

Abstract
Importance
Initial results of this intergroup trial of imatinib for patients with metastatic/ unresectable GIST were reported in 2008 (http://www.ncbi.nlm.nih.gov/pubmed/18235122). Updated results reported here show long-term survival with a significant subset of patients surviving 10 or more years, as well as new molecular disease insights.
Obective
To determine long-term survival of patients treated on SWOG study S0033, and to present new molecular data regarding treatment outcomes.
Design, Setting and Participants
Patients were required to have advanced GIST that was not surgically curable. Updated clinical information was obtained, including post-progression therapies. Using modern sequencing technologies, we analyzed 20 cases originally classified as “wild-type”. This intergroup study was coordinated by SWOG, a cooperative group member within the National Clinical Trials Network, with participation by member/affiliate institutions.
Interventions
Patients were randomly assigned to one of two dose levels of imatinib: 400 mg once daily vs. 400 mg twice daily, and were treated until disease progression or unacceptable toxicity.
Main Outcome Measure
The primary end point was overall survival. The primary aim of this report was to correlate updated survival with clinical and molecular factors, as well as to enumerate and describe patterns of post-imatinib therapies in long-term survivors.
Results
Of 695 eligible patients, 189 survived 8 years or longer, 95 on the 400 mg dose arm and 94 on the 800 mg arm. The 10-year estimate of overall survival (OS) is 23%. Among 142 long-term survivors, imatinib was the sole therapy administered in 49%, with additional systemic agents administered to 54 patients (38%). Resequencing studies of 20 cases originally classified as KIT/PDGFRA wild-type GIST revealed that 17 (85%) harboring a pathogenic mutation, most commonly a mutation of a subunit of the succinate dehydrogenase (SDH) complex. We report the first data on SDH-deficient GIST patients treated with imatinib in a prospective therapeutic study.
Conclusions and Relevance
A subset of patients with metastatic GIST enjoys durable long-term overall survival on imatinib. Although this study provides guidance for management of GIST harboring the most common KIT and PDGFRA mutations, optimal management of other genotypic subtypes remains unclear.
INTRODUCTION
Gastrointestinal stromal tumor (GIST) is the most common sarcoma of the gastro-intestinal tract. It comprises less than 1% of all gastrointestinal tumors and has an annual incidence of approximately 7-10 cases per million as determined by multiple population-based studies.1–5 A major breakthrough occurred with the discovery of activating mutations of the KIT gene resulting in oncogenic constitutive signaling in the majority of GISTs and the subsequent use of KIT (CD117) immunostaining as the first diagnostic marker for GIST.6,7
Prior to the year 2000, GIST was documented to be highly resistant to cytotoxic chemotherapy, with no available effective treatment and a uniformly grim prognosis for patients with metastatic or unresectable disease.8, 9,10 A brief report published in 2001 described the impressive effects of a pilot proof-of-concept protocol using the tyrosine kinase inhibitor imatinib mesylate in a patient with metastatic GIST harboring a KIT exon 11-mutation that had been previously refractory to chemotherapy.11 Since then, several phase II and III trials in metastatic disease were conducted, confirming the efficacy of imatinib in metastatic GIST.12–16
A phase II study (B2222) initially reported in 2002, was the first multicenter trial designed specifically for advanced GIST to report that imatinib produced high response rates and lasting disease control.12,15 A follow-up report from this study in 2008 showed an overall median survival of 4.75 years for the 147 patients treated, with 41 patients (28%) remaining on the drug long-term. The presence of a KIT exon 11 mutation was associated with better survival. Estimated median survival was 5.25 years for patients with KIT exon 11 mutations and 3.67 years for those with KIT exon 9 mutations.15, A subsequent analysis of this trial found that 26 (17.7%) of the total 147 patients entered onto this study had remained on imatinib therapy, with a median follow-up time of 9.4 years. 17 The estimated 9-year OS rate for all patients was 35%. No data was provided about other therapeutic modalities, such as surgical resection of metastatic lesions or other post-progression systemic therapies, which might have contributed to these overall survival results.
A large SWOG-directed randomized phase III intergroup study, S0033, was designed and conducted to compare the outcome of patients with metastatic and/or unresectable (“surgically incurable”) GIST randomized to be treated with imatinib either at an initial dose of 400 mg or 800 mg daily. We previously reported a median OS of 4.58 years for 345 patients treated with conventional dose imatinib, and 4.25 years for 349 patients treated on the high dose arm.14 Further long-term results of this study are now described in this paper, along with additional analyses of GIST tumors originally classified as “KIT/PDGFRA wild-type” (KIT/PDGFRA WT) GIST, using next generation sequencing techniques. We also have correlated GIST genotypes with clinical outcomes during treatment, including a cohort of patients with SDH-deficient GIST.
