfit the training data.
Figure 4.

Performance of PLncPRO on published lncRNA datasets using monocot and dicot specific models for lncRNA discovery. The datasets were obtained from the previous studies as given in Supplementary Table S2.
Performance comparison with other existing tools
We compared the performance of PLncPRO with other available lncRNA prediction tools, including PLEK, CPAT, CNCI and CPC, using all the plant test sets. For a fair comparison, we trained models for all plants using the same tool (wherever possible) with the datasets used in this study and used these models for species-specific prediction. For CNCI, we used its plant model and CPC was run using its web server. The performance of each tool in terms of accuracy along with sensitivity and specificity was compared (Table 2; Supplementary Table S9). We compared the MCC and area under ROC curve also to assess the quality of classification in each tool (Supplementary Table S9). In terms of prediction accuracy, PLncPRO performed better (90.7–97.9% accuracy) for 8 out of 10 plants. For O. sativa and S. moellendorffii, CNCI performed better by achieving marginally higher (0.18% and 1.38%, respectively) prediction accuracy than PLncPRO. Interestingly, we noted that PLncPRO had higher sensitivity and specificity as compared to other tools, which indicated good quality classification by reducing false positives and false negatives. However, CPC and CPAT showed higher sensitivity with low specificity, and CNCI had lower sensitivity with high specificity. MCC and area under ROC curve were also significantly higher for PLncPRO as compared to other tools. Overall, it is evident that PLncPRO performed much better showing higher prediction accuracy as compared to other existing tools.
Table 2. Performance comparison (percentage accuracy) of PLncPRO with existing tools on different plant data sets.
| Plant species | PLEK | CPAT | CNCI | CPC | PLncPRO |
|---|---|---|---|---|---|
| A. thaliana | 87.92 | 91.27 | 89.11 | 86.33 | 95.70 |
| G. max | 83.27 | 82.58 | 85.13 | 83.11 | 92.30 |
| V. vinifera | 85.62 | 85.45 | 86.08 | 88.06 | 93.64 |
| S. tuberosum | 74.85 | 76.06 | 81.87 | 60.52 | 83.05 |
| O. sativa | 87.24 | 89.81 | 94.11 | 83.78 | 93.93 |
| Z. mays | 84.10 | 80.93 | 82.18 | 77.88 | 91.22 |
| A. trichopoda | 88.72 | 97.53 | 83.36 | 58.51 | 97.92 |
| P. patens | 78.71 | 72.14 | 84.82 | 76.00 | 90.74 |
| S. moellendorffii | 86.94 | 87.47 | 95.08 | 83.90 | 93.70 |
| C. reinhardtii | 88.13 | 72.32 | 96.41 | 80.10 | 96.78 |
The highest accuracy achieved for each species is highlighted in bold.
Prediction of lncRNAs in human and mouse
To assess the performance of PLncPRO in vertebrates, we built human (OOB score: 0.95) and mouse (OOB score: 0.96) classification models. We plotted top 10 feature rankings for human and mouse models as computed by the random forest algorithm using PLncPRO (Figure 5A and B). FF-score was the most important feature in both the models. BLASTX based features also ranked high in both the models. Further, the performance of PLncPRO on human and mouse data was compared with the existing tools (Table 3). PLncPRO exhibited much better prediction accuracy in all the test cases for both human and mouse data sets (Table 3, Supplementary Table S10). The accuracy of PLncPRO ranged from 93.7% to 94.6%, while only lncRScan-SVM achieved an accuracy of >90% among other tools. The accuracy ranging from 75.7% to 89.6% was achieved for other tools, including PLEK, CPAT, CNCI and CPC. Apart from accuracy, we noticed that PLncPRO achieved highest sensitivity (>92%) along with high specificity (>96%) in all the test cases (Supplementary Table S10), which demonstrated that PLncPRO is a better quality classifier for vertebrates as well. However, all other tools showed high specificity as compared to the sensitivity on the test datasets. This implies that they may be biased towards classification of lncRNAs and may produce false negatives i.e. classify coding sequences wrongly as lncRNAs. Both MCC and area under ROC curve were also significantly higher in PLncPRO as compared to other tools (Supplementary Table S10). Overall, these results demonstrated PLncPRO as more accurate and reliable tool to differentiate between coding and long non-coding transcripts in human and mouse as well.
