

Abstract
Purpose of review:
Tuberous sclerosis complex (TSC) is a variably expressed neurocutaneous genetic disorder characterized by hamartomatous growths in multiple organ systems. Neurologic involvement often confers the most severe symptoms, and can include epilepsy, increased intracranial pressure from hydrocephalus, intellectual deficits, and autism. The purpose of this review is to provide a neurologically focused update in the diagnosis and treatment of these complications in patients with TSC.
Recent findings:
We highlight 5 new areas of understanding in TSC: the neurobiology of TSC and its translation into clinical practice, vigabatrin in the treatment of infantile spasms, the role of tubers and epilepsy surgery, the treatment of subependymal giant cell astrocytomas, and TSC-related neuropsychiatric disorders.
Summary:
These recent advances in diagnosis and treatment give our patients with TSC and their families hope for the future for improved care and possible preventive cures, to the end goal of improving quality of life.
Tuberous sclerosis complex (TSC), occurring in 1 in 6,000 people, is a variably expressed autosomal dominant neurocutaneous disorder characterized by hamartomatous growths in multiple organs. Neurologic involvement confers the most severe symptoms, and can include epilepsy, increased intracranial pressure, intellectual deficits, and autism. Early diagnosis and treatment are focused on treatment of symptoms and prevention of complications, to the end goal of improving quality of life.
The average age at diagnosis of TSC is 7.5 years, although this can range from birth to adulthood.1 The diagnostic criteria for TSC were recently revised with the notable changes being the inclusion of genetic testing and reduction of the clinical diagnostic classes to either definite or possible TSC.2 With the successful application of bench research, there has been an increasing understanding of the underlying mechanisms behind the clinical manifestations of TSC, which brings with it hope for novel treatments and improved outcomes. We are highlighting 5 new areas of understanding in TSC.
Neurobiology
TSC displays one of the most illustrative examples of how bench research can be translated into clinical care, specifically providing novel medical treatments of the known complications of TSC. TSC1 and TSC2 are the only known causative genes in TSC. Of the 85% of patients with detectable disease-causing mutations, 31% have mutations in TSC1, encoding for the protein hamartin, and 69% have mutations in TSC2, encoding for tuberin. Of the remaining patients with a clinical diagnosis of TSC, it is thought that they may be exhibiting somatic mosaicism or have a mutation in the TSC1 or TSC2 genes not detectable by conventional methods.3
Under normal physiologic states, hamartin and tuberin regulate cell differentiation and proliferation. They form a complex that activates GTPase, which keeps the Ras homolog enriched in brain protein inactive, keeping the mechanistic target of rapamycin (mTOR) pathway inhibited (figure 1). TSC1 and TSC2 mutations hyperactivate the mTOR pathway, leading to abnormalities in cell cycle progression.4 Despite some overlap, patients with TSC2 mutations tend to present earlier and are more likely to have complicated epilepsy and intellectual deficits, have a higher tuber to brain proportion, and develop subependymal giant-cell astrocytomas (SEGA).5,6
Figure 1. Mechanistic target of rapamycin (mTOR) pathway.

There are many components of the upstream and downstream pathways of the regulation of mTOR. Under normal physiologic conditions, some of the upstream pathways are activated by hypoxia and cellular stress. Serine/threonine kinase 11 (STK11) (also known as LKB1) is induced by hypoxia and cellular stress, which is further activated by adenosine monophosphate (AMP) and induces adenosine monophosphate-activated protein kinase (AMPK), which further activates the TSC1/TSC2 complex, which inhibits Ras homolog enriched in brain (Rheb) and therefore downregulates mTOR. Hypoxia-inducible factor 1-α (HIF1-α) is induced by hypoxia, which in turn activates regulated in development and DNA damage responses 1 (REDD1), which induces TSC1/TSC2. There are multiple growth factors and cytokines, which induce protein kinase B, also known as Akt (PKB), extracellular signal-regulated kinase (ERK), ribosomal protein S6 kinase (RSK), and inhibitor of nuclear factor κ-B kinase subunit β (IKKβ), and all inhibit the TSC1 and TSC2 complex, as seen in the diagram. The activation of mTOR leads to cell growth.
