

Key Points
Patients with SCD and severe organ damage can tolerate nonmyeloablative conditioning with no transplant-related mortality.
Posttransplant cyclophosphamide prevents severe GVHD, increases engraftment, and improves the success rate for haploidentical HSCT.
Abstract
Peripheral blood stem cell transplantation (PBSCT) offers a curative option for sickle cell disease (SCD). Although HLA-matched sibling transplantation is promising, the vast majority of patients lack such a donor. We sought to develop a novel nonmyeloablative HLA-haploidentical PBSCT approach that could safely be used for patients with severe organ damage. Based on findings in our preclinical model, we developed a phase 1/2 trial using alemtuzumab, 400 cGy total body irradiation, and escalating doses of posttransplant cyclophosphamide (PT-Cy): 0 mg/kg in cohort 1, 50 mg/kg in cohort 2, and 100 mg/kg in cohort 3. A total of 21 patients with SCD and 2 with β-thalassemia received a transplant. The mean hematopoietic cell transplant–specific comorbidity index of 6 reflected patients with cirrhosis, heart failure, and end-stage renal disease. The engraftment rate improved from 1 (33%) of 3 in cohort 1 to 5 (63%) of 8 in cohort 2 and 10 (83%) of 12 in cohort 3. Percentage of donor myeloid and CD3 chimerism also improved with subsequent cohorts. There was no transplant-related mortality, and overall survival was 87%. At present, 0% in cohort 1, 25% in cohort 2, and 50% in cohort 3 remain free of their disease. There was no grade 2 to 4 acute or extensive chronic graft-versus-host disease (GVHD). Therefore, PT-Cy improves engraftment and successfully prevents severe GVHD after nonmyeloablative conditioning in patients with SCD who are at high risk for early mortality. Additional strategies are necessary to decrease the graft rejection rate and achieve a widely available cure for all patients with SCD. This trial was registered at www.clinicaltrials.gov as #NCT00977691.
Visual Abstract
Introduction
Sickle cell disease (SCD) is an inherited red blood cell disorder in which patients experience severe complications, including debilitating recurrent painful crises, acute chest syndrome, kidney failure, stroke, pulmonary hypertension, and SCD-associated liver disease.1-3 The majority of patients experience a poor quality of life and significantly reduced lifespan.1,2,4 The most readily available curative option remains allogeneic hematopoietic stem cell transplantation (HSCT).5
The most common indications for transplant in pediatric patients with SCD and an HLA-matched sibling are stroke and recurrent vaso-occlusive crises and/or acute chest syndrome.6,7 A myeloablative approach that uses high doses of chemotherapy to replace the patient’s bone marrow with that of their donor is very efficacious, with a cure rate now expected to approach 95%7; however, there is a 10% to 20% risk of graft-versus-host disease (GVHD) and 6.9% transplant-related mortality.7 Because stable mixed chimerism with donor chimerism levels as low as 11% was found to reverse the sickle phenotype and was not associated with GVHD,8 and because adults with subclinical and overt organ damage cannot tolerate myeloablative conditioning, we and others sought to develop a successful nonmyeloablative conditioning regimen for SCD.9-14 Our nonmyeloablative HLA-matched sibling peripheral blood stem cell transplantation (PBSCT) regimen used alemtuzumab 300 cGy total body irradiation (TBI) and sirolimus, which led to the achievement of donor-type hemoglobin in 25 (83%) of 30 patients at 1 year posttransplant and the reversal of SCD in 26 (87%) of 30 patients and no GVHD at most recent follow-up.15
Although this HLA-matched sibling PBSCT protocol has been successful, the vast majority of patients with SCD do not have an HLA-matched sibling. In our study, 185 (64%) of 287 screened patients were excluded on that basis.15 However, many patients referred to the National Institutes of Health (NIH) were already known to have an HLA-matched sibling; the chance of a patient with SCD having an HLA-matched sibling in general practice is much lower at <15%.16 Therefore, we developed a mismatched murine transplant model and, on the basis of those results,17,18 we designed a nonmyeloablative haploidentical PBSCT conditioning regimen consisting of alemtuzumab, 400 cGy TBI, sirolimus, and a dose-escalation of posttransplant cyclophosphamide (PT-Cy). Here we report the results of our phase 1/2 study in which 21 adults with SCD and 2 adults with transfusion-dependent β-thalassemia underwent haploidentical PBSCT.
Patients and methods
Protocol specifics
This was a prospective phase 1/2 nonmyeloablative haploidentical PBSCT protocol for patients with severe SCD and β-thalassemia. The study was approved by the National Heart, Lung, and Blood Institute Institutional Review Board in 2009. All patients gave written informed consent. The study was monitored by an independent data and safety monitoring board.
Participants 18 years of age or older were eligible for the study. Sickle cell–specific criteria included homozygous SCD, compound heterozygous hemoglobin S and C disease or hemoglobin S and β0-thalassemia disease, stroke, sickle cell–related renal insufficiency (defined by creatinine ≥1.5 times the upper limit of normal and kidney biopsy consistent with sickle cell nephropathy, or nephrotic syndrome or creatinine clearance <50 mL/minute or requiring dialysis),19-21 tricuspid regurgitant jet velocity ≥2.5 m/s,22,23 sickle hepatopathy (defined as either ferritin ≥1000 μg/L or direct bilirubin >0.4 mg/dL and platelet count <250 000/μL at baseline),24 >1 hospitalization per year for vaso-occulsive crises or any acute chest syndrome while receiving a maximum tolerated dose of hydroxyurea for at least 6 months.1,25 β-thalassemia–specific criteria included Pesaro class II or III disease.26,27 Patients were excluded if they had an HLA-matched sibling donor available, Eastern Cooperative Oncology Group performance status of 3 or more, major ABO mismatched donor, and any antidonor HLA antibodies.28-30 Specific donors were selected primarily on the basis of the degree of HLA matching with the recipient.
The study treatment plan was written as a dose-escalation trial that evaluated the ability of PT-Cy to improve engraftment after haploidentical HSCT. The trial was designed to achieve a goal success rate of at least 70% of patients free of their primary disease such that regimen failure was defined as either graft rejection or severe GVHD (grade 3-4 acute GVHD [aGVHD] or extensive chronic GVHD [cGVHD]). If the regimen failure rate exceeded predefined stopping boundaries in a cohort, we advanced the study to the next dosing cohort. Cohort 1 used no PT-Cy, cohort 2 used 50 mg/kg PT-Cy, and cohort 3 used 100 mg/kg PT-Cy (Figure 1). Per our statistical plan, each cohort was monitored and accrual was stopped if the number of regimen failures reached the stopping boundaries.
Figure 1.