Figure 5.

Feature ranking and prediction accuracy of PLncPRO using human and mouse models. (A and B) Relative feature ranking in human (A) and mouse (B) models. (C) ROC curves for test datasets of human and mouse using human and mouse models, respectively. The accuracy of prediction for each test data has also been given.
Table 3. Performance comparison (percentage accuracy) of PLncPRO with existing tools on human and mouse data sets.
| Data set | PLEK | CPAT | CNCI | CPC | lncRScan | PLncPRO |
|---|---|---|---|---|---|---|
| Human Test A | 84.41 | 87.91 | 87.79 | 81.05 | 92.86 | 94.34 |
| Human Test B | 84.62 | 87.37 | 87.64 | 80.46 | 92.50 | 93.72 |
| Mouse Test A | 76.27 | 89.51 | 89.36 | 82.98 | 92.25 | 94.57 |
| Mouse Test B | 75.68 | 89.40 | 89.61 | 83.26 | 91.86 | 94.63 |
The highest accuracy achieved for each dataset is highlighted in bold.
Prediction of novel lncRNAs in chickpea and rice
To demonstrate the applicability of our tool, we predicted novel lncRNAs in rice and chickpea using the consensus monocot and dicot models, respectively. The transcriptomes of rice (42 064) and chickpea (33 179) were taken as input in PLncPRO with default parameters (probability cut-off ≥ 0.5 and minimum length of 200 bp) to annotate lncRNAs. A total of 7345 (17.5%) and 4969 (15%) transcripts in rice and chickpea, respectively, were annotated as lncRNAs. The sequences of all the lncRNAs identified in rice and chickpea are available at http://ccbb.jnu.ac.in/plncpro. A comparison with the previous studies on lncRNAs in rice (8,14,26–28) and chickpea (29) revealed the identification of at least 4815 (65%) and 4384 (87%) novel lncRNAs in rice and chickpea, respectively, in our study. We investigated various genomic features of the identified lncRNAs and compared them with mRNAs. The median length of mRNAs was significantly longer than lncRNAs in chickpea (1272 bp compared to 788 bp, P-value < 2.2e–16) and rice (1378 bp compared to 1171 bp, P-value < 0.05) (Figure 6). The median length of previously reported lncRNAs was also found to be shorter as compared to mRNAs (8,41). In contrast, the median exon length of lncRNA transcripts was longer as compared to mRNAs in chickpea (301 bp compared to 155 bp, P-value < 2.2e–16) and rice (256 bp compared to 169 bp, P-value < 2.2e–16), respectively (Figure 6). The lncRNAs were mostly single exonic. In rice, 44% of lncRNAs were single exonic (Figure 6A) with average of three exons per transcript as compared to 4.5 exons per transcript in protein coding transcripts. In chickpea, 55% of lncRNAs were single exonic (Figure 6B) with average of 2.13 exons per transcript. These results are consistent with previous studies of lncRNAs in rice and chickpea (8,29). It has been suggested that lesser number of exons in lncRNAs may result in higher exon length (38). The AU content distinctively differentiated lncRNAs from mRNAs in both rice and chickpea (Figure 6); as lncRNAs are considered to be AU rich, while mRNAs as GC rich. Similar observations have been reported in other plant species too (8,29,38).
Figure 6.

Comparative analysis of different characteristics of predicted lncRNAs and mRNAs in rice and chickpea. (A and B). The relative distribution of transcript length, exon length, AU content and exon number per transcript in lncRNAs and mRNAs in rice (A) and chickpea (B).