Sirolimus and everolimus are antiproliferative and immunosuppressive oral medications that inhibit the mTOR pathway. Sirolimus is approved by the US Food and Drug Administration (FDA) for the treatment of pulmonary lymphangioleiomyomatosis (LAM), which occurs in about a third of women with TSC. The current FDA approvals for everolimus in patients with TSC include pediatric and adult patients with SEGAs requiring therapeutic interventions but cannot be curatively resected, and adult patients with renal angiomyolipomas (AML) not requiring immediate surgery.
Clinical trials support the efficacy and safety of these mTOR inhibitors for the treatment of several neurologic and non-neurologic manifestations of TSC. Although initial case series reported SEGA regression with sirolimus treatment, subsequent studies have involved mainly everolimus. After positive phase I evidence, everolimus clinical trial C2485 enrolled 28 patients age 3 years and older with TSC-related SEGA, reporting at 6 months a decreased size of SEGAs.7 This was followed by the Examining Everolimus in a Study of TSC (EXIST-1) trial, which randomized patients ages 0–65 years, 39 to placebo and 78 to everolimus, reporting at 6 months at least a 50% decreased SEGA volume in 35% of the everolimus group vs none in the placebo group.8 The most common adverse events were mouth ulcerations (32% everolimus vs 5% placebo). In the open-label extension, persistent efficacy was noted at a median follow-up of 28 months.9 The positive clinical effects of mTOR inhibition using either sirolimus or everolimus are also reported in clinical trials and case series for renal AMLs, skin angiofibromas, cardiac rhabdomyomas, and lung LAM.10,11 Thus, with continued research comes the hope of furthering our understanding of how to best treat and prevent the multiorgan system complications of TSC.
Vigabatrin and infantile spasms (IS)
The syndrome of IS, usually consisting of flexor or extensor spasms, developmental plateau or regression, and an EEG pattern of hypsarrhythmia, is a common manifestation of epilepsy in TSC, occurring in over one-third of patients.12 Although IS is defined as occurring in infants, 5% of patients with TSC can manifest the seizure semiology of epileptic spasms after 2 years.13 The early diagnosis of patients with TSC has allowed the collection of prospective data, showing an association between the onset of IS and impairment in intellectual development.14 Retrospective data in infants with TSC support an association between an increased duration of time from treatment initiation until clinical spasm cessation and worse intellectual outcome.15 Thus, early recognition and successful treatment of IS can hopefully prevent subsequent long-term intellectual delays.
Vigabatrin (VGB), approved by the FDA in 2009, has shown remarkable efficacy for infants with TSC and IS. VGB irreversibly inhibits γ-aminobutyric acid (GABA) transaminase, which increases GABA levels, contributing to its anticonvulsant properties. Prior retrospective studies and case series have reported very high response rates (95%) of IS in TSC to VGB, although these high rates were based upon clinical cessation of spasms without EEG confirmation of hypsarrhythmia resolution.16 Although yielding less robust efficacy, prospective studies randomizing low-dose (18–36 mg/kg/d) to high-dose (100–148 mg/kg/d) VGB, using stricter criteria of EEG confirmation of hypsarrhythmia resolution in addition to clinical spasms cessation, still report higher response rates and a shorter time to response in patients with TSC vs other etiologies.17 VGB has been recommended as first-line treatment of IS by the 2012 International TSC Consensus Conference.18
VGB is available in both tablets and in sachets as a powder for oral solution. However, the distribution of VGB is controlled by the SHARE (Support, Help, and Resources for Epilepsy) program to ensure its safe use because VGB has been linked to irreversible visual field loss due to retinal toxicity, which can occur in 20%–30% of children.19 Longer durations of treatment are associated with a higher risk, thus VGB should be used for the shortest duration possible to ensure the cessation of spasms. For patients such as infants who are unable to cooperate with standard visual field testing, electroretinogram monitoring during treatment may predict toxicity prior to clinical changes.19 In addition, VGB has been associated with dose-dependent, reversible, and apparently asymptomatic MRI signal abnormalities in the basal ganglia and brainstem (figure 2).20 The underlying pathophysiology of this abnormal MRI signal is uncertain, but it may be related to intramyelinic edema. With these risks in mind, the goal of improving long-term cognitive outcomes underlies the need for the prompt diagnosis and treatment of IS in TSC.