Conditioning regimen. All patients received alemtuzumab, 400 cGy TBI, and sirolimus. Cohort 1 patients received no PT-Cy, cohort 2 patients received 50 mg/kg PT-Cy, and cohort 3 patients received 100 mg/kg PT-Cy. Sirolimus target level was 10 to 15 ng/dL. Sirolimus was started 1 day before PBSC infusion in patients 4 through 9 and 4 days posttransplant in patients 10 and 11. IV, intravenous; N/A, not applicable.
Donors received 5 to 6 days of granulocyte colony-stimulating factor at 10 to 16 μg/kg/d for mobilization followed by peripheral blood leukapheresis with a goal of at least 10 × 106 CD34 cells per kilogram of the recipient’s body weight, and the PBSC products were cryopreserved.31 All participants received an alemtuzumab infusion over 2 hours at 0.03 mg/kg 7 days before, 0.1 mg/kg 6 days before, and 0.3 mg/kg 5 through 3 days before PBSC infusion. Patients were treated with antiemetics, and then they received 400 cGy TBI in 2 divided doses on days –2 and –1 before PBSC infusion. Cyclophosphamide was infused over 60 minutes at a dose of 50 mg/kg on day +3 posttransplant in cohort 2 patients and at 100 mg/kg in 2 divided doses on days +3 and +4 posttransplant in cohort 3 patients. Patients were premedicated with antiemetics and received concurrent mesna and intravenous hydration if they made sufficient urine or were provided with a 3-way catheter with bladder irrigation if they did not make urine. Sirolimus was loaded 1 day before transplant in cohort 1 and in the first 6 patients who received a transplant in cohort 2 and 1 day after PT-Cy in the remaining cohort 2 patients (day +4) and in all of the cohort 3 patients (day +5). A trough level of 10 to 15 ng/mL was targeted until 3 to 4 months posttransplant, and then the level was decreased to 10 to 12 ng/mL until 1 year posttransplant and then 5 to 10 ng/mL thereafter in engrafted patients.
Recipients and donors from endemic countries were screened for malaria and recipients screened for tuberculosis and were treated as necessary. Hydroxyurea was given at the maximum tolerated dose until 1 day before conditioning. Erythropoietin was discontinued approximately 1 month before conditioning in patients with renal failure, and red blood cell (RBC) transfusions were provided to keep the hemoglobin at ≥8 g/dL. RBC exchange transfusions were performed within 1 week of conditioning with a target hemoglobin S level of <30%. Iron chelation was discontinued >48 hours before conditioning. Platelet transfusion thresholds were maintained at >50 000/μL when possible in patients with SCD, and hemoglobin was kept between 9 and 10 g/dL. The time for absolute neutrophil count recovery was defined as the first of 3 consecutive days during which absolute neutrophil count was ≥500/μL, and platelet recovery was defined as platelet count at least 1 week after the last platelet transfusion. Standardized supportive care guidelines were followed, including anti-herpes simplex virus and varicella-zoster virus, antifungal, and Pneumocystis jiroveci prophylaxis. Patients were monitored frequently for cytomegalovirus (CMV),32,33 Epstein-Barr virus (EBV),34 human herpesvirus 6, and adenovirus and treated if necessary. Penicillin prophylaxis was continued until pneumococcal vaccination was complete.
The primary end point of the study was the percentage of patients at 1 year posttransplant with sustained donor-type hemoglobin on hemoglobin electrophoresis for patients with SCD or for patients with β-thalassemia who were transfusion-independent and who did not have severe (grade 3-4) aGVHD or extensive cGVHD. Secondary end points included disease-free and overall survival, incidence of aGVHD and cGVHD, graft rejection rate, whether PT-Cy would reduce the incidence and severity of graft rejection or GVHD, and transplant-related mortality. Planned follow-up occurred 1 to 2 times per week until 100 days posttransplant, every 6 months until 3 years posttransplant, and once per year thereafter. The hematopoietic cell transplantation–specific comorbidity index (HCT-CI) consists of 16 different comorbidities, including cardiopulmonary, renal, gastrointestinal, and neurologic comorbidities. The HCT-CI score was calculated by using an online Web-based calculator (www.hctci.org).
Laboratory analysis
Laboratory studies including chemistries, hematologic studies, and lymphocyte subsets were run at the NIH Department of Laboratory Medicine. Lineage-specific chimerism studies (CD14/CD15 and CD3) were conducted as previously described.35,36 Donor red cell chimerism was assessed via hemoglobin electrophoresis and extended RBC phenotyping. RBC phenotyping and HLA antibody analysis were performed by the NIH Department of Transfusion Medicine.
Statistical analysis of chimerism and lymphocyte subsets
Descriptive statistics (primarily mean values, percentages, medians, and ranges) are presented for this small data set. Unless otherwise noted, mean values are shown ± standard deviation.
Results
Baseline characteristics
As of May 13, 2016, 23 patients had undergone haploidentical PBSCT, with a median follow-up of 3.17 years (range, 0.67 to 6.16 years). Median age at the time of transplant was 36 years (range, 20-56 years) (Table 1). Fourteen of the patients (61%) were male. The vast majority (19 [83%]) had homozygous SCD. Two of the patients had β-thalassemia, 1 had HbSC disease, and 1 had HbS β0-thalassemia disease.
Table 1.
