

Abstract
Psychological stress is thought to be an important trigger of cardiovascular events, yet the involved pathways and mediators are largely unknown. Elevated systemic levels of the pro-inflammatory alarmin S100A8/A9 correlate with poor prognosis in coronary artery disease (CAD) patients. Here, we investigated the links between S100A8/A9 release and parameters of anti-inflammatory glucocorticoid secretion in two different cohorts subjected to a psychological stress test. In the first cohort of 60 CAD patients, psychological stress induced a rapid increase of circulating S100A8/A9. This rapid S100A8/A9 response strongly correlated with elevated evening saliva cortisol levels, suggesting an association with a dysregulated hypothalamic–pituitary–adrenal (HPA) axis. In the second cohort of 27 CAD patients and 28 controls, elevated S100A8/A9 levels were still detectable 24 h after stress in 40% of patients and 36% of controls, with a tendency for higher levels in patients. The sustained S100A8/A9 response was associated with a poor rapid cortisol release after stress in patients, but not in the control group. Our findings reveal for the first time that acute psychological stress induces elevated levels of S100A8/A9. We also provide hypothesis-generating evidence that dysregulated cortisol secretion in CAD patients might be associated with an exaggerated pro-inflammatory S100A8/A9 response.
Introduction
Inflammation plays a central role in the development of cardiovascular disease (CVD). Immune and inflammatory cells mediate atherogenesis and plaque destabilization, and the circulating levels of inflammatory mediators have been linked with disease progression and prognosis1. The S100 proteins A8 and A9 are alarmins belonging to the S100 calcium-binding protein family. They are expressed in neutrophils, monocytes, thrombocytes and dendritic cells but also in activated macrophages, vascular endothelial cells and fibroblasts2,3. S100A8 and S100A9 exist as homodimers, but preferably form the heterodimer S100A8/A9, also known as calprotectin. S100A8/A9 constitutes approximately 45% of all cytosolic proteins in neutrophils and about 1% in monocytes4.
S100A8/A9 is an endogenous ligand of toll-like receptor 4 (TLR4)5,6 and of the receptor for advanced glycation end-products (RAGE)7–9, present on various cell types. S100A8/A9 binding to these receptors promotes the synthesis of pro-inflammatory mediators and has been shown to be involved in the pathogenesis of acute coronary syndrome (ACS)2. Systemic levels of S100A8/A9 correlate with established markers of systemic inflammation in patients with stable coronary artery disease (CAD)10, and with the severity of CAD in type 1 and type 2 diabetic patients2. Further, S100A8/A9 is elevated in vulnerable human carotid plaques11 and systemic levels are associated with the long-term risk of CV events and CV death in middle-aged individuals without previous CVD12. In ACS patients, S100A8/A9 levels are increased in blood and at the site of coronary occlusion, and recovered thrombi from coronary vessels contain S100A8/A9 positive cells13. These findings suggest that S100A8/A9 is released locally from the ischemic myocardium and diffuses into the systemic circulation. Moreover, the magnitude of S100A8/A9 response has prognostic value, as ACS patients with persistently elevated levels of S100A8/A9 30 days after the acute event have a significantly higher risk to suffer recurrent CV events14.
Acute psychological stress triggers the release of inflammatory mediators in humans15,16 and may thereby increase the risk for CV disease17,18. Glucocorticoids regulate immune and inflammatory pathways, and are essential for dampening the inflammatory response associated with acute stress19,20. Chronic inflammation in pathological conditions, such as autoimmune diseases and depression, has been linked to impaired HPA axis function21. This is also proposed as a potential mechanism for low-grade chronic inflammation in CAD22. Compared to healthy controls, CAD patients have a flat diurnal cortisol rhythm and a blunted cortisol response to acute stress, a cortisol pattern that is associated with elevated levels of pro-inflammatory markers23. An imbalance between pro- and anti-inflammatory pathways in CAD patients is generally believed to promote disease progression and lead to worse clinical outcome24. Laboratory psychological stress tests may therefore be useful to reveal whether such an inflammatory imbalance exists.
S100A8/A9 is readily produced and stored in neutrophils and quickly released upon activation, serving as an ideal biomarker for monitoring rapid inflammatory responses. However, it has not previously been studied whether acute psychological stress triggers S100A8/A9 release. Here, we investigated the S100A8/A9 response to acute psychological stress in two different cohorts of CAD patients and related the findings to cortisol reactivity as well as to diurnal cortisol rhythm. We hypothesized that S100A8/A9 release is an integral part of the inflammatory response to stress, and that this response may be exacerbated in CAD patients with a dysregulated cortisol metabolism.