Figure 2. Vigabatrin-related brain MRI abnormalities.
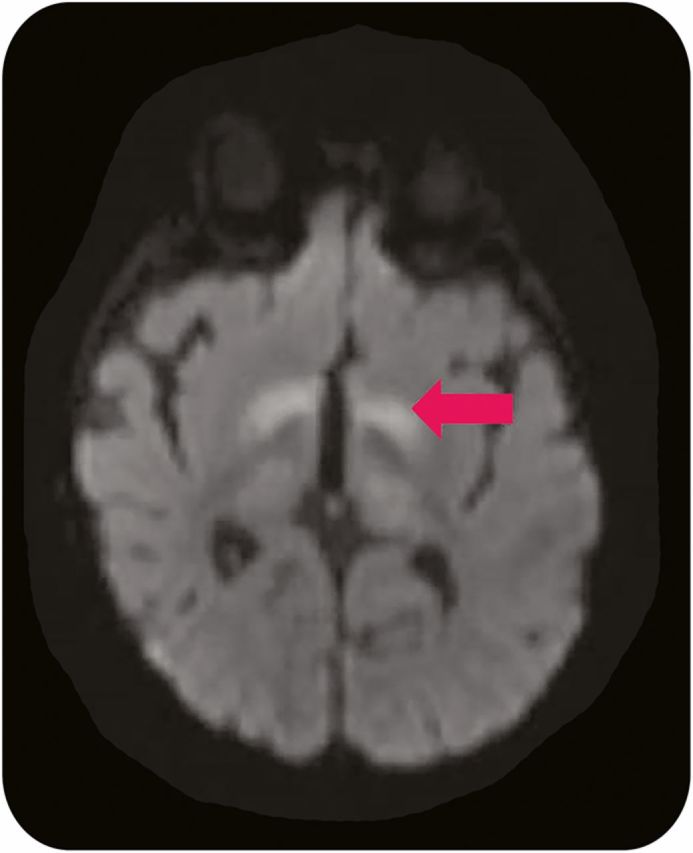
Axial diffusion-weighted 1.5 T MRI of a 5-month-old boy with tuberous sclerosis complex and infantile spasms treated with vigabatrin reveals abnormal signal in the bilateral basal ganglia (arrow).
Tubers and epilepsy surgery
Epilepsy, occurring in up to 85% of patients, is a major cause of neurologic morbidity in patients with TSC, with poor control correlating with a worse cognitive outcome.12 Although treatment is indicated at the clinical onset of seizures, it is possible that treatment at the onset of EEG abnormalities, prior to clinical seizures, might improve long-term outcome,21 but this is a concept in need of further research. Epilepsy in TSC is often characterized by multiple seizure types, in particular focal onset seizures, and is medication-refractory in 30%–60% of patients.12,22 Patients with a history of IS are more likely to develop refractory epilepsy (75%) vs those without a history of IS (40%).12 Medication-refractory patients should be considered for alternative treatments, often epilepsy surgery, but also should include dietary therapy and neurostimulation. Although neurobiology-based treatment of epilepsy using mTOR inhibitors also holds promise,23 long-term studies are needed to assess the safety and viability of this treatment option.
Multiple types of dysgenetic brain findings occur in TSC, to include cortical tubers, subependymal nodules, radial glial lines, and abnormal white matter diffusivity. In particular, the cortical tuber holds an important role in the development of epilepsy and also as a target of surgical resection. Although most cortical tubers act as stable congenital lesions, tubers may occasionally change over time. Microstructural changes in tubers can be detected with diffusion tensor imaging (DTI) with age,24 and on MRI, tubers sometimes evolve into a cyst-like appearance. These cyst-like tubers (figure 3) are often present in TSC patients with more severe epilepsy phenotypes, and thus are often looked at closely as surgical targets.25 However, the exact role of cortical tubers in epilepsy is still being defined, as it appears that although many tubers are epileptogenic as proven by intracranial, intratuber depth electrodes,26 in other TSC patients tubers have been found to be electrically silent.27
Figure 3. Cystic tuber.