METHODS
S0033 Study Population
The S0033 trial accrued patients from December 15, 2000 to September 1, 2001 from four cancer clinical trial cooperative groups (SWOG, CALGB, ECOG and NCIC-CTG) and from the UT MD Anderson Cancer Center. For the original analysis, approval by the Institutional Review Board of each participating institution was obtained, with written informed consent obtained from each participant. Data collection and analyses for the trial, registered at ClinicalTrials.gov Identifier: NCT00009906, were performed by the SWOG Statistical Center (Figure 1, Consort Figure).
Figure 1.
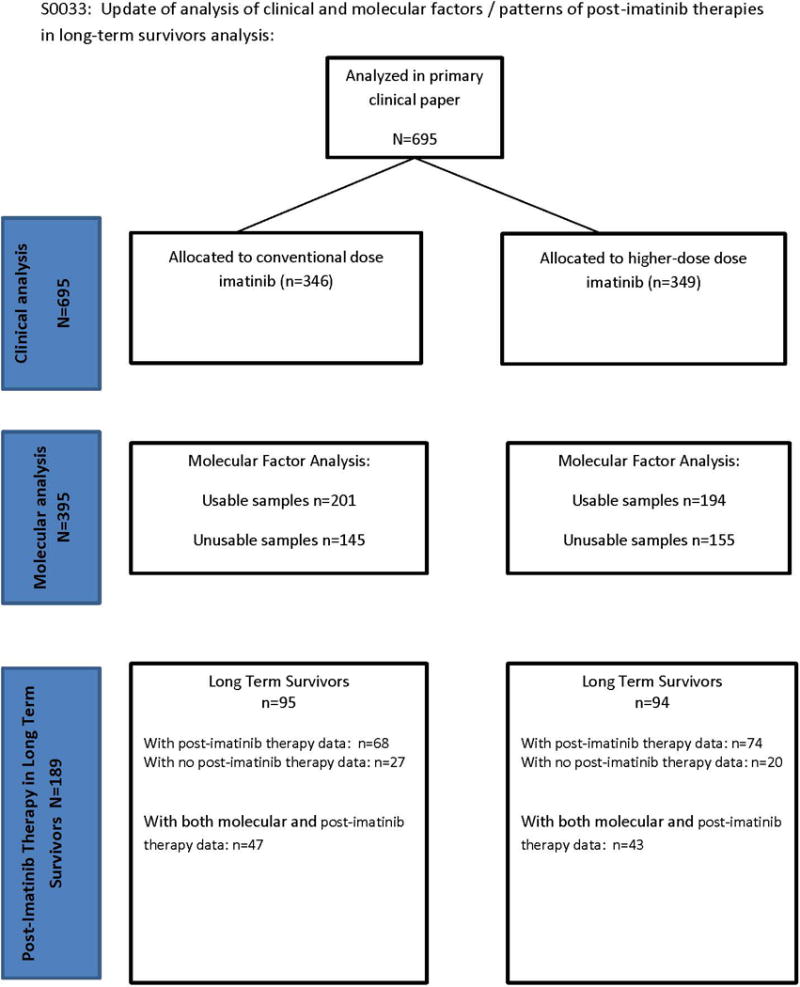
Consort flow chart of clinical study.
Patients were required to have biopsy proven metastatic and/or unresectable GIST of visceral or abdominal origin and with immunohistochemical demonstration of KIT expression documented by DAKO antibody staining. Complete details and results from the clinical study were previously reported.14 Tumor samples were sent to the Oregon Health & Science University where tumor genotyping was centrally assessed. Initial results of the KIT and PDGFRA genotyping and correlation with clinical outcomes were also previously published.18
Ten years after initiation of accrual on this study, investigators following patients last known to be alive were contacted to update follow-up. Patients known to have lived eight years or more were defined as long-term survivors. A two page data form was created to obtain fadditional information about these long-term survivors. Use of additional therapies after discontinuation of imatinib on this study was tabulated for these long-term survivors. The primary aim of this report was to correlate updated survival with clinical and molecular factors, as well as to enumerate and describe patterns of post-imatinib therapies of the long-term survivors.