Results
Rapid S100A8/A9 response to acute psychological stress
In Study I, we measured the early changes in systemic S100A8/A9 levels induced by acute psychological stress in 60 CAD patients. The clinical and biochemical characteristics of the study group are presented in Table 1. Psychological stress induced a significant elevation in plasma S100A8/A9 measured at 20 min after test completion compared to baseline [median (IQR) 750 (615–1190) ng/mL pre-stress vs 910 (720–1452) ng/mL post-stress; p = 1.8 × 10−8] (Fig. 1). There was a strong positive association between baseline and post-stress S100A8/A9 levels (r = 0.880, p = 1.9 × 10−20) (Fig. 2a), and both correlated positively with blood neutrophil counts (r = 0.301, p = 0.019 and r = 0.302, p = 0.019, respectively). However, the relative S100A8/A9 increase expressed as percent of baseline levels did not correlate with pre-stress S100A8/A9 (r = −0.114, p = 0.385) (Fig. 2b) or with the number of circulating neutrophils (Table 2), suggesting that the individual reactivity to psychological stress was independent of the habitual S100A8/A9 levels and of the neutrophil counts in blood. We found a positive correlation between the relative S100A8/A9 increase and systolic blood pressure at 10 minutes after the test (r = 0.287, p = 0.016), suggesting that individuals with a stronger S100A8/A9 response had an impaired systolic blood pressure recovery after the stressor. There were no correlations between the S100A8/A9 response and diastolic blood pressure or heart rate at any time point.
Table 1.
Clinical and biochemical parameters of the CAD patients and controls included in studies I and II
| CAD patients Study I (N = 60) | CAD patients Study II (N = 27) | Controls Study II (N = 28) | P* Study II | |
|---|---|---|---|---|
| Age (years) | 65.1 (8.9) | 61 (6.0) | 61 (6.0) | n.s. |
| Male sex, n (%) | 51 (85.0) | 22 (81.4) | 23 (82.1) | n.s. |
| Smoking, n (%) | 5 (8.3) | 13 (48.1) | 9 (32.1) | n.s. |
| Diabetes, n (%) | 11 (18.3) | 1 (3.7) | 1 (3.6) | n.s. |
| Systolic blood pressure (mmHg) | 138 (15) | 136 (21) | 144 (16) | n.s. |
| Diastolic blood pressure (mmHg) | 79 (9) | 80 (10) | 85 (8) | 0.012 |
| Heart rate (beats/min) | 60 (10) | 64 (11) | 71 (13) | 0.041 |
| BMI (kg/m2) | 27.4 (3.3) | 27 (3) | 27 (3) | n.s. |
| Plasma lipids (mmoL/L) | ||||
| Total cholesterol | 3.76 (0.78) | 4.44 (0.78) | 5.89 (0.92) | 6.9 × 10−8 |
| LDL | 1.99 (0.55) | 2.40 (0.63) | 3.80 (0.92) | 4.6 × 10−8 |
| HDL | 1.17 (0.35) | 1.27 (0.33) | 1.38 (0.31) | n.s. |
| TG | 1.39 (0.70) | 1.72 (0.75) | 1.75 (0.91) | n.s. |
| Circulating cell populations (million/dL) | ||||
| Leukocytes | 6.6 (2.1) | 6.4 (1.3) | 6.6 (1.5) | n.s. |
| Neutrophils | 3.7 (1.4) | 3.7 (1.1) | 3.7 (1.1) | n.s. |
| Monocytes | 0.5 (0.2) | 0.7 (0.3) | 0.5 (0.2) | n.s. |
| Lymphocytes | 2.1 (0.9) | 1.9 (0.6) | 2.2 (0.6) | n.s. |
| Medication | ||||
| Statin, n (%) | 59 (98.3) | 25 (92.6) | 3 (10.7) | 3.3 × 10−10 |
| ACE/ARB, n (%) | 42 (70.0) | 13 (48.1) | 3 (10.7) | 0.003 |
| Betablocker, n (%) | 46 (76.7) | 24 (88.9) | 5 (17.9) | 9.9 × 10−8 |
| Calcium channel blockers, n (%) | 21 (35.0) | 4 (14.8) | 3 (10.7) | n.s. |
Continuous variables are presented as mean (SD).
*Comparison between CAD patients and controls in Study II. Student’s T-test was used for continuous variables. The chi-square test or Fischer’s exact test were used to compare categorical variables, as appropriate.
Figure 1.