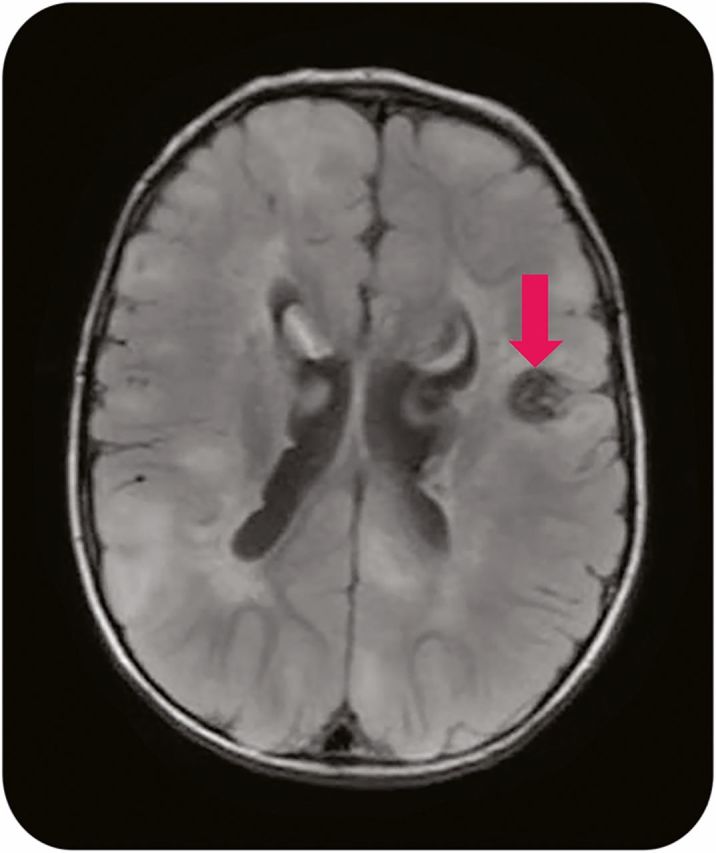
Axial fluid-attenuated inversion recovery 1.5 T MRI of the brain reveals multiple cortical tubers to include a cystic-appearing tuber (arrow).
An epilepsy surgery evaluation to include ictal scalp or intracranial EEG, MRI, functional imaging, and neuropsychological testing is needed to define those patients with TSC as potential candidates. This is often challenging in patients with TSC due to the frequent occurrence of multiple tubers, the possibility of epileptogenic tissue extending outside of tubers, and also the changing appearance and epileptogenicity of other tubers. However, despite these challenges, a single epileptogenic cortical tuber, causing a predominant seizure semiology, is often determined to be a suitable surgical target, but with perituberal cortex also being evaluated to define the extent of resection.27,28 When surgical candidates are chosen carefully, epilepsy surgery can result in good outcomes with seizure freedom in about 60% when followed for at least 1–2 years29–31 (as a comparison, the success rate of pediatric temporal lobe resection approaches 80%).32 Several factors have been associated with better outcomes, to include unilateral focality in interictal or ictal EEG, lobectomy, removal of epileptogenic tissue by MRI and intracranial EEG, and agreement of interictal and ictal EEG localization.29,30 Poorer outcomes have been associated with younger age at seizure onset, history of IS, and interictal bilateral focality.31 Most importantly, successful epilepsy surgery is associated with improvement in quality of life measures in patients with TSC.33,34
SEGAs
SEGAs are the most common CNS tumor in patients with TSC, developing in 10%–15% of patients. Per the 2012 Washington Consensus Conference, they are defined as a lesion >1 cm at the caudothalamic groove or a subependymal lesion showing serial growth, with most showing gadolinium enhancement.35 SEGAs commonly present in the first 2 decades of life with symptoms of headache, increased seizure frequency, and emesis due to obstructive hydrocephalus, although symptoms can initially be very subtle. SEGAs rarely become symptomatic during adulthood. Current clinical consensus is for screening neuroimaging every 1–3 years during childhood to identify SEGAs before they become symptomatic; once diagnosed, more frequent imaging is indicated to monitor evidence of growth.