Differential expression analysis of lncRNAs under stress conditions
Recent studies have revealed the role of lncRNAs in different biological contexts including response to different stresses in plants (39,42–45). To reveal the potential role of lncRNAs in abiotic stress responses, we analysed their differential expression under drought and/or salinity stress conditions in rice and chickpea. We extracted expression level (FPKM) of all the lncRNAs from rice and chickpea in different samples using Cufflinks (Supplementary Tables S11 and S12), and compared with that of mRNAs (Supplementary Figure S2). The expression level of lncRNAs in most of the samples (except few in chickpea) was found to be less as compared to mRNAs. However, this difference was much higher in rice samples as compared to the chickpea samples (Supplementary Figure S2). The lncRNAs were found to be expressed at low levels as compared to mRNAs in previous studies too (8,39,42). Further, we analysed RNA-seq data of stress-sensitive and tolerant of rice cultivars (IR64, N22 and Pokkali) at the seedling stage and chickpea genotypes (ICC 1882, ICC 4958, ICCV 2 and JG 62) at the vegetative (Veg), early reproductive (ER) or late reproductive stages (Rep/LR) under control and stress conditions (Supplementary Table S4) to identify differentially expressed lncRNAs under drought and/or salinity stress. For differential expression analysis, we analyzed a total of 3714 and 3457 high-confidence lncRNAs (probability cut-off of ≥ 0.8) in rice and chickpea, respectively. The analysis revealed differential expression of a large number of lncRNAs under stress conditions as compared to control condition. In chickpea, a total of 1503 (43.5%) high-confidence lncRNAs exhibited differential expression under at least one of the stress condition/developmental stage/genotype analysed. This fraction is similar to the fraction (43.1%) of total gene loci that showed differential expression under these conditions in our previous study (24). The number of lncRNAs showing differential expression varied under different stress conditions/developmental stage/genotype (Figure 7A; Supplementary Figure S3A). Drought-sensitive chickpea genotype (ICC 1882) exhibited least number of differentially expressed lncRNAs (126) at ER stage (Dsen-ER-DS), while largest number (455) of lncRNAs were differentially expressed in salinity-tolerant genotype (JG 62) at the reproductive stage (Stol-Rep-SS) (Supplementary Figure S3A). A larger fraction of lncRNAs were found to be down-regulated on exposure to stress in all the samples (except salinity-sensitive genotype). Further, we identified at least 827 lncRNAs that showed differential expression between stress-sensitive and stress-tolerant chickpea genotypes under control conditions. A larger number of lncRNAs depicted differential expression in drought-tolerant genotype at early reproductive stage, whereas a larger number of lncRNAs were differentially expressed in salinity-tolerant genotype at the late reproductive stage (Supplementary Figure S3B). These results are consistent with our previous study based on the whole transcriptome analysis (25).
Figure 7.

Differential expression analysis and validation of lncRNAs. (A and B) Venn diagram showing differentially expressed lncRNAs in different samples in chickpea (A), and rice (B). (C) Scatter-plot showing correlation of expression pattern of lncRNAs obtained from RNA-seq and qRT-PCR analyses for chickpea and rice. Each data point in the scatter plot shows average log2 fold change (from at least two biological replicates) of the selected lncRNAs under the stress condition(s) in different genotypes/cultivars/developmental stage via RT-qPCR and RNA-seq.
Further, a major fraction (38.1–76.5%) of lncRNAs was differentially expressed in a specific sample (genotype/developmental stage/stress condition) (Figure 7A). A significantly larger fraction of the differentially expressed lncRNAs in the drought-tolerant genotype at the ER stage (73.8%) and salinity-tolerant genotype at the Rep stage (76.5%) revealed specific differential expression (Figure 7A; Supplementary Figure S3). These results suggested developmental stage/genotype specific role of lncRNAs in determining stress response in chickpea.