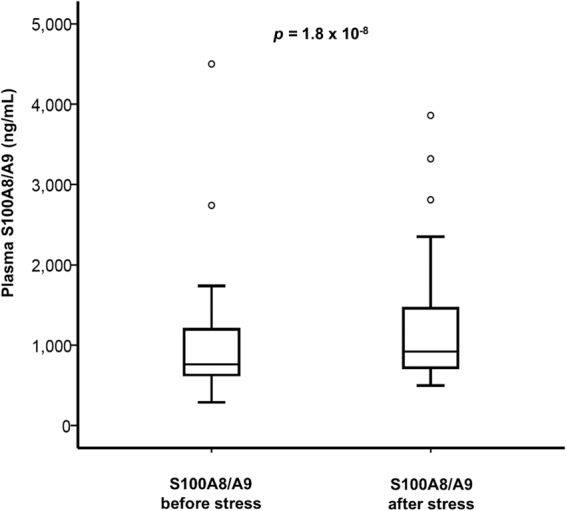
Circulating S100A8/A9 before and after acute psychological stress (Study I). Plasma S100A8/A9 increased significantly in CAD patients (n = 60) subjected to acute psychological stress. Plasma samples were collected before and at 20 minutes after the completion of the psychological stress test. The difference between the groups was calculated using the paired Wilcoxon Signed Ranks test. °Denotes outliers.
Figure 2.

Correlation between baseline S100A8/A9 and the rapid S100A8/A9 response induced by acute psychological stress (Study I). (a) Correlation between S100A8/A9 in plasma before and 20 minutes after the end of the psychological stress test. (b) Correlation between S100A8/A9 in plasma before the test and the percentage S100A8/A9 increase compared to baseline induced by acute psychological stress. The correlations were examined using the Spearman test.
Table 2.
Potential determinants of the rapid S100A8/A9 response induced by acute psychological stress in CAD patients (Study I).
| Spearman correlation | Multivariable linear regression* | |||
|---|---|---|---|---|
| r | P | Beta coefficient | P | |
| Age | 0.029 | n.s. | −0.100 | n.s. |
| Sex | — | — | 0.098 | n.s. |
| BMI | −0.148 | n.s. | −0.274 | n.s. |
| Diabetes | — | — | 0.068 | n.s. |
| Hypertension | — | — | −0.138 | n.s. |
| Smoking | — | — | −0.029 | n.s. |
| Plasma lipids | ||||
| Total cholesterol | −0.033 | n.s. | — | — |
| LDL | −0.087 | n.s. | −0.211 | n.s. |
| HDL | −0.061 | n.s. | −0.124 | n.s. |
| TG | −0.100 | n.s. | −0.050 | n.s. |
| Circulating cell populations | ||||
| Leukocytes | 0.113 | n.s. | — | — |
| Neutrophils | 0.154 | n.s. | −0.106 | n.s. |
| Monocytes | 0.048 | n.s. | 0.041 | n.s. |
| Lymphocytes | 0.011 | n.s. | −0.101 | n.s. |
| Saliva cortisol | ||||
| Morning | −0.006 | n.s. | 0.165 | n.s. |
| Evening | 0.315 | 0.016 | 0.635 | 0.004 |
| Cortisol response to stress# | 0.032 | n.s. | 0.218 | n.s. |
| Medication | ||||
| Statin | — | — | −0.049 | n.s. |
| Betablocker | — | — | 0.082 | n.s. |
| ACE/ARB | — | — | 0.205 | n.s. |
| Calcium-channel blockers | — | — | 0.038 | n.s. |
*Multivariable linear regression with stress-induced S100A8/A9 increase, expressed as percentage of baseline, as dependent variable.
#Percent cortisol increase at 20 minutes after the stress test compared to baseline.
Further, we assessed whether the rapid stress-induced increase of S100A8/A9 in CAD patients was associated with the presence of CV risk factors, blood immune cell counts and parameters of cortisol homeostasis (Table 2). In a bivariate Spearman correlation analysis, the relative S100A8/A9 increase was not associated with any of the considered CV risk factors or cell counts, and did not correlate with the rapid cortisol response to psychological stress. However, the S100A8/A9 release strongly correlated with evening saliva cortisol (Table 2). After adjustment for age, sex, BMI, diabetes, hypertension, smoking, plasma lipids, blood immune cell counts, morning saliva cortisol, cortisol response to the stress test and medication in a multivariable linear regression analysis, evening saliva cortisol remained a potent determinant of stress-induced S100A8/A9 increase (Table 2).
Sustained S100A8/A9 response to acute psychological stress
In Study II, we measured the changes in systemic S100A8/A9 levels 24 hours after the psychological stress test in 27 CAD patients and 28 healthy controls. The clinical and biochemical characteristics of the participants, previously described by Nijm et al.23 are shown in Table 1. The patients had significantly lower levels of total cholesterol and LDL cholesterol, as well as lower diastolic blood pressure and heart rate compared to controls, likely due to medication. However, both patients and controls reacted to the psychological stress test with significant increases in blood pressure and heart rate, as we have previously described in this cohort23. When all subjects were considered, there were no significant differences between patients and controls regarding the stress-induced changes in S100A8/A9 levels relative to baseline [median (IQR) 7 (−34–106)% vs 33 (−19–62)%, p = 0.960]. However, there was a large variability in the relative change in S100A8/A9 from baseline to 24 h within both groups, indicating that the S100A8/A9 response to stress was highly heterogeneous among individuals. A sustained S100A8/A9 response, defined as an increase of at least 50% at 24 h compared to baseline, was seen in 11 (40%) of the patients and 10 (36%) of the controls. Among these subjects, patients tended to have a slightly stronger S100A8/A9 response compared to controls [median (IQR) 155 (88–268)% vs 69 (57–205)%, p = 0.049].