Currently, early surgical resection remains the standard treatment for SEGAs showing serial growth on imaging. For smaller lesions, complete resection can be curative with rare associated surgical complications.36 However, in larger (>3 cm), bilateral, multiple, or previously resected lesions, the risk for surgical complications is high, with one series of 57 TSC patients reporting 0% complications with SEGAs <2 cm vs 46% in SEGAs 2–3 cm, and 83% in 3–4 cm, and a 67% complication risk in patients with bilateral SEGAs.36
With the discovery that genetic lesions in TSC patients lead to unregulated mTOR activation, there has been renewed interest in possible medical management of complex patients. Medical management with mTOR inhibitors is currently indicated for patients with symptomatic SEGA who are not amendable to surgery (figure 4).37 There may also be a role to reduce tumor size prior to surgical resection. Duration of treatment is currently unknown as regrowth has been documented with cessation of therapy. Finally, further research is needed to better understand the possible long-term side effects of pharmacologic mTOR inhibition, as there have been conflicting reports regarding effects on linear growth, pubertal progression, and immunosuppressive effects in the post-transplant population.38
Figure 4. Multiple subependymal giant-cell astrocytomas (SEGAs).
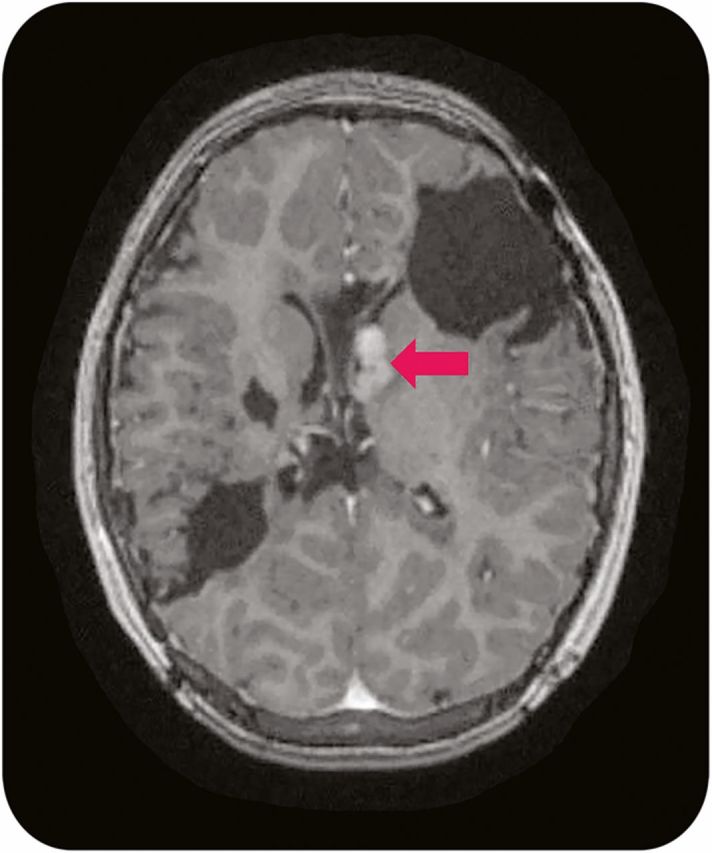
Axial T1-weighted 1.5 T MRI with gadolinium contrast reveals multiple SEGAs near the left foramen of Monroe (arrow), without evident hydrocephalus, in a patient with prior resectional epilepsy surgeries.
TSC-associated neuropsychiatric disorders (TANDs)
TAND is a recently agreed-upon term that encompasses a spectrum of disorders commonly present in patients with TSC to include autism spectrum disorders, intellectual and learning disabilities, and psychiatric and behavioral disorders.19 Over 90% of patients with TSC exhibit difficulties in areas within the realm of TANDs,39,40 particularly autism spectrum disorders and intellectual deficits, which each occur in up to 50% of patients.40 The TSC Neuropsychiatry Panel recommends regular screening of patients in order to assist in the identification and proper treatment of these common issues.39 A TAND checklist has been developed and has undergone pilot validation.40 In addition to regular screening, a sudden change in symptoms within the spectrum of TANDs should also prompt the consideration of TSC-related medical complications, such as hydrocephalus secondary to SEGA growth.