In rice, a total of 1010 (27.2%) lncRNAs exhibited differential expression under at least one of the stress condition/cultivar analysed. This fraction is quite higher than the fraction (17.4%) of total transcripts that showed differential expression under these conditions in the previous study (23), suggesting an important role of lncRNAs in regulation of abiotic stress responses in rice. The stress-sensitive cultivar (IR64) revealed the differential expression of at least 249 and 63 lncRNAs under desiccation and salinity stress, respectively (Figure 7B; Supplementary Figure S4A). Drought-tolerant cultivar (N22) exhibited 227 differentially expressed lncRNAs and 81 lncRNAs were differentially expressed in salinity-tolerant cultivar (Pokkali) after exposure to desiccation and salinity stress, respectively (Figure 7B, Supplementary Figure S4A). A larger fraction of lncRNAs were up-regulated upon exposure to stress in the rice cultivars (Supplementary Figure S4a). Further, a larger number of differentially expressed lncRNAs were identified under drought stress as compared to salinity stress. Likewise, a larger fraction of lncRNAs showed differential expression in IR64/N22 (408) as compared to IR64/Pokkali (227) under control conditions too (Supplementary Figure S4B). Here also, we found a large fraction (38.1–73.3%) of lncRNAs to be differentially expressed in cultivar/stress condition specific manner (Figure 7B). The overall result suggested that differentially expressed lncRNAs might be involved in regulating stress/genotype/developmental stage specific responses in rice and chickpea.
Validation of differential expression of lncRNAs by RT-qPCR
The differential expression of randomly selected 21 and 13 lncRNAs from rice and chickpea, respectively, were validated in different genotypes/cultivars under control and stress (drought and/or salinity) conditions by RT-qPCR. The results showed high correlation between RNA-seq and RT-qPCR analysis. Even though correlation between RNA-seq and RT-qPCR was variable (0.43–1.00 for rice and 0.45–1.00 for chickpea) for individual lncRNAs (Supplementary Figure S5), overall correlation was 0.70 and 0.72 in chickpea and rice, respectively (Figure 7C). At least 17 and nine lncRNAs in rice and chickpea, respectively, showed correlation of >0.70 between RNA-seq and RT-qPCR analysis. The results confirmed the lncRNA prediction via demonstrating their expression and differential expression as predicted from the RNA-seq data.
DISCUSSION
Recent studies have discovered various roles of lncRNAs in diverse cellular and developmental processes including transcription regulation (4,7). Researchers are interested to study lncRNAs in great detail to understand the underlying mechanism by which lncRNAs perform their functions. However, the accurate identification of lncRNAs still remains a challenging task especially in plants (13,39,46). In this study, we have developed a new tool, PLncPRO, for lncRNA discovery in plants and vertebrates. This tool performed much better than other available lncRNA prediction tools, including CPC, CNCI, CPAT, PLEK and lncRScan-SVM (9–13). We found that other tools performed inconsistently with plant datasets, whereas PLncPRO was consistent with an accuracy of over 90% in most of the plants analyzed. We built consensus models for dicot and monocot species and found that they perform quite well with dicot and monocot plants, respectively. These consensus models will be helpful in the prediction of lncRNAs for any poorly annotated and/or newly sequenced dicot/monocot species. PLncPRO performed better on human and mouse data as well with an accuracy of >94% in all test sets, whereas only lncRScan-SVM was able to achieve an accuracy of 93% among other tools. However, lncRScan-SVM showed high specificity and low sensitivity, which implies that it may predict many false negatives, whereas PLncPRO demonstrated both high specificity and sensitivity to give an overall good quality classification.
The performance of any machine learning based classifier depends on the training data and features of the data (23). Several features, such as sequence homology, ORF quality and codon usage bias, individually and/or in combination, have been shown to be informative to predict coding potential of transcript sequences (17,18). PLncPRO extracts all these features together to facilitate better prediction accuracy for lncRNAs. In addition, the implemented random forest algorithm also offers several advantages, including robustness, suitability for both numerical and categorical data, non-requirement of cross-validation set, and high sensitivity and specificity. This is well supported by the consistent performance of PLncPRO, i.e. balanced sensitivity and specificity, on all the test datasets analysed for plants and vertebrates in our study. PLncRPO revealed high accuracy with the published lncRNA datasets from different plant species, which were identified using different programs and pipelines (8,28,34–40) This implied that models generated by PLncPRO did not overfit the training data and were able to learn the general features of lncRNAs.