Baseline characteristics
| Patient | Cohort | Age (y) | Sex | Disease type | Disease-related complications/comorbidities | HCT-CI | Donor | HLA match | CD34 dose (× 106/kg) | CD3 dose (× 108/kg) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (y) | Sex | Relation | HbS (%) | ||||||||||
| 1 | 1 | 44 | F | HbSS | ESRD on PD, pHTN, diastolic dysfunction, iron overload, history of ACS/VOC | 6 | 38 | F | Sister | 39 | 5/10 | 15.9 | 5.50 |
| 2 | 1 | 20 | M | HbSS | pHTN, iron overload, stroke, moyamoya | 10 | 47 | F | Mother | 40 | 6/10 | 15.0 | 2.65 |
| 3 | 1 | 36 | F | HbSS | pHTN, history of VOC, RA | 6 | 46 | F | Sister | 0 | 7/10 | 9.76 | 2.83 |
| 4 | 2 | 37 | F | HbSS | Iron overload, history of VOC, MS | 7 | 66 | F | Mother | 39 | 8/10 | 10.2 | 3.78 |
| 5 | 2 | 47 | M | HbSS | Systolic dysfunction, cirrhosis/NRH, history of ACS/VOC, stroke | 7 | 60 | F | Sister | 38 | 6/10 | 11.9 | 7.93 |
| 6 | 2 | 36 | M | HbSS | Systolic dysfunction, iron overload, history of ACS/VOC, stroke | 8 | 28 | M | Brother | 38 | 7/10 | 28.0 | 5.01 |
| 7 | 2 | 31 | F | HbSS | Iron overload, moyamoya | 3 | 60 | F | Mother | 38 | 7/10 | 13.0 | 8.07 |
| 8 | 2 | 35 | M | β-thal | Iron overload | 3 | 67 | F | Mother | 0 | 6/10 | 8.79 | 3.75 |
| 9 | 2 | 21 | M | HbSS | Iron overload, history of ACS/VOC, stroke, moyamoya | 7 | 51 | F | Mother | 39 | 5/10 | 12.2 | 3.51 |
| 10 | 2 | 25 | M | β-thal | Iron overload | 7 | 55 | F | Mother | 0 | 7/10 | 7.04 | 1.86 |
| 11 | 2 | 24 | M | HbSS | ESRD on HD, systolic dysfunction, iron overload, history of ACS/VOC | 6 | 20 | M | Brother | 39 | 7/10 | 13.4 | 2.59 |
| 12 | 3 | 37 | M | HbSS | ESRD on PD, iron overload, history of VOC | 6 | 61 | F | Mother | 34 | 8/10 | 25.6 | 4.73 |
| 13 | 3 | 41 | M | HbSS | ESRD on PD, systolic and diastolic dysfunction, iron overload, history of VOC | 10 | 45 | F | Sister | 0 | 8/10 | 15.9 | 5.08 |
| 14 | 3 | 37 | M | HbSS | CRI, iron overload, history of ACS/VOC, stroke | 3 | 56 | F | Mother | 40 | 7/10 | 10.2 | 2.98 |
| 15 | 3 | 31 | F | HbSS | pHTN, iron overload, history of ACS/VOC, TIA | 7 | 30 | F | Sister | 42 | 7/10 | 15.1 | 4.00 |
| 16 | 3 | 56 | F | HbSC | Iron overload, history of ACS/VOC | 3 | 31 | M | Son | 40 | 8/10 | 29.7 | 3.78 |
| 17 | 3 | 26 | M | HbSS | History of ACS/VOC | 3 | 51 | F | Mother | 39 | 6/10 | 16.8 | 3.948 |
| 18 | 3 | 20 | F | HbSS | Iron overload, history of ACS/VOC | 2 | 51 | F | Mother | 40 | 6/10 | 10.2 | 6.14 |
| 19 | 3 | 47 | F | HbSS | CRI, diastolic dysfunction, iron overload, history of ACS/VOC | 9 | 23 | F | Sister | 0 | 5/10 | 16.6 | 2.95 |
| 20 | 3 | 34 | F | HbS β0-thal | Cirrhosis, history of VOC | 8 | 30 | M | Brother | 0 | 8/10 | 9.70 | 5.28 |
| 21 | 3 | 27 | M | HbSS | Iron overload, history of ACS/VOC, stroke, moyamoya | 6 | 52 | F | Mother | 39 | 5/10 | 11.5 | 3.65 |
| 22 | 3 | 36 | M | HbSS | Iron overload, history of ACS/VOC | 5 | 64 | M | Father | 35 | 5/10 | 12.2 | 2.42 |
| 23 | 3 | 42 | M | HbSS | pHTN, systolic dysfunction, history of ACS | 6 | 23 | F | Sister | 0 | 7/10 | 10.1 | 6.12 |
ACS, acute chest syndrome; β-thal, β-thalassemia; CRI, chronic renal insufficiency; ESRD, end-stage renal disease; F, female; HbS, hemoglobin S; HbSC, compound heterozygous HbS and HbC disease; HbSS, homozygous sickle cell disease; HbS β0-thal, compound heterozygous HbS and β0-thalassemia disease; HCT-CI, hematopoietic cell transplantation–specific comorbidity index; HD, hemodialysis; M, male; MS, multiple sclerosis; NRH, nodular regenerative hyperplasia; PD, peritoneal dialysis; pHTN, pulmonary hypertension; RA, rheumatoid arthritis; TIA, transient ischemic attack; VOC, vaso-occlusive crisis.
Four of the donors were minor ABO-mismatched to the recipients. Median donor age was 51 years (range, 20-67 years), 5 of the donors (22%) were male, and 16 of the donors (70%) had sickle cell trait. Five of the donors were 5/10 HLA-matched, 5 were 6/10 HLA-matched, 8 were 7/10 HLA-matched, and 5 were 8/10 HLA-matched. Median CD34 cell dose was 12.2 × 106 cells per kilogram of body weight (range, 7.0-29.7 × 106 cells per kilogram of body weight), and median CD3 cell dose was 3.8 × 108 cells per kilogram of body weight (range, 1.86-8.1 × 108 cells per kilogram of body weight).
The vast majority of the patients had a severe disease-related complication and/or comorbidity at baseline (Tables 1 and 2). Almost all of the patients (87%) had a hepatic complication. Five patients (22%) had systolic dysfunction with left ventricular ejection fractions ranging from 36% to 52%, and 3 patients (13%) had diastolic dysfunction. Six patients (26%) had severe renal disease, and 5 (22%) had right heart catheterization–documented pulmonary hypertension. The majority of patients had at least 3 complications or comorbidities: 4 (17%) had 3, 9 (39%) had 4, and 1 (4%) had 7. Mean HCT-CI score was 6 ± 2.3: 1 of the patients had an HCT-CI score of 2, 5 had a score of 3, and 17 had a score ≥5 (Table 1). None of the patients had a score below 2.
Table 2.
Disease-related complications and comorbidities
| Complication or comorbidity | No. | % |
|---|---|---|
| Hepatic | 20 | 87 |
| Iron overload | 18 | 78 |
| Cirrhosis | 2 | 9 |
| Recurrent acute chest syndrome and/or vaso-occlusive crisis | 19 | 83 |
| Neurologic* | 8 | 35 |
| Stroke | 6 | 26 |
| Moyamoya syndrome | 4 | 17 |
| Transient ischemic attack | 1 | 4 |
| Cardiac* | 7 | 30 |
| Systolic dysfunction | 5 | 22 |
| Diastolic dysfunction | 3 | 13 |
| Renal | 6 | 26 |
| End-stage renal disease on peritoneal dialysis | 3 | 13 |
| End-stage renal disease on hemodialysis | 1 | 4 |
| CRI with baseline creatinine 2.5-5.0 mg/dL | 2 | 9 |
| Pulmonary hypertension | 5 | 22 |
| Autoimmune | 2 | 9 |
| Multiple sclerosis | 1 | 4 |
| Rheumatoid arthritis | 1 | 4 |
The number of events do not add up to the total number of events because at least 1 patient in each group experienced more than 1 complication.