The underlying etiology of TANDs is uncertain, but probably multifactorial, to include contributions from genetic, functional, and structural factors. Genetically, patients with TSC2 mutations tend to have worse cognitive outcomes than patients with TSC1.6 Functionally, an earlier onset of seizures is associated with a worse long-term cognitive outcome,41,42 thus the goal to diagnose and treat seizures early.43 Structurally, there are inconsistent data for whether tuber burden is related to cognitive outcomes, although the tuber-to-brain proportion may be a better predictor than tuber quantification.41,42 However, there are other factors involved, exemplified by examples of TSC patients without tubers with cognitive delays and epilepsy.44 One possible contributor could be abnormal white matter in TSC, as evidenced by DTI, which can detect abnormalities of microscopic tissue structures by indirectly measuring the direction and degree of water diffusion. Patients with TSC can have either decreased fractional anisotropy or increased mean diffusivity within normal-appearing white matter, suggesting abnormalities in the structural integrity of the imaged white matter.45 Furthermore, these white matter abnormalities appear to be modifiable using treatments with mTOR inhibitors, as evidenced in subgroup analysis of studies of everolimus for the treatment of SEGAs.46 It is uncertain what the clinical implications of these findings are, but further research is ongoing to study whether these changes might have a positive effect on TANDs.
Tuberous sclerosis complex: Five new things
Understanding of the neurobiology of TSC has successfully translated into clinical practice with novel medical treatments using mTOR inhibitors.
Vigabatrin, approved by the FDA in 2009, has shown remarkable efficacy for infants with TSC and infantile spasms.
Epilepsy surgery in patients with TSC with medication-refractory epilepsy can improve quality of life, when candidates are chosen carefully.
The roles of new medical treatments with mTOR inhibitors and traditional resectional surgical treatments to treat subependymal giant cell astrocytomas are currently being defined.
TAND are extremely common and regular screening and early identification and treatment are an essential component of care in patients with TSC.
AUTHOR CONTRIBUTIONS
David T. Hsieh: conceptualized the manuscript and authored the first draft and revisions. Susan L. Whiteway: authored the first draft and revisions. Luis O. Rohena: authored the first draft and revisions. Elizabeth A. Thiele: reviewed and edited the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
D.T. Hsieh and S. L. Whiteway report no disclosures. L.O. Rohena is Chief Editor of Medscape: Genetics and Metabolic Diseases. E.A. Thiele has received funding for travel or speaker honoraria from Tuberous Sclerosis Alliance, Tuberous Sclerosis Australia, and GW Pharma; receives publishing royalties for Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics (Wiley-Blackwell, 2010); serves as a consultant for GW Pharma, Eisai Pharmaceuticals, Novartis, and Zogenix Pharmaceuticals; and receives research support from GW Pharma and Lennox Gastaut Foundation. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
DISCLAIMER
The views expressed herein are those of the authors and do not reflect the official policy or position of Brooke Army Medical Center, the US Army Medical Department, the US Army Office of the Surgeon General, the Department of the Army, the Department of the Air Force, the Department of Defense, or the US Government.

REFERENCES
- 1.Staley BA, Vail EA, Thiele EA. Tuberous sclerosis complex: diagnostic challenges, presenting symptoms, and commonly missed signs. Pediatrics 2011;127:e117–e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tyburczy ME, Dies KA, Glass J, et al. . Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet 2015;11:e1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Napolioni V, Curatolo P. Genetics and molecular biology of tuberous sclerosis complex. Curr Genomics 2008;9:475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kothare SV, Singh K, Chalifoux JR, et al. . Severity of manifestations in tuberous sclerosis complex in relation to genotype. Epilepsia 2014;55:1025–1029. [DOI] [PubMed] [Google Scholar]