Statistical Analysis
The distribution of overall survival (OS) was estimated by the Kaplan-Meier method.19 Proportional hazards regression models were used to investigate the prognostic impact of the following variables on OS: age, sex, performance status (0, 1, 2, 3), time from initial diagnosis (in years), primary disease site (small bowel vs. other), maximum diameter of largest tumor, prior surgery, chemotherapy or RT, baseline WBC, hemoglobin, ANC, platelets, bilirubin, albumin and creatinine. For baseline WBC, HGB, ANC, PLT, bilirubin and creatinine, a log transformation was used in the regression models. Initially, each factor was assessed in a univariate fashion. Subsequently, multivariable models were performed using the factors found to be significant in the univariate models at the p=0.05 using a backward selection model. Associations between genotype and patient characteristics were tested using Chi-Square of Fisher’s exact test. P-values are unadjusted for multiple comparisons.
Targeted Exome Sequencing methods
These analyses were performed as previously described. 20 Additional details are included in our supplemental methods section.
RESULTS
Our initial clinical report found no statistical difference in OS, PFS or response between the treatment arms of imatinib 400 mg daily vs. 400 mg twice daily.14 Similarly, neither the B2222 phase II study reported by Blanke et al. nor the EORTC phase III study using an identical study design reported by Verweij et al. found an OS difference in outcomes between standard and higher doses of imatinib. However, Verweij et al. reported a PFS difference in the KIT exon 9 mutant subset in favor of the high-dose imatinib treatment arm.13, 16 In our study of 695 patients, 556 have died with a median OS of 52 months (95% CI: 48 to 61 months) (Figure 2A, Supplemental Table 1). The 10-year OS estimate is 23% (95% CI: 20% – 26%). The 10 year PFS estimate is 7% (95 CI: 6% - 10%), Figure 2B, Supplemental Table 1).
Figure 2.
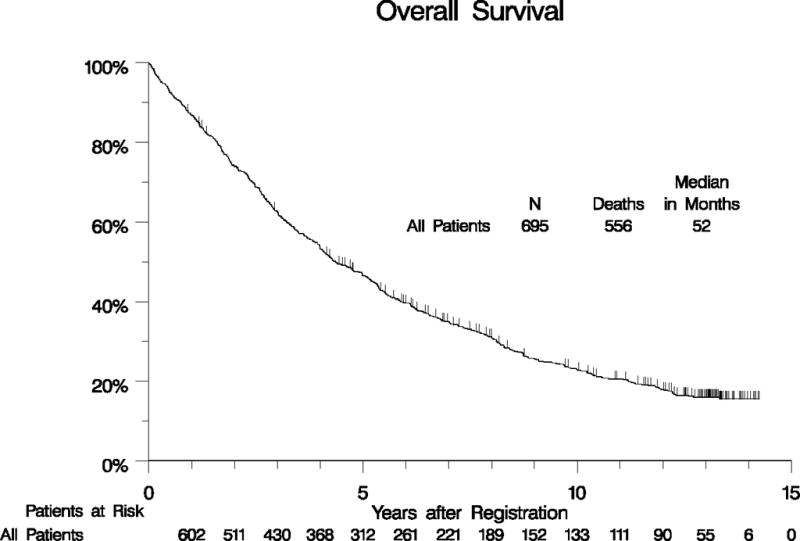
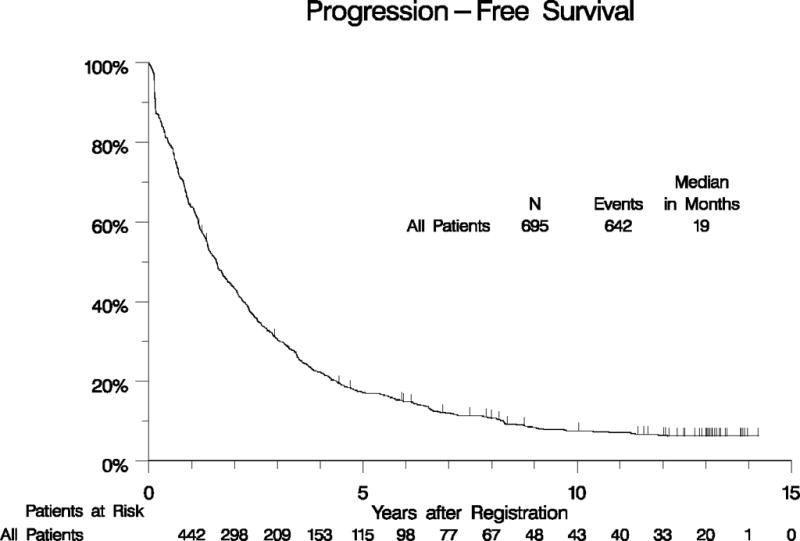
Long-term OS (panel A) and PFS (panel B) for all patients.