PBSCT outcomes
Engraftment and success rates improve with posttransplant cyclophosphamide.
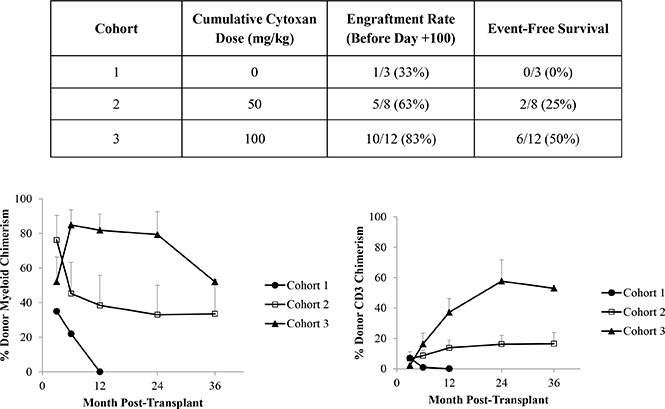
Three patients received a transplant in cohort 1 in which no PT-Cy was given (Table 3). One patient (33%) engrafted, and she rejected her graft 7 months posttransplant. Therefore, stopping rules were reached and the study was advanced to cohort 2 in which 50 mg/kg PT-Cy was administered. Eight patients received a transplant: 5 (63%) engrafted and 3 were determined to have insufficient donor chimerism at a median of 1.30 years posttransplant (range, 127 days to 2.35 years posttransplant). One of the patients acutely rejected his graft, and the other 2 (including a patient who discontinued sirolimus approximately 12 and again 17 months posttransplant) had persistent low levels of donor myeloid chimerism (ranging from 7% to 13%) that were inadequate to reverse the sickle phenotype. Therefore, 25% of the second cohort patients remain free of SCD. When stopping rules were met, the study was advanced to cohort 3, the last cohort. Twelve patients received a transplant, and 10 (83%) engrafted. Four patients rejected their grafts at a median of 73.5 days posttransplant (range, 63-90 days posttransplant), and 50% of patients are free of SCD. Stopping rules were reached and the study is now closed to further accrual.
Table 3.
Engraftment rate and disease-free survival according to cohort number
| Cohort | Cumulative cyclophosphamide dose (mg/kg) | Engraftment rate (before day +100) | Disease-free survival | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| 1 | 0 | 1/3 | 33 | 0/3 | 0 |
| 2 | 50 | 5/8 | 63 | 2/8 | 25 |
| 3 | 100 | 10/12 | 83 | 6/12 | 50 |
No. refers to the number of patients who engrafted or remain disease-free over the total number of patients transplanted in that cohort.
Twenty of the 23 patients are alive with an overall survival rate of 87%. There was no transplant-related mortality. All deaths occurred in patients who had experienced graft rejection. One patient (patient 5) died of sepsis 6 months posttransplant after unexpectedly receiving a contaminated platelet transfusion, 1 patient (patient 14) died 3 years posttransplant from pulmonary hypertension and congestive heart failure, and a third patient (patient 1) died 5 years posttransplant from a surgery-related infectious complication. At 1 year post-PBSCT, 0 patients (0%) in cohort 1, 3 patients (38%) in cohort 2, and 6 patients (50%) in cohort 3 maintained donor-type hemoglobin without severe GVHD, our primary end point. Mean HbS in the 6 patients whose donor had sickle cell trait (HbAS) was 41.2% ± 3.1%, and mean HbS in the 3 patients whose donor had normal hemoglobin (HbAA) was 1.9% ± 3.3%. The median duration of neutropenia for 22 of the patients was 45 days (range, 16-57 days) in cohort 1, 19 days (range, 18-89 days) in cohort 2, and 27 days (range, 19-51 days) in cohort 3. One patient with β-thalassemia in cohort 2 rejected his graft and unexpectedly did not recover autologous cells. Therefore, he received a second transplant that used a donor with a different haplotype 4 months after the first transplant, which used Hopkins’ regimen,37 and he remains engrafted with 100% donor chimerism. The protocol was subsequently amended to require plerixafor-mobilized autologous PBSC collection for patients with β-thalassemia before transplant.38 The median time for reaching a platelet count >50 000/μL in 18 of the patients was 31 days (range, 17-64 days); 6 patients, including the previously mentioned patient with β-thalassemia, required >100 days to reach a platelet count of >50 000/μL. Seven patients experienced primary graft failure, and 8 patients experienced secondary graft failure at a median of 108.5 days posttransplant (range, 63 days to 2.35 years posttransplant).
Donor leukocyte chimerism improves with posttransplant cyclophosphamide.
Percentage of donor myeloid and CD3 chimerism in the 1 patient in cohort 1 with successful engraftment is shown in Figure 2. Mean percentage (± the standard error of the mean) of donor myeloid chimerism in the successful cohort 2 patients was 76.2% (± 14.2%) at month 3, and this decreased to 33% (± 17.0%) at 2 years and 33.5% (± 19.8%) at 3 years posttransplant. Mean percentages of donor CD3 at these time points were 6.8% (± 4.6%), 16.2% (± 5.8%), and 16.5% (± 7.4%). Mean percentage of donor myeloid chimerism in the successful cohort 3 patients was originally lower at 3 months posttransplant than that in the cohort 2 patients at 52.1% (± 14.4%). However, by 6 months, mean donor myeloid chimerism was 84.8% (± 8.8%). At 12 and 24 months posttransplant, mean donor myeloid chimerism was 81.8% (± 9.5%) and 79.3% (± 13.3%). Donor myeloid chimerism in the single cohort 3 patient who is at least 3 years posttransplant was 52%. Mean donor CD3 chimerism levels increased from 2.2% (± 1.3%) at 3 months posttransplant to a mean of 57.7% (± 14.2%) by 2 years posttransplant. Unlike the study reported by the Hopkins group,37 no patient transplanted with our regimen achieved complete 100% donor chimerism. Therefore, all engrafted patients are still receiving immunosuppression.
Figure 2.

Donor chimerism levels in the 3 patient cohorts posttransplant over time. Percentage of (A) donor myeloid chimerism and (B) donor CD3 chimerism. Chimerism levels are reported only for patients who initially engrafted. Data are reported as mean plus standard error of the mean for each time point.
GVHD.