- 6.Jansen FE, Braams O, Vincken KL, et al. . Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology 2008;70:908–915. [DOI] [PubMed] [Google Scholar]
- 7.Krueger DA, Care MM, Holland K, et al. . Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010;363:1801–1811. [DOI] [PubMed] [Google Scholar]
- 8.Franz DN, Belousova E, Sparagana S, et al. . Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013;381:125–132. [DOI] [PubMed] [Google Scholar]
- 9.Franz DN, Belousova E, Sparagana S, et al. . Everolimus for subependymal giant cell astrocytoma in patients with tuberous sclerosis complex: 2-year open-label extension of the randomised EXIST-1 study. Lancet Oncol 2014;15:1513–1520. [DOI] [PubMed] [Google Scholar]
- 10.Curatolo P, Moavero R. mTOR inhibitors in tuberous sclerosis complex. Curr Neuropharmacol 2012;10:404–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goyer I, Dahdah N, Major P. Use of mTOR inhibitor everolimus in three neonates for treatment of tumors associated with tuberous sclerosis complex. Pediatr Neurol 2015;52:450–453. [DOI] [PubMed] [Google Scholar]
- 12.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010;51:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh DT, Jennesson MM, Thiele EA. Epileptic spasms in tuberous sclerosis complex. Epilepsy Res 2013;106:200–210. [DOI] [PubMed] [Google Scholar]
- 14.Humphrey A, MacLean C, Ploubidis GB, et al. . Intellectual development before and after the onset of infantile spasms: a controlled prospective longitudinal study in tuberous sclerosis. Epilepsia 2014;55:108–116. [DOI] [PubMed] [Google Scholar]
- 15.Goh S, Kwiatkowsi DJ, Dorer DJ, Thiele EA. Infantile spasms and intellectual outcomes in children with tuberous sclerosis complex. Neurology 2005;65:235–238. [DOI] [PubMed] [Google Scholar]
- 16.Hancock E, Osborne JP. Vigabatrin in the treatment of infantile spasms in tuberous sclerosis: literature review. J Child Neurol 1999;14:71–74. [DOI] [PubMed] [Google Scholar]
- 17.Elterman RD, Shields WD, Bittman RM, Torri SA, Sagar SM, Collins SD. Vigabatrin for the treatment of infantile spasms: final report of a randomized trial. J Child Neurol 2010;25:1340–1347. [DOI] [PubMed] [Google Scholar]
- 18.Krueger DA, Northrup H; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013;49:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westall CA, Wright T, Cortese F, Kumarappah A, Snead OC, Buncic JR. Vigabatrin retinal toxicity in children with infantile spasms: an observational cohort study. Neurology 2014;83:2262–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh DT, Thiele EA. Vigabatrin-related magnetic resonance imaging abnormalities in an infant with tuberous sclerosis complex and infantile spasms. J Pediatr 2013;162:215. [DOI] [PubMed] [Google Scholar]
- 21.Jozwiak S, Kotulaska K, Domanaska-Pakiela D, et al. . Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol 2011;15:424–431. [DOI] [PubMed] [Google Scholar]
- 22.Vignoli A, La Briola F, Turner K, et al. . Epilepsy in TSC: certain etiology does not mean certain prognosis. Epilepsia 2013;54:2134–2142. [DOI] [PubMed] [Google Scholar]
- 23.Krueger DA, Wilfong AA, Holland-Bouley K, et al. . Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol 2013;74:679–687. [DOI] [PubMed] [Google Scholar]
- 24.Peters JM, Prohl AK, Tomas-Fernandez XK, et al. . Tubers are neither static nor discrete: evidence from serial diffusion tensor imaging. Neurology 2015;85:1536–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu-Shore CJ, Major P, Montenegro M, Thiele EA. Cyst-like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology 2009;72:1165–1169. [DOI] [PubMed] [Google Scholar]
- 26.Mohamed AR, Bailey CA, Freeman JL, Maixner W, Jackson GD, Harvey AS. Intrinsic epileptogenicity of cortical tubers revealed by intracranial EEG monitoring. Neurology 2012;79:2249–2257. [DOI] [PubMed] [Google Scholar]