We demonstrated the applicability of PLncPRO via identification of the lncRNAs from rice and chickpea transcriptome data related to abiotic stress response. A significant fraction of these lncRNAs were found to be differentially expressed under drought/salinity stress conditions. Recent studies suggested the roles of lncRNAs in abiotic stress responses in various plants, such as Arabidopsis, maize, cotton, Medicago and Populus (36,39,41,42,47). In Arabidopsis, npc536 lncRNA was found to be involved in salt stress and increased primary and secondary root growth (41). In cotton, lincRNAs, XLOC_063105 and XLOC_115463, were involved in drought stress response by regulating neighbouring genes (42). Several lncRNAs were identified in leaf and root of Medicago and were predicted to regulate expression of stress-responsive genes involved in oxidation/reduction reaction, transcription, energy synthesis and signal transduction (39). The lncRNAs have been shown to regulate drought stress response by regulating miRNAs either acting as precursors of miRNAs in maize (47) or acting as target mimics of miRNAs in Populus (36). Several genotype/cultivar-specific and stress-specific lncRNAs were identified in both rice and chickpea. Several genes encoding for regulatory proteins and metabolic pathways were found to be regulated under drought/salinity stresses in rice and chickpea (23,24). The differential coexpression of a large number of lncRNAs and mRNAs under stress conditions suggest their interaction and complex regulatory network, which needs to be studied further. This study provides a comprehensive list of lncRNAs expressed under drought and/or salinity stress, which can serve as a very useful resource to analyse their exact function in abiotic stress responses and other biological process.
In this study, we reported a novel tool, PLncPRO, which allows the discovery of lncRNAs and is particularly well-suited for plants. PLncPRO outperforms other existing tools for lncRNA prediction on several parameters. We demonstrated the utility/accuracy of this tool with human and mouse data as well. We discovered novel lncRNAs in chickpea and rice expressed under drought and salinity stresses and revealed their differential expression to identify the stress-responsive lncRNAs. With the huge amount of RNA-seq data being generated, PLncPRO will be a useful tool for discovery of lncRNAs especially in plants. Further. The lncRNAs identified in rice and chickpea will provide a resource to elucidate their exact function in abiotic stress responses in future studies.
DATA AVAILABILITY
PLncPRO has been implemented using python 2.7.11. It uses python libraries like scikit-learn, bio-python and sci-py. We used scikit-learn's random forest to implement our method. PLncPRO is freely available for non-commercial purposes and can be downloaded from http://ccbb.jnu.ac.in/plncpro/. A comprehensive user manual describing the usage guidelines is also available along with the software.
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
FUNDING
Department of Science & Technology, Government of India, under the Promotion of University Research and Scientific Excellence (PURSE) grant (Phase II) scheme to the Jawaharlal Nehru University, New Delhi and Department of Biotechnology, Government of India under Centre of Excellence in Bioinformatics to the School of Computational & Integrative Sciences. Funding for open access charge: Department of Science & Technology, Government of India, under the Promotion of University Research and Scientific Excellence (PURSE) grant (Phase II) scheme.
Conflict of interest statement. None declared.