Of the 346 patients initially assigned to the 400 mg daily dose arm, there were 95 (27%) long-term survivors; this included 72 subjects that remained on low dose imatinib, and 23 subjects who were crossed over to the 800 mg/day dose arm. A total of 130 patients crossed over during study conduct. Amongst the 349 patients initially assigned to the 800 mg daily dose arm, there were 94 (27%) long-term survivors.
The following prognostic factors were identified by univariate analysis as statistically significant with respect to OS: age, sex, performance status, prior chemotherapy, maximum tumor diameter, baseline platelets, hemoglobin, ANC, WBC, bilirubin and albumin. In the multivariate model, 551 of the 695 eligible patients had complete data for all baseline prognostic variables. Using backward selection, multivariate analyses showed that younger age, female gender, good performance status, smaller tumor diameters, lower WBC values and higher albumin values were associated with significantly improved OS. The p-values and hazard ratios are given below (Supplemental Table 2) for both univariate and multivariate analyses. The influence of these prognostic factors on the estimate of 10-year survival is listed in Supplemental Table 3. For example, the 10 year survival estimate for patients with a performance status of 0-1 at the time of treatment initiation was 26% (95% CI: 22-30%) compared with 7% (95% CI: 2-13%) for patients with a performance status of 2-3 at the time of starting imatinib therapy.
Genotype results from our original analysis were available for 395 eligible patients, of which 282 (71%) had KIT exon 11 mutations, 67 (17%) had KIT/PDGFRA wild-type (WT) genotype, 32 (8%) had KIT exon 9 mutations, and 14 (4%) had other KIT or PDGFRA mutations. A univariate analysis of OS by KIT exon 11 mutant, KIT exon 9 mutant, and KIT/PDGFRA WT genotypes was performed and revealed median survival times of 66, 38, and 40 months, respectively. When adjusted for all prognostically significant factors found in the analysis of eligible patients, patients with KIT exon 11 mutation had significantly longer OS than those with KIT/PDGFRA WT (p=0.004; Figure 3A, Supplemental Table 1). We also analyzed survival for different classes of KIT exon 11 mutations, including any type of KIT exon 11 deletion vs. insertion/duplication mutations vs. point mutations. No significant difference in OS was found for patients whose GIST harbored different subsets of KIT exon 11 mutations (Figure 3B, Supplemental Table 1).
Figure 3. Overall survival by tumor genotype.
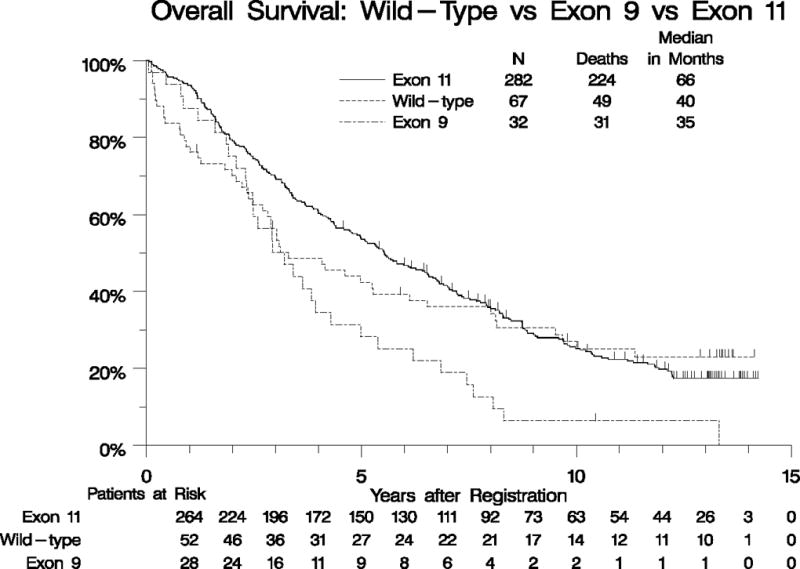
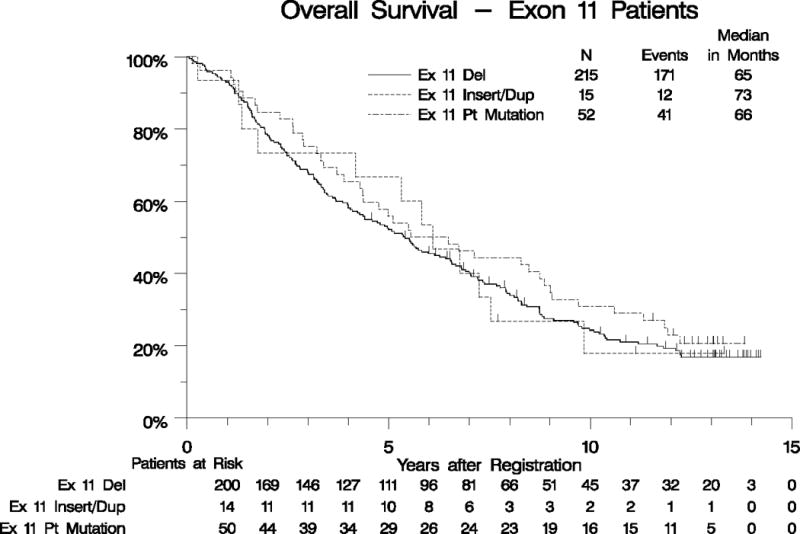
Panel A: OS for patients with KIT Exon11-mutant vs. KIT/PDGFRA WT vs. KIT exon 9-mutant GIST. Panel B: OS for patients with different classes of KIT exon 11-mutant GIST.