The acute cortisol response to stress, defined as percentage cortisol increase at 20 minutes after the test relative to baseline values, was significantly lower in CAD patients compared to controls [median (IQR) 25 (0.7–31)% vs. 41 (17–88)%, p = 0.029]. The variation in cortisol levels correlated negatively with the S100A8/A9 response at 24 h after stress in CAD patients (r = −0.486, p = 0.016) (Fig. 3a), but no such correlation was present in healthy controls (r = −0.154, p = 0.442) (Fig. 3b). The data suggest that CAD patients with a poor cortisol reaction to acute psychological stress maintain a pro-inflammatory status characterized by elevated levels of S100A8/A9 that persist for at least 24 hours after the stressor.
Figure 3.

Correlation between the rapid cortisol response to psychological stress and the sustained S100A8/A9 response at 24 hours after the test (Study II). Correlation between the rapid cortisol response measured in saliva at 20 min after the psychological stress test and the S100A8/A9 increase at 24 hours after the stress test, expressed as percentage increase relative to baseline, in CAD patients (a) and healthy controls (b). The correlations were examined using the Spearman test.
Discussion
In this study we evaluated for the first time the stress-induced response of the pro-inflammatory alarmin S100A8/A9 in humans. We found that acute psychological stress induced a rapid S100A8/A9 increase in a population of CAD patients, already detectable at 20 minutes after the end of the stressor. In a second cohort of CAD patients and healthy controls, elevated S100A8/A9 levels were still present 24 h after stress in a substantial fraction of participants from both groups, indicating that the release of S100A8/A9 in response to stress is not only restricted to CAD patients. Among individuals with a sustained S100A8/A9 increase at 24 h after stress, there was a slight tendency for a higher increase in patients compared with controls, but the S100A8/A9 response was highly heterogeneous within each group. Interestingly, the magnitude of S100A8/A9 rise at 24 h correlated inversely with the ability of CAD patients to mount an adequate rapid cortisol response to stress, while no such correlation was seen in controls.
Psychological stress, both chronic and acute, has long been identified as a risk factor for CVD18. The underlying mechanisms are not completely understood but several studies have shown that stress causes potent inflammatory reactions in peripheral blood mononuclear cells and elevated levels of pro-inflammatory cytokines15,22,25. CAD patients are often characterized by the presence of low-grade systemic inflammation and it has also been reported that they have a more potent increase of inflammatory mediators in response to stress compared to healthy individuals23,26. Additionally, patients who had reported emotional distress immediately before the onset of myocardial infarction were characterized by a significant increase of monocyte-platelet and neutrophil-platelet aggregation in response to a mental stress test, compared with patients who had not reported any emotional trigger27. The patients with an identifiable emotional trigger also presented delayed recovery of systolic blood pressure after stress27, which is in line with our findings of a correlation between a strong S100A8/A9 response and elevated blood pressure levels immediately after stress.
S100A8/A9 is a biomarker of innate immune activation and an active mediator in the pathogenesis of CVD2. Experimental and clinical studies have identified S100A8/A9 as an important pro-atherogenic factor11,28,29, and elevated S100A8/A9 levels have been correlated with increased risk for coronary events and mortality in healthy middle-aged individuals12. In myocardial infarction patients, S100A8/A9 is elevated both systemically and at the site of coronary occlusion13 and high S100A8/A9 levels correlate with poor prognosis14. In experimental studies, S100A8/A9 has been shown to promote cardiomyocyte dysfunction and post-ischemic heart failure9,30. Moreover, S100A8/A9 expression induced by angiotensin II in myocardial fibroblasts induces cardiac inflammation, fibrosis, hypertrophy and dysfunction31. Taken together, the data indicate that elevated S100A8/A9 levels contribute to accelerated CAD development and a worse prognosis once an acute coronary event has occurred. Thus, it is reasonable to speculate that CAD patients with repeated and sustained elevations of S100A8/A9 in response to daily stressors are at higher risk to suffer recurrent coronary events and heart failure. Adequately powered studies, designed for long-term follow-up of CAD patients, are required to address this hypothesis.
Interestingly, we found no correlation between the rapid S100A8/A9 response to psychological stress and the baseline S100A8/A9 concentration or blood neutrophil counts. It is unlikely that the rapid S100A8/A9 increase detected at 20 minutes after the stress test is due to a fast rise in circulating neutrophil numbers within this short time frame. Instead, we speculate that the magnitude of S100A8/A9 increase is determined by neutrophil responsiveness, i.e. the ability of neutrophils to become activated and release S100A8/A9 in response to psychological stress. In support of this hypothesis, previous studies have shown that neutrophils from CAD patients are more prone to ex-vivo stimulation compared to neutrophils from controls32. However, we cannot exclude the contribution of other cell types to the rise in S100A8/A9 observed in our study.