No patients in cohort 1 developed GVHD. One patient in cohort 2 with 63% donor myeloid and 4% donor CD3 chimerism developed questionable grade 1 aGVHD at 4 months posttransplant which resolved with systemic steroids. In addition, 1 patient in cohort 3 developed grade 1 aGVHD (100% donor myeloid chimerism and unquantifiable CD3 chimerism as a result of low lymphocyte count), which cleared with topical steroids 3 weeks posttransplant, and a second patient was diagnosed with limited ocular cGVHD (99% donor myeloid and 83% donor CD3 chimerism) at 2 years posttransplant, which has responded to topical therapy.
Transplant-related complications.
There were no acute sickle cell–related complications peritransplant. No patient developed sinusoidal obstructive syndrome. Fifteen patients developed bacteremia that responded to intravenous antibiotics. One patient in cohort 2 developed an adenovirus upper respiratory infection, transaminitis, and viremia that quickly cleared spontaneously at 1.3 years posttransplant. One patient in cohort 2 developed EBV-associated lymphoproliferative disorder at 40 days posttransplant, which was treated effectively with 6 doses of rituximab. No patients in cohorts 1 and 2 developed CMV infection. However, 4 patients in cohort 3 experienced CMV reactivation 27 to 90 days posttransplant, and an additional patient developed CMV colitis at 18 days posttransplant; all patients were successfully managed with foscarnet. Two patients in cohort 3 maintained chronic EBV viremia that did not require treatment. Two patients in cohort 2 and 1 patient in cohort 3 were treated for presumed fungal pulmonary nodules. Two patients (1 in cohort 1 and 1 in cohort 3, both of whom had initially engrafted and subsequently rejected their grafts) developed high-grade myelodysplastic syndrome with fibrosis 2 and 5 years posttransplant. One died 1 year after diagnosis as a result of pulmonary hypertension and congestive heart failure, and the second died 2 months after diagnosis as a result of an infectious complication of surgery.
Seven severe adverse events occurred possibly or definitely as a result of sirolimus: 3 experienced bone or joint pain and/or swelling primarily in the upper and lower extremities, 2 developed nausea and/or abdominal pain, 1 developed rhabdomyolysis, and 1 developed recurrent gastric ulcer–associated bleeding. The latter patient who also had end-stage renal disease was transitioned to tacrolimus and then developed posterior reversible encephalopathy syndrome. She now remains engrafted while receiving mycophenolate mofetil. An additional patient developed sirolimus-associated nephrotic syndrome peritransplant, which resolved after appropriate treatment and transitioning her sirolimus to low-dose tacrolimus and mycophenolate mofetil. All other engrafted patients remain on sirolimus. Two patients developed possible sirolimus-associated pneumonitis, and another patient who was platelet transfusion refractory and who developed a coagulopathy had diffuse alveolar hemorrhage to which sirolimus possibly contributed. Finally, 1 patient developed atrial fibrillation, and a second patient developed nonsustained ventricular tachycardia that may be related to alemtuzumab; they did not require chronic therapy.
Immune reconstitution.
Although numbers were small, we noticed that immune recovery differed between cohorts but was consistent with previously published data.39,40 B cells were increased at baseline and at all time points in cohorts 2 and 3 and stayed within the normal range in cohort 1 posttransplant. Mean CD3 recovery was limited in cohort 1 by slow CD3+CD4+ T-cell recovery. CD4 counts normalized within 1 year in cohorts 2 and 3 and after 2 years in cohort 1. CD8 counts returned to the normal range within the first 6 months. Natural killer cells remained within the normal range at all time points.
Discussion
Allogeneic HSCT has increasingly been used as a modality to cure patients with SCD. Although the success rate is 95% with HLA-matched siblings using a myeloablative approach for pediatric patients7 and 87% with a nonmyeloablative approach for adults,15 the vast majority of patients do not have an HLA-matched sibling donor. The potential donor pool is further reduced because of the genetics of the disease such that siblings may also have SCD. Conversely, the majority of patients have a half-matched parent, child, or sibling who can serve as a donor.37 Because the immunologic barrier in the haploidentical setting is greater, we hypothesized that additional immunomodulation above what is given in our HLA-matched sibling protocol would be necessary. We believe that sirolimus is critical to the success seen in our HLA-matched sibling protocol because it has been shown to induce tolerance both in vitro and in vivo.41,42 Because sirolimus decreases lymphocyte proliferation, we performed murine studies by using a mismatched transplant model to determine whether PT-Cy would improve engraftment when administered with sirolimus, because PT-Cy is thought to exert its beneficial effect at least in part by deleting alloreactive lymphocytes and preserving regulatory T cells.43-47 We found that in the lymphocyte-replete setting, sirolimus and PT-Cy acted synergistically to induce and maintain stable mixed chimerism.17 However, in the setting of profound lymphocyte depletion, PT-Cy did not improve engraftment.18 We used these data as the basis for a modest increase in conditioning radiation dose and the addition of PT-Cy.
Because there is no graft-versus-sickle effect and therefore no benefit obtained from GVHD in patients with SCD, our goal was to absolutely avoid GVHD and accept graft rejection as the worst outcome. With regard to GVHD, the study was successful, with only 2 cases of mild aGVHD and 1 case of limited cGVHD among 23 patients, but this resulted in part at the cost of a higher-than-targeted rate of graft rejection, which was ultimately 50% in our cohort 3 patients. We believe that SCD poses a particularly robust engraftment barrier associated with a hyperactive marrow and potential donor alloreactivity that develops in patients who are recurrently transfused. PT-Cy has been shown to decrease the risk of both graft rejection and GVHD in mice and humans.43,44,48 Our clinical outcomes in the trial also show that engraftment rate and steady-state donor chimerism progressively improve in cohorts treated with increasing doses of PT-Cy. We hypothesize that our treatment failures reflect incomplete depletion of host lymphocytes by alemtuzumab and TBI, leaving a sufficient number to expand in response to donor HLA and induce graft rejection. Of note, our success rate is similar to that reported in the largest haploidentical trial to date for patients with SCD in which patients were conditioned with pre- and posttransplant cyclophosphamide, fludarabine, 200 cGy TBI, and rabbit antithymocyte globulin.37 However, because our regimen does not include fludarabine, it could be extended to patients with severe organ damage, including renal failure.