When mutation status in long-term survivors was correlated with response [complete response (CR) + partial response (PR)], more responses were seen in the KIT exon 11 mutant genotype group (n=92) than in the KIT/PDGFRA wild-type (WT) (n=21) or KIT exon 9 (n=4) groups: 64 patients (70%), 10 (48%) and 2 (50%), respectively. Median progression free survival (PFS) estimates by arm and mutation status are summarized in Supplemental Table 1. The median PFS for patients with KIT exon 11 mutations was 25 months (95% C.I.: 21-28 months) compared to 17 months (95% C.I.: 9-25 months) for patients with KIT exon 9 mutations and 13 months (95% C.I.:8-18 months) for those with WT genotype (Supplemental Figure 1A). As with our OS analysis, no difference in PFS were found amongst patients whose GIST had different classes of KIT exon 11 mutations (Supplemental Figure 1B, Supplemental Table 1).
Post-Imatinib Progression Therapies during Survival Follow-up
Descriptions of additional therapies received after discontinuation of imatinib on this protocol were obtained for patients defined as long-term survivors (i.e., those known to have lived for at least eight years after enrollment on this trial). This additional follow-up was not planned as part of the initial study. Of the 189 long-term survivors, additional treatment information obtained from 142 patients indicates that 73 (51%) received other therapies. Fifty-four (38%) patients received additional systemic agents; sunitinib (41/142; 30%) and sorafenib (17/142; 12%) were the most commonly utilized additional therapies among the long-term survivors. Subsequent surgeries, radiofrequency ablation and radiation therapy were also reported in 29%, 7%, and 4%, respectively (Supplemental Table 4). Of the 395 eligible patients with a known GIST genotype, 117 are long-term survivors (survival known to be at least eight years after randomization). Of these, 90 (81%) had information on additional non-protocol therapy.
Resequencing studies from a subset of cases previously identified as KIT/PDGFRA WT
At the time of study initiation, KIT mutations were the only known pathogenic abnormality associated with GIST. Following the completion of enrollment of patients to this study (September, 2001), a number of additional pathogenic events in GIST were described, including PDGFRA, RAS, and BRAF mutations, loss of SDHB protein expression due to mutation or epimutation, and the association of GIST with neurofibromatosis.21–24 KIT and PDGFRA mutations were the only analyses performed in our original report, due to limitations of testing that existed at that time. However, significant advances have improved our ability to sequence tumor DNA from formalin-fixed, paraffin embedded tumors and to test for multiple genomic abnormalities in the same analysis. We identified 20 KIT/PDGFRA WT cases for which there was sufficient residual DNA from our original studies for re-analysis using a targeted exome panel for GIST-associated pathogenic abnormalities. We identified the putative causative mutation in 17 of the 20 cases (85%) (Supplemental Table 5). The most commonly mutated pathway in our analysis was the succinate dehydrogenase complex (SDH), with mutation in 12/20 cases (60%; SDHA 9, SDHB 2, SDHC 1). We also identified two cases with NF1 loss as the presumed pathogenic mutation. Finally, we found three cases with KIT mutations that were missed in our original analysis (KIT exon 13 K642E, KIT exon 11 V559D, and KIT exon 9 K509I, 1 case each). Three cases (15%) of our original KIT/PDGFRA WT cohort did not contain an identifiable pathogenic mutation using our targeted panel. Due to the lack of residual unstained slides, we could not perform SDHB immunohistochemistry on these three cases. Therefore, we do not know whether these represent so called “quadruple WT GIST” or cases with SDH deficiency with alterations that could not be detected using our methods (e.g. epimutation).25,26
Our collection of 12 patients with SDH-deficient GIST represents the largest series of such patients treated uniformly with imatinib as part of a prospective clinical study. The demographics and baseline characteristics of these patients are detailed in Table 1, and compared to patients with KIT exon 11-mutant GIST. As expected, the patients with SDH-mutant GIST were younger than the patients with KIT exon 11-mutant GIST treated patients, and 100% of the primary tumors arose outside the small bowel (presumably all gastric), compared with 66% non-small bowel for KIT exon 11-mutant tumors (p=0.02). The response rate of the SDH-mutant GIST patients was 8% (0% CR, 8% PR) versus 66% (6% CR, 59% PR, p=0.0001), for the patients with KIT exon 11-mutant tumors. Median OS was similar for both groups, although the confidence interval for the small SDH-deficient population was quite broad (116 months vs. 66 months, p= 0.38; Table 1, Figure 4A). Likewise, median PFS was not significantly different for patients with SDH-deficient tumors compared with KIT exon 11-mutant GIST (9 vs. 24 months, p= 0.07; adjusted for all prognostically significant variables; Table 1, Figure 4B).