In Study I, we found a significant relationship between the rapid release of S100A8/A9 in response to stress and evening cortisol levels. Cortisol regulates the secretion of inflammatory mediators by inhibiting pro-inflammatory cytokine production and by stimulating the production of anti-inflammatory cytokines in leukocytes33–35. In line with this, S100A8/A9 has been shown to be negatively regulated by glucocorticoids36. Cortisol reactivity is a vital adaptive physiological mechanism that enables the organism to cope with stressful situations19. A flat cortisol diurnal slope due to high evening levels is generally considered to be indicative of long-term overstimulation of the HPA axis, which gradually becomes hypo-reactive in response to acute stimuli37. A flat diurnal cortisol slope has been associated with a higher prevalence of coronary calcifications38, and with increased risk of coronary events39. Moreover, in a large population-based study, high evening cortisol levels and a flatter diurnal cortisol slope were clearly linked to increased risk for cardiovascular disease mortality, independently of traditional risk factors40. In patients with established CVD, a flat diurnal cortisol slope has been shown to predict worse outcome after coronary by-pass surgery41, and elevated evening cortisol has also been associated with increased mortality in patients with systolic heart failure42. In Study I, we did not find any correlation between the rapid cortisol and S100A8/A9 responses at 20 min after stress, suggesting that the anti-inflammatory effects of cortisol do not occur fast enough to counteract the rapid S100A8/A9 increase. It is known that cortisol mainly acts at gene and transcription factor level21, and thus requires longer time (4–24 h) to reach an adequate effect. Interestingly, in study II we found a significant inverse relationship between the acute cortisol response to stress and the sustained 24 h increase in S100A8/A9 levels in CAD patients, but not in the control group. However, the study population was small and data should be interpreted with caution. It is important to note that the results indicate an association but do not allow us to draw any conclusions about a causal relationship between a dysfunctional HPA axis and S100A8/A9 release in response to stress. The potential inability of a blunted cortisol response to inhibit the pro-inflammatory S100A8/A9 rise will have to be tested in experimental models.
Our study has some important limitations that need mentioning. We were unable to assess whether there were any differences in the rapid S100A8/A9 response to psychological stress between CAD patients and healthy controls, as Study I only included patients. Similarly, we could not conclude whether the participants with a higher early increase in S100A8/A9 also maintained higher S100A8/A9 levels over a 24 h period, since early and late measurements of stress-induced S100A8/A9 were performed in different experimental settings. In the second study, we cannot exclude that the disparities in baseline medication, diastolic blood pressure, heart rate and plasma lipids between patients and controls might have contributed to differences in S100A8/A9 secretion between the two groups. Considering the small number of participants and the large variability in S100A8/A9 response, the differences in S100A8/A9 increase at 24 h after stress between patients and controls have to be interpreted with due caution. Additionally, our study was not powered to detect a prospective link between S100A8/A9 response and increased risk for recurrent coronary events and mortality. Studies in larger cohorts are needed in order to confirm whether there is a significant difference in S100A8/A9 response between patients and controls, and to establish whether the S100A8/A9 response to psychological stress can be used to risk-stratify CAD patients.
In conclusion, we demonstrate for the first time that acute psychological stress triggers systemic release of the pro-inflammatory alarmin S100A8/A9. Our data also indicate that the S100A8/A9 response to stress may be associated with dysregulated cortisol secretion in CAD patients, thereby providing further insight into the link between HPA axis dysfunction, inflammation and CVD.
Methods
Study populations
Two cohorts of CAD patients have been used in this study. In Study I, we included 60 CAD patients at 6–12 months after an index cardiac event. The patients had been diagnosed in accordance with the consensus document of the Joint European Society of Cardiology/American College of Cardiology Committee43. Fourty-four patients had suffered an acute coronary syndrome [24 non-ST-segment elevation myocardial infarction (NSTEMI), 20 ST-segment-elevation myocardial infarction (STEMI)], and 16 had undergone percutaneous coronary intervention (PCI) due to stable angina. The exclusion criteria were age >75 years, on-going infection, worsened or recurrent angina or readmission to hospital within the previous 10 weeks, moderate to severe heart failure, chronic inflammatory or immunologic disorders, neoplasm, continuous treatment with immunosuppressive/anti-inflammatory agents, drug or alcohol abuse or poor mental function.