Two other recent studies of HSCT for SCD explored the use of alternative donor sources and conditioning regimens. Shenoy et al49 reported 29 patients with SCD who underwent matched unrelated donor HSCT. Patients received alemtuzumab starting 22 days before transplant, fludarabine, and melphalan. The 1- and 2-year disease-free survival rates were 76% and 69%, respectively, with 28% grade 2 to 4 aGVHD and 38% extensive cGVHD; 7 patients died as a result of GVHD. A pediatric haploidentical trial for SCD in which patients were conditioned with busulfan, pretransplant cyclophosphamide, and horse antithymocyte globulin reported GVHD rates ranging from 20% to 30% and a success rate of 38%.50 Importantly, these 2 trials, which used greater conditioning intensity, did not achieve substantially better long-term outcomes because of the increased incidence of severe GVHD. In contrast, our aGVHD and cGVHD rates were <10%, and none of our patients has developed grade 2 to 4 aGVHD or extensive cGHVD. We believe that PT-Cy is a critical contributor to this difference. The addition of PT-Cy has been shown to decrease the incidence of GVHD in the malignant and nonmalignant setting.37,48,51-53 We acknowledge the fact that higher graft rejection is a competing risk against GVHD but predict that a modest intensification of conditioning will not significantly increase GVHD incidence if PT-Cy remains part of the conditioning.
Toxicities of our regimen overall proved to be manageable. High sirolimus levels used peritransplant likely led to complications, including pneumonitis and nephrotic syndrome. Besides nephrotic syndrome and gastric ulcer–associated bleeding, which led to transition to a different immunosuppressive agent, all other complications resolved when the sirolimus level was reduced. We also saw more viral reactivation compared with the HLA-matched sibling setting and in cohorts 1 and 2, suggesting that the increased incidence was related to 2 doses of PT-Cy. Lymphocyte subpopulations were normal at all time points measured; however, CMV reactivation was diagnosed in all patients <100 days posttransplant, whereas the first assessment of lymphocyte phenotyping posttransplant was at 6 months. All patients with CMV reactivation or disease were treated with foscarnet because of the concern for the myelosuppressive effect of ganciclovir in the nonmyeloablative setting, and CMV was effectively treated in all patients. Therefore, close viral monitoring and early preemptive treatment should be initiated in patients treated with similar regimens. In addition, 2 patients developed high-grade myelodysplastic syndrome 2 and 5 years posttransplant likely as a result of TBI and/or PT-Cy; 1 patient did not receive PT-Cy. Therefore, protocols that monitor patients for 1 to 2 years are not sufficient; instead, long-term follow-up of patients is imperative for evaluating the late effects of therapy.
Although our success rate of 50% is not ideal, it is important to interpret our success rate in the context of the very severe disease phenotype of our patients who received a transplant and the wide availability of donors. Fifty of 100 patients, including those with significant organ damage, could potentially be cured compared with 14 of 100 patients in the HLA-matched sibling setting in which the success rate is closer to 90% but the chance of having an HLA-matched sibling is 14%.16 When viewed in this context, our results represent a step forward. In contrast with another recent trial of haploidentical HSCT for SCD that used more typical exclusion criteria,37 our patients were much sicker: 2 of our patients had cirrhosis, 8 had systolic and/or diastolic heart dysfunction, 2 had stage IV kidney disease, 4 were receiving dialysis, and 5 had pulmonary hypertension. And most of our patients (61%) had at least 3 severe SCD-related complications and comorbidities at baseline. In fact, some of our patients had a predicted survival of ≤60% within 4 years,24,54,55 which approximates the 5-year survival rate for patients diagnosed with leukemia (59.7%), non-Hodgkin lymphoma (70.7%), and colon and rectum cancer (65.1%) between 2006 and 2012 based on data from Surveillance, Epidemiology, and End Results (SEER) (http://seer.cancer.gov). Risks that might be unique to haploidentical HSCT for SCD were not well known at the time this trial opened in 2009, so patients with significant sickle cell–related morbidity were enrolled when the potential benefits of a transplant outweighed potentially higher risks, and an enrollment age of 18 years was chosen so that all patients could provide consent for themselves. The HCT-CI score, which categorizes patient organ dysfunction before a transplant, is inversely correlated with nonrelapse mortality at 2 years after a transplant, with the highest mortality rate if the HCT-CI score was ≥3 in patients with malignancies.56,57 The HCT-CI score has some limitations in our patient population because SCD is inherently associated with increased bilirubin and aspartate aminotransferase that would automatically assign a score of 2 in the majority of patients. In addition, pulmonary hypertension and moyamoya syndrome without stroke are not included in HCT-CI, but they contribute significantly to SCD-related mortality. Despite the severity of disease in 74% of our patients who had an HCT-CI score of at least 5, only 1 died <2 years posttransplant.
Our trial has some limitations. Two patients with thalassemia who received a transplant may have had a different inherent risk for graft rejection because of ineffective hematopoiesis. This was a single-institution study, so the results may not be generalizable. The sample size was small because of the high rate of graft rejection and predetermined stopping rules. And there was no control group, so rates of morbidity and mortality could not be compared with a group of patients who did not receive a transplant.
In summary, we have established that a regimen of alemtuzumab, 400 cGy TBI, PT-Cy, and sirolimus is tolerated by adults with severe end-organ damage from SCD and can achieve control of SCD in 50% of patients with low risk of aGVHD and cGVHD. We enrolled patients with established severe disease and believe a similar approach should be used in other clinical trials in which a high risk of toxicity is possible. Additional studies will be needed to establish a regimen that is sufficient to overcome the engraftment barrier presented by SCD while minimizing the risk for GVHD and other transplant-related complications. Anurathapan et al58 recently reported in the haploidentical setting for patients with thalassemia the benefits associated with additional first-line immunosuppression to target recipient lymphocytes while giving PT-Cy to decrease the risk for GVHD, and the same should be considered for patients with SCD as the next logical step forward. Establishing a more effective regimen while retaining a favorable safety profile is indicated such that the ratio of benefit to risk will favor providing a transplant for patients before severe organ damage occurs. Furthermore, the transplant approach in children should be sufficiently safe to justify providing transplants for a patient population in which 99% are expected to reach 18 years of age.59,60 Such an approach can serve to maximize the chance that a successful transplant will truly improve not only the quality but also the quantity of life.
Acknowledgments
The authors acknowledge Roger Kurlander and Jodie Keary for chimerism analysis; Sharon Adams for help with HLA analysis; William Flegel and the Department of Transfusion Medicine for red cell phenotyping, antibody analysis, and protocol support; Stephanie Housel, Terri Wakefield, and Adriana Byrnes for protocol support, and the Radiation Oncology department, the clinical staff, and the patients and their families.
This work was funded by the intramural research program of the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health.
Authorship
Contribution: C.D.F., M.M.H., J.P., L.L., and J.F.T. designed the study; C.D.F., D.W., E.W., and N.J. performed data analysis; C.D.F., M.M.H., T.T., W.C., K.R., and J.F.T. provided protocol support; C.D.F., M.M.H., and J.F.T. wrote the manuscript; and C.D.F., M.M.H., T.T., W.C., K.R., D.W., E.W., N.J., C.J.G., J.P., L.L., and J.F.T. reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Courtney D. Fitzhugh, National Heart, Lung and Blood Institute, National Institutes of Health, 10 Center Dr, MSC 1589, Building 10, Room 6N-240A, Bethesda, MD 20892; e-mail: fitzhughc@nhlbi.nih.gov.