Table 1. Patient Characteristics for Cases with SDH Mutations vs Original Exon 11 Mutations.
| SDH-mutant (n=12) |
Original KIT Exon 11 (n=282) |
p-value | |||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Treatment Arm | |||||
| Low-Dose | 7 | 58% | 140 | 50% | |
| High-Dose | 5 | 42% | 142 | 50% | |
| AGE AT STUDY ENTRY | <0.0001 | ||||
| Median (range) | |||||
| < 40 years | 7 | 58% | 24 | 8% | |
| 40 to <50 years | 2 | 17% | 36 | 13% | |
| 50 to <60 years | 3 | 25% | 67 | 24% | |
| 60 to <70 years | 0 | 0% | 74 | 26% | |
| ≥ 70 years | 0 | 0% | 81 | 29% | |
| GENDER | 0.03 | ||||
| Female | 9 | 75% | 120 | 43% | |
| Male | 3 | 25% | 162 | 57% | |
| RACE | 0.45 | ||||
| White | 9 | 75% | 231 | 82% | |
| Black | 1 | 8% | 30 | 11% | |
| Asian | 2 | 17% | 16 | 6% | |
| Pacific Islander | 0 | 0% | 1 | <1% | |
| Native American | 0 | 0% | 1 | <1% | |
| Unknown | |||||
| Ethnicity | 0.14 | ||||
| Non-Hispanic | 10 | 83% | 266 | 94% | |
| Hispanic | 2 | 17% | 8 | 3% | |
| Unknown | 0 | 0% | 8 | 3% | |
| PERFORMANCE STATUS | 0.23 | ||||
| 0-1 | 12 | 100% | 237 | 85% | |
| 2-3 | 0 | 0% | 43 | 15% | |
| TYPE OF DISEASE | 0.34 | ||||
| Measurable | 12 | 100% | 262 | 93%% | |
| Non-measurable | 0 | 0% | 20 | 7% | |
| PRIOR CHEMOTHERAPY | 0.75 | ||||
| No | 9 | 82% | 206 | 78% | |
| Yes | 2 | 18% | 59 | 22% | |
| PRIOR SURGERY | 0.89 | ||||
| No | 1 | 8% | 20 | 7% | |
| Yes | 11 | 92% | 256 | 98% | |
| FAMILY HISTORY OF GIST | 0.08 | ||||
| No/unknown | 11 | 92% | 281 | 100% | |
| Yes | 1 | 8% | 1 | <1% | |
| PRIOR RT | 0.21 | ||||
| No | 11 | 100% | 238 | 88% | |
| Yes | 0 | 0% | 33 | 12% | |
| SMALL BOWEL ORIGIN | 0.02 | ||||
| No | 11 | 100% | 172 | 66% | |