In Study II, we included 30 patients at 12–14 weeks after a first-time ACS and 30 healthy controls with equal age and gender distribution randomly selected from the population register. Eleven of the patients had been diagnosed with NSTEMI, and 19 with STEMI. Twenty-eight patients had been treated with thrombolysis and/or PCI, and two had an acute coronary artery by-pass grafting operation (CABG). The controls were all clinically healthy, with neither history nor clinical signs of CAD or other CVD. The exclusion criteria were age >70 years, but otherwise similar to Cohort I. In controls, antihypertensive drugs and statins as primary preventive treatments were allowed. A complete set of blood samples was available from 27 patients and 28 controls in Study II. Written informed consent was obtained from all study participants, the Ethical Review Board of Linköping University approved the research protocol, and the study was conducted according to the ethical guidelines of the Declaration of Helsinki.
Psychological stress test and sample collection
In both studies, the participants came to the hospital in a fasting state, and were instructed to take prescribed drugs as usual. Testing always started between 07.30–08.00 a.m. Baseline blood pressure and heart rate were recorded with subjects comfortably lying down on a bed. Saliva samples for baseline cortisol were taken before venipuncture and collection of baseline blood samples.
The psychological stress test included two parts with different stressors, an anger recall test and an arithmetic test, with a pause of 2 minutes in-between16,23. In the first part, the participant was instructed to recall an event that made him/her angry, frustrated or upset, and to discuss for 6 min what had happened and how that made him/her feel. In the second part, the participants were instructed to count backwards from 700 minus 7 as quickly and accurately as possible, and to reach zero within 4 min. Blood pressure and pulse rate were measured before stress and every 2 min until the end of the test.
In Study I, the second blood sample was collected at 34 min after the start of the first stressor, i.e. 20 min after the completion of the second stressor. In Study II, the second blood sample was collected at 24 h after the stress test. In both cohorts, saliva samples were collected at baseline and at 20 min after stress. For measurement of diurnal cortisol variation, additional saliva samples were collected from Study I participants twice daily on three consecutive days, 30 minutes after awakening and in the evening before going to bed. Saliva collection was performed by placing salivette cotton swabs (Sarstedt, Nürnbrecht, Germany) under the tongue for 2 min. The swabs were then immediately frozen at −20 °C.
Biochemical analyses
S100A8/A9 ELISA
S100A8/A9 was measured by using a previously described ELISA technique44. Microtiter plates (Maxisorp, Nunc, Roskilde, Denmark) were coated at 4 °C overnight with 5 μg/ml monoclonal antibody against S100A8/A9 (27E10, BMA Biomedicals AB, August, Switzerland) diluted in PBS pH 7.2, followed by a blocking step using 150 μl of 1% BSA (ICN Biomedicals Inc., Aurora, OH, USA). A buffer containing 0.15M NaCl, 10mM HEPES (Invitrogen, Carlsbad, CA, USA), 1 mM CaCl2, 0.02 mM ZnCl2, 0.05% Tween 20 and 0.1% BSA was used for dilution of serum samples, detection antibody and streptavidin. Serum samples diluted 1/100 were added to the microtiter plates and incubated for 2h. The wells were thereafter washed three times with PBS containing 0.05% Tween 20, followed by overnight incubation with biotinylated polyclonal antibodies against S100A8/A9 (chicken polyclonal antibody MRP8/14, Abcam, Cambridge, UK) diluted 1/2000. After washing, the antibodies were labeled with ALP-bound streptavidin (Dako, Glostrup, Denmark) diluted 1/1000. Bound streptavidin was visualized with disodium-p-nitrophenyl phosphate (Sigma, St Louis, MO, USA) 1 mg/ml dissolved in 10% (w/v) diethanolamine pH 9.8 containing 50 mM MgCl2. The absorbance was measured at 405 nm. All samples were run in duplicate. Optical density values of uncoated wells were used as background and subtracted from the values of the samples. Titration curves obtained from one serum with known concentration were used to calculate the S100A8/A9 concentrations. The lower detection level was 3 ng/mL.
Saliva cortisol
Measurement of free cortisol levels in saliva was performed at the accredited Clinical Chemical Laboratory at Karolinska University Hospital, Sweden, using a commercial radioimmunoassay assay, CORT-CT2 (Cisbio, Bioanalyser, Codolet, France), with a limit of detection of 3.0 nmol/L and limit of quantitation of 100 nmol/L. The interassay coefficient of variance was less than 10% according to repeatedly performed quality assessments.
Statistical analysis
We have used the Student’s t-test to compare the values of normally distributed continuous variables between groups, and the Mann-Whitney U-test for non-normally distributed continuous variables. Differences in categorical variables were assessed with the chi-square test or Fischer’s exact test, as appropriate. Variations in values between different time-points when serial measurements were performed in the same subjects were examined by the Wilcoxon paired test for non-normally distributed variables. Spearman’s rank test and a multivariable linear regression analysis model were used to test associations between percentage S100A8/A9 increase relative to baseline and other clinical and biochemical parameters in the population. Non-normally distributed continuous variables were logarithmated before being used in the linear regression model. A p-value under 0.05 was considered to be statistically significant. Values are presented as mean (SD) if normally distributed and median (inter-quartile range) if not normally distributed. All calculations were made using SPSS 22.0 (IBM software, Armonk, NY).