- 27.Major P, Rakowski S, Simon MV, et al. . Are cortical tubers epileptogenic? Evidence from electrocorticography. Epilepsia 2009;50:147–154. [DOI] [PubMed] [Google Scholar]
- 28.Ruppe V, Dilsiz P, Reiss CS, et al. . Developmental brain abnormalities in tuberous sclerosis complex: a comparative tissue analysis of cortical tubers and perituberal cortex. Epilepsia 2014;55:539–550. [DOI] [PubMed] [Google Scholar]
- 29.Zhang K, Hu WH, Zhang C, Meng FG, Chen N, Zhang JG. Predictors of seizure freedom after surgical management of tuberous sclerosis complex: a systematic review and meta-analysis. Epilepsy Res 2013;105:377–383. [DOI] [PubMed] [Google Scholar]
- 30.Krsek P, Jahodova A, Kyncl M, et al. . Predictors of seizure-free outcome after epilepsy surgery for pediatric tuberous sclerosis complex. Epilepsia 2013;54:1913–1921. [DOI] [PubMed] [Google Scholar]
- 31.Madhavan D, Schaffer S, Yankovsky A, et al. . Surgical outcome in tuberous sclerosis complex: a multicenter survey. Epilepsia 2007;48:1625–1628. [DOI] [PubMed] [Google Scholar]
- 32.Wyllie E, Comair YG, Kotagal P, Bulacio J, Bingaman W, Ruggieri P. Seizure outcome after epilepsy surgery in children and adolescents. Ann Neurol 1998;44:740–748. [DOI] [PubMed] [Google Scholar]
- 33.Liang S, Li A, Zhao M, et al. . Epilepsy surgery in tuberous sclerosis complex: emphasis on surgical candidate and neuropsychology. Epilepsia 2010;51:2316–2321. [DOI] [PubMed] [Google Scholar]
- 34.Roth J, Olasunkanmi A, MacAllister WS, et al. . Quality of life following epilepsy surgery for children with tuberous sclerosis complex. Epilepsy Behav 2011;20:561–565. [DOI] [PubMed] [Google Scholar]
- 35.Roth J, Roach ES, Bartels U, et al. . Subependymal giant cell astrocytoma: diagnosis, screening, and treatment: recommendations from the International Tuberous Sclerosis Complex Consensus Conference 2012. Pediatr Neurol 2013;49:439–444. [DOI] [PubMed] [Google Scholar]
- 36.Kotulska K, Borkowska J, Roszkowski M, et al. . Surgical treatment of subependymal giant cell astrocytoma in tuberous sclerosis complex patients. Pediatr Neurol 2014;50:307–312. [DOI] [PubMed] [Google Scholar]
- 37.Jozwiak S, Nabbout R, Curatolo P. Management of subependymal giant cell astrocytoma (SEGA) associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol 2013;17:348–352. [DOI] [PubMed] [Google Scholar]
- 38.Somers MJ, Paul E. Safety consideration of mammalian target of rapamycin inhibitors in tuberous sclerosis complex and renal transplantation. J Clin Pharmacol 2015;55:368–376. [DOI] [PubMed] [Google Scholar]
- 39.de Vries PJ, Whittemore VH, Leclezio L, et al. . Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND checklist. Pediatr Neurol 2015;52:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leclezio L, Jansen A, Whittemore VH, de Vries PJ. Pilot validation of the tuberous sclerosis-associated neuropsychiatric disorders (TAND) checklist. Pediatr Neurol 2015;52:16–24. [DOI] [PubMed] [Google Scholar]
- 41.Jansen FE, Vincken KL, Algra A, et al. . Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology 2008;70:916–923. [DOI] [PubMed] [Google Scholar]
- 42.Kaczorowska M, Jurkiewicz E, Domanska-Pakiela D, et al. . Cerebral tuber count and its impact on mental outcome of patients with tuberous sclerosis complex. Epilepsia 2011;52:22–27. [DOI] [PubMed] [Google Scholar]
- 43.Bombardieri R, Pinci M, Moavero R, Cerminara C, Curatolo P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol 2010;14:146–149. [DOI] [PubMed] [Google Scholar]
- 44.Kaufmann R, Kornreich L, Goldberg-Stern H. Unusual clinical presentation of tuberless tuberous sclerosis complex. J Child Neurol 2009;24:361–364. [DOI] [PubMed] [Google Scholar]
- 45.Makki MI, Chugani DC, Janisse J, Chugani HT. Characteristics of abnormal diffusivity in normal-appearing white matter investigated with diffusion tensor MR imaging in tuberous sclerosis complex. AJNR Am J Neuroradiol 2007;28:1662–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tillema JM, Leach JL, Krueger DA, Franz DN. Everolimus alters white matter diffusion in tuberous sclerosis complex. Neurology 2012;78:526–531. [DOI] [PubMed] [Google Scholar]