REFERENCES
- 1. Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F. et al. . Landscape of transcription in human cells. Nature. 2012; 489:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wade J.T., Grainger D.C.. Pervasive transcription: illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014; 12:647–653. [DOI] [PubMed] [Google Scholar]
- 3. Cech T.R., Steitz J.A.. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014; 157:77–94. [DOI] [PubMed] [Google Scholar]
- 4. Rinn J.L., Chang H.Y.. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012; 81:145–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu X., Hao L., Li D., Zhu L., Hu S.. Long non-coding RNAs and their biological roles in plants. Genomics. Proteomics Bioinformatics. 2015; 13:137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quinodoz S., Guttman M.. Long noncoding RNAs: an emerging link between gene regulation and nuclear organization. Trends Cell Biol. 2014; 24:651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fatica A., Bozzoni I.. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 2014; 15:7–21. [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y.C., Liao J.Y., Li Z.Y., Yu Y., Zhang J.P., Li Q.F., Qu L.H., Shu W.S., Chen Y.Q.. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014; 15:512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kong L., Zhang Y., Ye Z.Q., Liu X.Q., Zhao S.Q., Wei L., Gao G.. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007; 35:W345–W349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W.. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013; 41:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sun L., Liu H., Zhang L., Meng J.. lncRScan-SVM: a tool for predicting long non-coding RNAs using support vector machine. PLoS One. 2015; 10:e0139654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li A., Zhang J., Zhou Z.. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics. 2014; 15:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., Liu Y., Chen R., Zhao Y.. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013; 41:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Szcześniak M.W., Rosikiewicz W., Makałowska I.. CANTATAdb: a collection of plant long non-coding RNAs. Plant Cell Physiol. 2016; 57:e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N. et al. . Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012; 40:1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al. . GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fickett J.W., Tung C.S.. Assessment of protein coding measures. Nucleic Acids Res. 1992; 20:6441–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dinger M.E., Pang K.C., Mercer T.R., Mattick J.S.. Differentiating protein-coding and noncoding RNA: Challenges and ambiguities. PLoS Comput. Biol. 2008; 4:e1000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Slater G. Algorithms for the analysis of expressed sequence tags. 2000; University of Cambridge, U.K.
- 20. Altschul S. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O’Donovan C., Martin M.J., Gattiker A., Gasteiger E., Bairoch A., Apweiler R., O’donovan C., Martin M.J., Gattiker A., Gasteiger E. et al. . High-quality protein knowledge resource: SWISS-PROT and TrEMBL. Brief. Bioinform. 2002; 3:275–284. [DOI] [PubMed] [Google Scholar]
- 22. Shannon C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948; 27:379–423. [Google Scholar]
- 23. Breiman L. Random forests. Mach. Learn. 2001; 45:5–32. [Google Scholar]
- 24. Shankar R., Bhattacharjee A., Jain M.. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Sci. Rep. 2016; 6:23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garg R., Shankar R., Thakkar B., Kudapa H., Krishnamurthy L., Mantri N., Varshney R.K., Bhatia S., Jain M.. Transcriptome analyses reveal genotype and developmental stage-specific molecular responses to drought and salinity stresses in chickpea. Sci. Rep. 2016; 6:19228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yi X., Zhang Z., Ling Y., Xu W., Su Z.. PNRD: a plant non-coding RNA database. Nucleic Acids Res. 2015; 43:D982–D989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paytuví Gallart A., Hermoso Pulido A., Anzar Martínez de Lagrán I., Sanseverino W., Aiese Cigliano R.. GREENC: a Wiki-based database of plant lncRNAs. Nucleic Acids Res. 2016; 44:D1161–D1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H., Niu Q.-W., Wu H.-W., Liu J., Ye J., Yu N., Chua N.-H.. Analysis of non-coding transcriptome in rice and maize uncovers roles of conserved lncRNAs associated with agriculture traits. Plant J. 2015; 84:404–416. [DOI] [PubMed] [Google Scholar]