Data availability statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author Contributions
L.J. has participated in study conception and design, data acquisition and interpretation, and critical revision of the manuscript for intellectual content. H.G.L. has been involved in data analysis and interpretation, and manuscript drafting. A.K.L., B.G. and A.A.B. have participated in data acquisition and critical revision of the manuscript for intellectual content. A.S. contributed in study conception and design, data analysis and interpretation, and manuscript drafting. All authors have approved the final version of the manuscript to be submitted.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. 2013;2013:828354. doi: 10.1155/2013/828354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lood C, et al. Platelet-Derived S100A8/A9 and Cardiovascular Disease in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016;68:1970–1980. doi: 10.1002/art.39656. [DOI] [PubMed] [Google Scholar]
- 4.Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identificationofp8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266:7706–7713. [PubMed] [Google Scholar]
- 5.Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 6.Ehrchen JM, Sunderkotter C, Foell D, Vogl T, Roth J. The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol. 2009;86:557–566. doi: 10.1189/jlb.1008647. [DOI] [PubMed] [Google Scholar]
- 7.Hofmann MA, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/S0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 8.Yan L, et al. Beneficial effects of quinoline-3-carboxamide (ABR-215757) on atherosclerotic plaque morphology in S100A12 transgenic ApoE null mice. Atherosclerosis. 2013;228:69–79. doi: 10.1016/j.atherosclerosis.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102:1239–1246. doi: 10.1161/CIRCRESAHA.107.167544. [DOI] [PubMed] [Google Scholar]
- 10.Baumann M, et al. MRP8/14 is associated with systemic inflammation in stable coronary atherosclerosis in men. Eur J Clin Invest. 2011;41:1261–1267. doi: 10.1111/j.1365-2362.2011.02530.x. [DOI] [PubMed] [Google Scholar]
- 11.Ionita M, et al. High levels of myeloid-related protein 14 in human atherosclerotic plaques correlate with the characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol. 2009;29:1220–1227. doi: 10.1161/ATVBAHA.109.190314. [DOI] [PubMed] [Google Scholar]
- 12.Cotoi OS, et al. Plasma S100A8/A9 correlates with blood neutrophil counts, traditional risk factors, and cardiovascular disease in middle-aged healthy individuals. Arterioscler Thromb Vasc Biol. 2014;34:202–210. doi: 10.1161/ATVBAHA.113.302432. [DOI] [PubMed] [Google Scholar]
- 13.Altwegg LA, et al. Myeloid-related protein 8/14 complex is released by monocytes and granulocytes at the site of coronary occlusion: a novel, early, and sensitive marker of acute coronary syndromes. Eur Heart J. 2007;28:941–948. doi: 10.1093/eurheartj/ehm078. [DOI] [PubMed] [Google Scholar]
- 14.Morrow DA, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the Pravastatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155:49–55. doi: 10.1016/j.ahj.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steptoe A, Hamer M, Chida Y. The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Behav Immun. 2007;21:901–912. doi: 10.1016/j.bbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 16.Lundberg AK, Jonsson S, Stenmark J, Kristenson M, Jonasson L. Stress-induced release of matrix metalloproteinase-9 in patients with coronary artery disease: The possible influence of cortisol. Psychoneuroendocrinology. 2016;73:117–124. doi: 10.1016/j.psyneuen.2016.07.219. [DOI] [PubMed] [Google Scholar]
- 17.Hammoudeh AJ, Alhaddad IA. Triggers and the onset of acute myocardial infarction. Cardiol Rev. 2009;17:270–274. doi: 10.1097/CRD.0b013e3181bdba75. [DOI] [PubMed] [Google Scholar]
- 18.Lagraauw HM, Kuiper J, Bot I. Acute and chronic psychological stress as risk factors for cardiovascular disease: Insights gained from epidemiological, clinical and experimental studies. Brain Behav Immun. 2015;50:18–30. doi: 10.1016/j.bbi.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 20.Kunz-Ebrecht SR, Mohamed-Ali V, Feldman PJ, Kirschbaum C, Steptoe A. Cortisol responses to mild psychological stress are inversely associated with proinflammatory cytokines. Brain Behav Immun. 2003;17:373–383. doi: 10.1016/S0889-1591(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 21.Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. 2012;1261:55–63. doi: 10.1111/j.1749-6632.2012.06633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohleder N. Stimulation of systemic low-grade inflammation by psychosocial stress. Psychosom Med. 2014;76:181–189. doi: 10.1097/PSY.0000000000000049. [DOI] [PubMed] [Google Scholar]