| Yes | 0 | 0% | 90 | 34% | |
| Abdomen | 2 | 18% | 56 | 21% | |
| Stomach | 9 | 82% | 108 | 41% | |
| Small Bowel | 0 | 0% | 90 | 34% | |
| Other | 0 | 0% | 8 | 3% | |
| MAXIMUM DIAMETER | 0.35 | ||||
| < 4.0 cm | 2 | 17% | 63 | 22% | |
| 4.0 - 8.0 cm | 6 | 50% | 72 | 26% | |
| 8.0 - 12.0 cm | 1 | 8% | 67 | 24% | |
| ≥ 12.0 cm | 3 | 25% | 70 | 25% | |
| unknown | 0 | 0% | 10 | 3% | |
| TIME SINCE INITIAL DIAGNOSIS | 0.58 | ||||
| < 12 months | 0 | 0% | 7 | 2% | |
| ≥ 12 months | 12 | 100% | 275 | 98% | |
| Hemoglobin | 12.4 (9.6 – 14.0) | 12.8 (8.4 – 19.5) | |||
| ANC | 3.7 (1.7 – 5.7) | 4.6 (1.3 – 18.5) | |||
| WBC | 5.5 (3.7 – 8.2) | 7.1 (3.5 – 21.5) | |||
| Platelets | 233 (131 – 1188) | 271 (103 – 890) | |||
| Albumin | 4.2 (3.7 – 4.7) | 3.8 (2.0 – 8.3) | |||
| Bilirubin | 0.35 (0.2 – 0.6) | 0.52 (0.10 – 8.00) | |||
| Creatinine | 0.78 (0.09 – 1.10) | 0.9 (0.12 – 2.00) | |||
| Response Rate | 8% (0% – 48%) | 66% (60% – 71%) | 0.0001 | ||
| CR (confirmed and unconfirmed) | 0% (0% – 26%) | 6% (3% – 10%) | |||
| PR (confirmed and unconfirmed) | 8% (0% – 48%) | 59% (53% – 65%) | |||
| Stable | 50% (21% – 79%) | 18% (14% – 23%) | |||
| Increasing Disease / Not Assessable | 33% (10% – 65%) | 16% (12% – 21%) | |||
| Median OS | 116 mo. (22 mo. – 116 mo.) | 66 mo. (57 mo. – 78 mo.) | p=0.38* | ||
| Median PFS | 9 mo. (3 mo. – 58 mo.) | 24 (21mo. – 28 mo.) | p=0.07* | ||
Adjusted for all prognostically significant factors found in the analysis of eligible patients.
Figure 4.
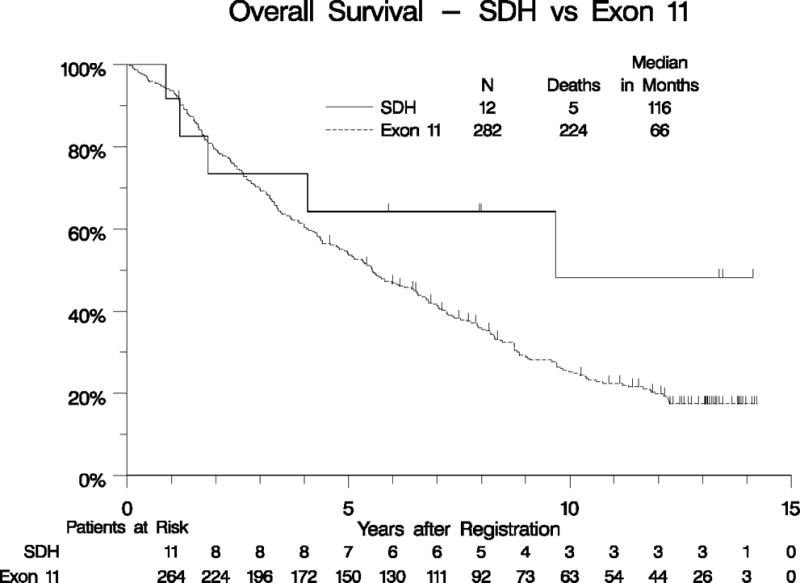
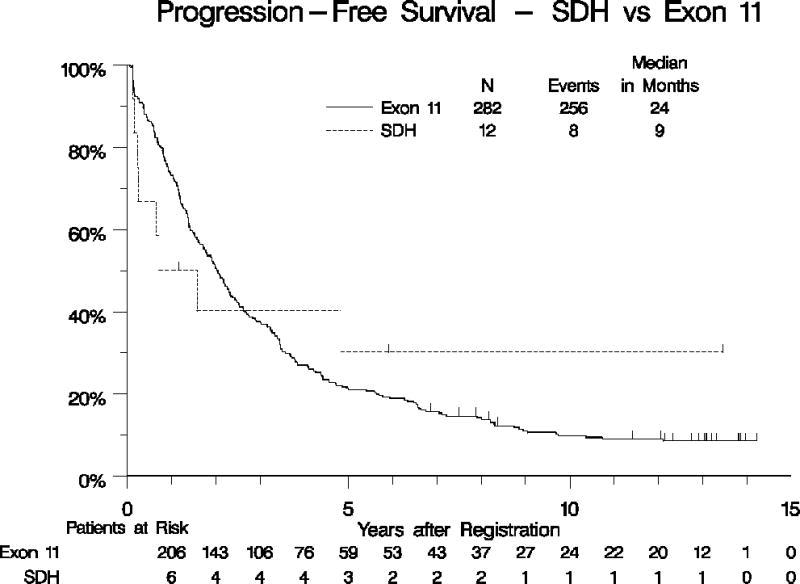
Overall survival (panel A) and PFS (panel B) for patients with KIT exon 11-mutant GIST vs. SDH-mutant GIST.