References
- 1.Platt OS, Brambilla DJ, Rosse WF, et al. . Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644. [DOI] [PubMed] [Google Scholar]
- 2.Fitzhugh CD, Hsieh MM, Allen D, et al. . Hydroxyurea-increased fetal hemoglobin is associated with less organ damage and longer survival in adults with sickle cell anemia. PLoS One. 2015;10(11):e0141706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore). 2005;84(6):363-376. [DOI] [PubMed] [Google Scholar]
- 4.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 Suppl):S512-S521. [DOI] [PubMed] [Google Scholar]
- 5.Hsieh MM, Fitzhugh CD, Tisdale JF. Allogeneic hematopoietic stem cell transplantation for sickle cell disease: the time is now. Blood. 2011;118(5):1197-1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walters MC, Patience M, Leisenring W, et al. . Bone marrow transplantation for sickle cell disease. N Engl J Med. 1996;335(6):369-376. [DOI] [PubMed] [Google Scholar]
- 7.Bernaudin F, Socie G, Kuentz M, et al. ; SFGM-TC. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110(7):2749-2756. [DOI] [PubMed] [Google Scholar]
- 8.Walters MC, Patience M, Leisenring W, et al. ; Multicenter Investigation of Bone Marrow Transplantation for Sickle Cell Disease. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001;7(12):665-673. [DOI] [PubMed] [Google Scholar]
- 9.Iannone R, Casella JF, Fuchs EJ, et al. . Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9(8):519-528. [DOI] [PubMed] [Google Scholar]
- 10.van Besien K, Bartholomew A, Stock W, et al. . Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant. 2000;26(4):445-449. [DOI] [PubMed] [Google Scholar]
- 11.Horwitz ME, Spasojevic I, Morris A, et al. . Fludarabine-based nonmyeloablative stem cell transplantation for sickle cell disease with and without renal failure: clinical outcome and pharmacokinetics. Biol Blood Marrow Transplant. 2007;13(12):1422-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krishnamurti L, Kharbanda S, Biernacki MA, et al. . Stable long-term donor engraftment following reduced-intensity hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2008;14(11):1270-1278. [DOI] [PubMed] [Google Scholar]
- 13.Matthes-Martin S, Lawitschka A, Fritsch G, et al. . Stem cell transplantation after reduced-intensity conditioning for sickle cell disease. Eur J Haematol. 2013;90(4):308-312. [DOI] [PubMed] [Google Scholar]
- 14.Bhatia M, Jin Z, Baker C, et al. . Reduced toxicity, myeloablative conditioning with BU, fludarabine, alemtuzumab and SCT from sibling donors in children with sickle cell disease. Bone Marrow Transplant. 2014;49(7):913-920. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. . Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312(1):48-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walters MC, Patience M, Leisenring W, et al. . Barriers to bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 1996;2(2):100-104. [PubMed] [Google Scholar]
- 17.Fitzhugh CD, Weitzel RP, Hsieh MM, et al. . Sirolimus and post transplant Cy synergistically maintain mixed chimerism in a mismatched murine model. Bone Marrow Transplant. 2013;48(10):1335-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fitzhugh C, Hsieh MM, Phang O, et al. . Post-transplant cyclophosphamide and sirolimus are synergistic in preventing rejection and inducing stable mixed chimerism independently of regulatory T cells [abstract]. Blood 2009;114(22). Abstract 3540. [Google Scholar]
- 19.Powars DR. Sickle cell anemia and major organ failure. Hemoglobin. 1990;14(6):573-598. [DOI] [PubMed] [Google Scholar]
- 20.Bakir AA, Hathiwala SC, Ainis H, et al. . Prognosis of the nephrotic syndrome in sickle glomerulopathy. A retrospective study. Am J Nephrol. 1987;7(2):110-115. [DOI] [PubMed] [Google Scholar]
- 21.Abbott KC, Hypolite IO, Agodoa LY. Sickle cell nephropathy at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol. 2002;58(1):9-15. [DOI] [PubMed] [Google Scholar]
- 22.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101(4):1257-1261. [DOI] [PubMed] [Google Scholar]
- 23.Gladwin MT, Sachdev V, Jison ML, et al. . Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med. 2004;350(9):886-895. [DOI] [PubMed] [Google Scholar]
- 24.Feld JJ, Kato GJ, Koh C, et al. . Liver injury is associated with mortality in sickle cell disease. Aliment Pharmacol Ther. 2015;42(7):912-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castro O, Brambilla DJ, Thorington B, et al. ; The Cooperative Study of Sickle Cell Disease. The acute chest syndrome in sickle cell disease: incidence and risk factors. Blood. 1994;84(2):643-649. [PubMed] [Google Scholar]
- 26.Lucarelli G, Clift RA, Galimberti M, et al. . Bone marrow transplantation in adult thalassemic patients. Blood. 1999;93(4):1164-1167. [PubMed] [Google Scholar]
- 27.La Nasa G, Argiolu F, Giardini C, et al. . Unrelated bone marrow transplantation for beta-thalassemia patients: The experience of the Italian Bone Marrow Transplant Group. Ann N Y Acad Sci. 2005;1054:186-195. [DOI] [PubMed] [Google Scholar]
- 28.Chang YJ, Zhao XY, Xu LP, et al. . Donor-specific anti-human leukocyte antigen antibodies were associated with primary graft failure after unmanipulated haploidentical blood and marrow transplantation: a prospective study with randomly assigned training and validation sets. J Hematol Oncol. 2015;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshihara S, Maruya E, Taniguchi K, et al. . Risk and prevention of graft failure in patients with preexisting donor-specific HLA antibodies undergoing unmanipulated haploidentical SCT. Bone Marrow Transplant. 2012;47(4):508-515. [DOI] [PubMed] [Google Scholar]
- 30.Ciurea SO, de Lima M, Cano P, et al. . High risk of graft failure in patients with anti-HLA antibodies undergoing haploidentical stem-cell transplantation. Transplantation. 2009;88(8):1019-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang EM, Areman EM, David-Ocampo V, et al. . Mobilization, collection, and processing of peripheral blood stem cells in individuals with sickle cell trait. Blood. 2002;99(3):850-855. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura R, Cortez K, Solomon S, et al. . High-dose acyclovir and pre-emptive ganciclovir to prevent cytomegalovirus disease in myeloablative and non-myeloablative allogeneic stem cell transplantation. Bone Marrow Transplant. 2002;30(4):235-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomblyn M, Chiller T, Einsele H, et al. ; Center for International Blood and Marrow Research; National Marrow Donor program; European Blood and Marrow Transplant Group; American Society of Blood and Marrow Transplantation; Canadian Blood and Marrow Transplant Group; Infectious Diseases Society of America; Society for Healthcare Epidemiology of America; Association of Medical Microbiology and Infectious Disease Canada; Centers for Disease Control and Prevention. Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant. 2009;15(10):1143-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reddy N, Rezvani K, Barrett AJ, Savani BN. Strategies to prevent EBV reactivation and posttransplant lymphoproliferative disorders (PTLD) after allogeneic stem cell transplantation in high-risk patients. Biol Blood Marrow Transplant. 2011;17(5):591-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nollet F, Billiet J, Selleslag D, Criel A. Standardisation of multiplex fluorescent short tandem repeat analysis for chimerism testing. Bone Marrow Transplant. 2001;28(5):511-518. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh MM, Kang EM, Fitzhugh CD, et al. . Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361(24):2309-2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolaños-Meade J, Fuchs EJ, Luznik L, et al. . HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012;120(22):4285-4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yannaki E, Papayannopoulou T, Jonlin E, et al. . Hematopoietic stem cell mobilization for gene therapy of adult patients with severe β-thalassemia: results of clinical trials using G-CSF or plerixafor in splenectomized and nonsplenectomized subjects. Mol Ther. 2012;20(1):230-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanakry CG, Coffey DG, Towlerton AM, et al. . Origin and evolution of the T cell repertoire after posttransplantation cyclophosphamide. JCI Insight. 2016;1(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Di Stasi A, Milton DR, Poon LM, et al. . Similar transplantation outcomes for acute myeloid leukemia and myelodysplastic syndrome patients with haploidentical versus 10/10 human leukocyte antigen-matched unrelated and related donors. Biol Blood Marrow Transplant. 2014;20(12):1975-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powell JD, Lerner CG, Schwartz RH. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J Immunol. 1999;162(5):2775-2784. [PubMed] [Google Scholar]
- 42.Powell JD, Fitzhugh C, Kang EM, Hsieh M, Schwartz RH, Tisdale JF. Low-dose radiation plus rapamycin promotes long-term bone marrow chimerism. Transplantation. 2005;80(11):1541-1545. [DOI] [PubMed] [Google Scholar]
- 43.O’Donnell PV, Luznik L, Jones RJ, et al. . Nonmyeloablative bone marrow transplantation from partially HLA-mismatched related donors using posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2002;8(7):377-386. [DOI] [PubMed] [Google Scholar]
- 44.Luznik L, Jalla S, Engstrom LW, Iannone R, Fuchs EJ. Durable engraftment of major histocompatibility complex-incompatible cells after nonmyeloablative conditioning with fludarabine, low-dose total body irradiation, and posttransplantation cyclophosphamide. Blood. 2001;98(12):3456-3464. [DOI] [PubMed] [Google Scholar]
- 45.Colson YL, Wren SM, Schuchert MJ, et al. . A nonlethal conditioning approach to achieve durable multilineage mixed chimerism and tolerance across major, minor, and hematopoietic histocompatibility barriers. J Immunol. 1995;155(9):4179-4188. [PubMed] [Google Scholar]
- 46.Colson YL, Li H, Boggs SS, Patrene KD, Johnson PC, Ildstad ST. Durable mixed allogeneic chimerism and tolerance by a nonlethal radiation-based cytoreductive approach. J Immunol. 1996;157(7):2820-2829. [PubMed] [Google Scholar]
- 47.Kanakry CG, Ganguly S, Zahurak M, et al. . Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci Transl Med. 2013;5(211):211ra157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luznik L, O’Donnell PV, Symons HJ, et al. . HLA-haploidentical bone marrow transplantation for hematologic malignancies using nonmyeloablative conditioning and high-dose, posttransplantation cyclophosphamide. Biol Blood Marrow Transplant. 2008;14(6):641-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shenoy S, Eapen M, Panepinto JA, et al. . A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. 2016;128(21):2561-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dallas MH, Triplett B, Shook DR, et al. . Long-term outcome and evaluation of organ function in pediatric patients undergoing haploidentical and matched related hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant. 2013;19(5):820-830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ciurea SO, Mulanovich V, Saliba RM, et al. . Improved early outcomes using a T cell replete graft compared with T cell depleted haploidentical hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2012;18(12):1835-1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solomon SR, Sizemore CA, Sanacore M, et al. . Haploidentical transplantation using T cell replete peripheral blood stem cells and myeloablative conditioning in patients with high-risk hematologic malignancies who lack conventional donors is well tolerated and produces excellent relapse-free survival: results of a prospective phase II trial. Biol Blood Marrow Transplant. 2012;18(12):1859-1866. [DOI] [PubMed] [Google Scholar]
- 53.Esteves I, Bonfim C, Pasquini R, et al. . Haploidentical BMT and post-transplant Cy for severe aplastic anemia: a multicenter retrospective study. Bone Marrow Transplant. 2015;50(5):685-689. [DOI] [PubMed] [Google Scholar]
- 54.Machado RF. Sickle cell anemia-associated pulmonary arterial hypertension. J Bras Pneumol. 2007;33(5):583-591. [DOI] [PubMed] [Google Scholar]
- 55.McClellan AC, Luthi JC, Lynch JR, et al. . High one year mortality in adults with sickle cell disease and end-stage renal disease. Br J Haematol. 2012;159(3):360-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raimondi R, Tosetto A, Oneto R, et al. . Validation of the Hematopoietic Cell Transplantation-Specific Comorbidity Index: a prospective, multicenter GITMO study. Blood. 2012;120(6):1327-1333. [DOI] [PubMed] [Google Scholar]
- 57.Sorror ML, Maris MB, Storb R, et al. . Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106(8):2912-2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anurathapan U, Hongeng S, Pakakasama S, et al. . Hematopoietic stem cell transplantation for homozygous β-thalassemia and β-thalassemia/hemoglobin E patients from haploidentical donors. Bone Marrow Transplant. 2016;51(6):813-818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lê PQ, Gulbis B, Dedeken L, et al. . Survival among children and adults with sickle cell disease in Belgium: Benefit from hydroxyurea treatment. Pediatr Blood Cancer. 2015;62(11):1956-1961. [DOI] [PubMed] [Google Scholar]
- 60.Couque N, Girard D, Ducrocq R, et al. . Improvement of medical care in a cohort of newborns with sickle-cell disease in North Paris: impact of national guidelines. Br J Haematol. 2016;173(6):927-937. [DOI] [PubMed] [Google Scholar]