- 29. Khemka N., Singh V.K., Garg R., Jain M.. Genome-wide analysis of long intergenic non-coding RNAs in chickpea and their potential role in flower development. Sci. Rep. 2016; 6:33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fu L., Niu B., Zhu Z., Wu S., Li W.. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012; 28:3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ihaka R., Gentleman R.. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 1996; 5:299–314. [Google Scholar]
- 32. Garg R., Sahoo A., Tyagi A.K., Jain M.. Validation of internal control genes for quantitative gene expression studies in chickpea (Cicer arietinum L.). Biochem. Biophys. Res. Commun. 2010; 396:283–288. [DOI] [PubMed] [Google Scholar]
- 33. Jain M., Nijhawan A., Tyagi A.K., Khurana J.P.. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem. Biophys. Res. Commun. 2006; 345:646–651. [DOI] [PubMed] [Google Scholar]
- 34. Di C., Yuan J., Wu Y., Li J., Lin H., Hu L., Zhang T., Qi Y., Gerstein M.B., Guo Y. et al. . Characterization of stress-responsive lncRNAs in Arabidopsis thaliana by integrating expression, epigenetic and structural features. Plant J. 2014; 80:848–861. [DOI] [PubMed] [Google Scholar]
- 35. Wang J., Yu W., Yang Y., Li X., Chen T., Liu T., Ma N., Yang X., Liu R., Zhang B.. Genome-wide analysis of tomato long non-coding RNAs and identification as endogenous target mimic for microRNA in response to TYLCV infection. Sci. Rep. 2015; 5:16946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shuai P., Liang D., Tang S., Zhang Z., Ye C.Y., Su Y., Xia X., Yin W.. Genome-wide identification and functional prediction of novel and drought-responsive lincRNAs in Populus trichocarpa. J. Exp. Bot. 2014; 65:4975–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang M., Yuan D., Tu L., Gao W., He Y., Hu H., Wang P., Liu N., Lindsey K., Zhang X.. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015; 207:1181–1197. [DOI] [PubMed] [Google Scholar]
- 38. Hao Z., Fan C., Cheng T., Su Y., Wei Q., Li G.. Genome-wide identification, characterization and evolutionary analysis of long intergenic noncoding RNAs in cucumber. PLoS One. 2015; 10:e0121800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang T.Z., Liu M., Zhao M.G., Chen R., Zhang W.H.. Identification and characterization of long non-coding RNAs involved in osmotic and salt stress in Medicago truncatula using genome-wide high-throughput sequencing. BMC Plant Biol. 2015; 15:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li L., Eichten S.R., Shimizu R., Petsch K., Yeh C.T., Wu W., Chettoor A.M., Givan S.A., Cole R.A., Fowler J.E. et al. . Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014; 15:R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Amor B. Ben, Wirth S., Merchan F., Laporte P., D’Aubenton-Carafa Y., Hirsch J., Maizel A., Mallory A., Lucas A., Deragon J.M. et al. . Novel long non-protein coding RNAs involved in Arabidopsis differentiation and stress responses. Genome Res. 2009; 19:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lu X., Chen X., Mu M., Wang J., Wang X., Wang D., Yin Z., Fan W., Wang S., Guo L. et al. . Genome-wide analysis of long non-coding RNAs and their responses to drought stress in cotton (Gossypium hirsutum L.). PLoS One. 2016; 11:e0156723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu J., Jung C., Xu J., Wang H., Deng S., Bernad L., Arenas-Huertero C., Chua N. H.. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell. 2012; 24:4333–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Franco Zorrilla J.M., Valli A., Todesco M., Mateos I., Puga M.I., Rubio-Somoza I., Leyva A., Weigel D., García J.A., Paz-Ares J.. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007; 39:1033–1037. [DOI] [PubMed] [Google Scholar]
- 45. Heo J.B., Sung S.. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2011; 331:76–79. [DOI] [PubMed] [Google Scholar]
- 46. St. Laurent G., Wahlestedt C., Kapranov P.. The landscape of long noncoding RNA classification. Trends Genet. 2015; 31:239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang W., Han Z., Guo Q., Liu Y., Zheng Y., Wu F., Jin W.. Identification of maize long non-coding RNAs responsive to drought stress. PLoS One. 2014; 9:e98958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
PLncPRO has been implemented using python 2.7.11. It uses python libraries like scikit-learn, bio-python and sci-py. We used scikit-learn's random forest to implement our method. PLncPRO is freely available for non-commercial purposes and can be downloaded from http://ccbb.jnu.ac.in/plncpro/. A comprehensive user manual describing the usage guidelines is also available along with the software.