- 23.Nijm J, Kristenson M, Olsson AG, Jonasson L. Impaired cortisol response to acute stressors in patients with coronary disease. Implications for inflammatory activity. J Intern Med. 2007;262:375–384. doi: 10.1111/j.1365-2796.2007.01817.x. [DOI] [PubMed] [Google Scholar]
- 24.Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114:1867–1879. doi: 10.1161/CIRCRESAHA.114.302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuebler U, et al. Stress-induced modulation of NF-kappaB activation, inflammation-associated gene expression, and cytokine levels in blood of healthy men. Brain Behav Immun. 2015;46:87–95. doi: 10.1016/j.bbi.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 26.Nijm J, Jonasson L. Inflammation and cortisol response in coronary artery disease. Ann Med. 2009;41:224–233. doi: 10.1080/07853890802508934. [DOI] [PubMed] [Google Scholar]
- 27.Strike PC, et al. Pathophysiological processes underlying emotional triggering of acute cardiac events. Proc Natl Acad Sci USA. 2006;103:4322–4327. doi: 10.1073/pnas.0507097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peng WH, et al. Increased serum myeloid-related protein 8/14 level is associated with atherosclerosis in type 2 diabetic patients. Cardiovasc Diabetol. 2011;10:41. doi: 10.1186/1475-2840-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croce K, et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–436. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Volz HC, et al. S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic Res Cardiol. 2012;107:250. doi: 10.1007/s00395-012-0250-z. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, et al. S100a8/a9 released by CD11b+Gr1+neutrophils activates cardiac fibroblasts to initiate angiotensin II-Induced cardiac inflammation and injury. Hypertension. 2014;63:1241–1250. doi: 10.1161/HYPERTENSIONAHA.113.02843. [DOI] [PubMed] [Google Scholar]
- 32.Jonsson S, et al. Increased levels of leukocyte-derived MMP-9 in patients with stable angina pectoris. PLoS One. 2011;6:e19340. doi: 10.1371/journal.pone.0019340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirsch G, Lavoie-Lamoureux A, Beauchamp G, Lavoie JP. Neutrophils are not less sensitive than other blood leukocytes to the genomic effects of glucocorticoids. PLoS One. 2012;7:e44606. doi: 10.1371/journal.pone.0044606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids–new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 35.Bierhaus A, et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci USA. 2003;100:1920–1925. doi: 10.1073/pnas.0438019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gebhardt C, et al. Calgranulins S100A8 and S100A9 are negatively regulated by glucocorticoids in a c-Fos-dependent manner and overexpressed throughout skin carcinogenesis. Oncogene. 2002;21:4266–4276. doi: 10.1038/sj.onc.1205521. [DOI] [PubMed] [Google Scholar]
- 37.Heim C, Ehlert U, Hellhammer DH. The potential role of hypocortisolism in the pathophysiology of stress-related bodily disorders. Psychoneuroendocrinology. 2000;25:1–35. doi: 10.1016/S0306-4530(99)00035-9. [DOI] [PubMed] [Google Scholar]
- 38.Matthews K, Schwartz J, Cohen S, Seeman T. Diurnal cortisol decline is related to coronary calcification: CARDIA study. Psychosom Med. 2006;68:657–661. doi: 10.1097/01.psy.0000244071.42939.0e. [DOI] [PubMed] [Google Scholar]
- 39.Criqui MH, et al. Calcium density of coronary artery plaque and risk of incident cardiovascular events. JAMA. 2014;311:271–278. doi: 10.1001/jama.2013.282535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumari M, Shipley M, Stafford M, Kivimaki M. Association of diurnal patterns in salivary cortisol with all-cause and cardiovascular mortality: findings from the Whitehall II study. J Clin Endocrinol Metab. 2011;96:1478–1485. doi: 10.1210/jc.2010-2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ronaldson A, et al. Diurnal Cortisol Rhythm Is Associated With Adverse Cardiac Events and Mortality in Coronary Artery Bypass Patients. J Clin Endocrinol Metab. 2015;100:3676–3682. doi: 10.1210/jc.2015-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammer F, et al. High evening salivary cortisol is an independent predictor of increased mortality risk in patients with systolic heart failure. Int J Cardiol. 2016;203:69–73. doi: 10.1016/j.ijcard.2015.10.084. [DOI] [PubMed] [Google Scholar]
- 43.Seeman T. Myocardial infarction redefined–a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. Eur Heart J. 2000;21:1502–1513. doi: 10.1053/euhj.2000.2305. [DOI] [PubMed] [Google Scholar]
- 44.Tyden, H. et al. Increased serum levels of S100A8/A9 and S100A12 are associated with cardiovascular disease in patients with inactive systemic lupus erythematosus. Rheumatology, 10.1093/rheumatology/ket263 (2013). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.