DISCUSSION
As previously noted, prior to the clinical development of imatinib, metastatic GIST was a uniformly fatal disease with a rapidly progressive course and a median overall survival of less than two years. In the initial publication of the results from S0033, we reported on median survival and molecular biomarkers associated with response to imatinib. With a much longer follow up period, we now report that a significant subset of GIST patients experience long-term survival with front-line imatinib treatment of advanced and metastatic disease, with an estimated 8-year and 10-year survival of 31% and 23%, respectively. Notably, patients with advanced GIST treated with imatinib alone have experienced survival without report of progression for periods that approach, and even exceed, one decade. In the subset of patients who have survived more than 8 years since study entry, 51% received additional non-protocol therapy, with less than 40% receiving further systemic therapies beyond imatinib.
In this study, tumor bulk at the time of initiation of imatinib therapy significantly influences PFS and OS. This finding is in agreement with previous studies of imatinib therapy of advanced GIST.28,29 The influence of tumor bulk (CR vs PR vs SD) on PFS was also noted in the randomized imatinib discontinuation studies reported by the French Sarcoma Group.30 Taken together, these observations raise the hypothesis that surgical tumor debulking may enhance the duration of imatinib response and disease control, at least in some patients. Several non-randomized, retrospective surgical series have suggested a benefit to surgical removal of metastatic lesions in imatinib-treated patients. This hypothesis was tested in a randomized phase III study, reported by Du et al.31 In their study, patients were randomized between 3 and 13 months after starting imatinib to eitherundergo surgery for residual disease with the goal of removing macroscopic disease as completely as possible or to remain on imatinib therapy without surgical intervention. The primary endpoint was 2-year PFS. Unfortunately, the study was closed early due to poor accrual with only 41 patients enrolled out of a planned 210. There was no advantage to surgery in terms of PFS (2-year PFS 88% for combined treatment vs. 58% for medical therapy only, p=0.09). However, the secondary endpoint of OS was significantly improved by combination treatment (median OS not reached for surgery vs. 49 months for imatinib-only treatment, p=0.024). These data suggest that surgical debulking may maximize the imatinib response for some patients, but it remains unclear how to optimally integrate surgery and imatinib treatment in the management of patients with metastatic GIST.
The resequencing studies reported here confirm the hypothesis that the vast majority of GISTs harbor identifiable pathogenic mutations. In our original report, we were able to identify the presumed causative mutation in 85% of GIST (KIT and PDGFRA analysis only). Using next-generation sequencing technologies, such as those described above, we now estimate that 97.5% of GIST can be assigned a pathogenic genotype. The remaining 2.5% of GIST represent either GIST with SDH deficiency due to mutations outside of the four SDH subunits that affect SDH complex function or GIST with undiscovered pathogenic mechanisms (quadruple WT GIST). The objective response rate to imatinib treatment was significantly lower for SDH-mutant GIST when compared with KIT exon 11-mutant GIST (8% vs. 66%, Fisher’s exact p=0.001) based on post-hoc analyses. The single responding SDH-mutant GIST patient had a verified 62% decrease in tumor size which lasted for 4 years before progression was noted. These results are consistent with a recent retrospective case series that reported 1 partial response out of 49 SDH-deficient GIST patients treated with imatinib. Our results also identify GIST with KIT exon 11 mutations or lacking KIT/PDGFRA mutations (most of which are SDH-deficient) as having longer median OS when compared with KIT exon 9-mutant GIST. Given prior reports of cases of indolent behavior of untreated SDH-deficient metastatic GIST, it is unclear whether imatinib alters the natural history of most cases of KIT/PDGFRA WT GIST. Further studies are required to better define the role of imatinib therapy in the treatment of this type of GIST.
CONCLUSION
In summary, our results provide important data on the long-term outcomes of patients with metastatic GIST treated with imatinib. Further studies are needed to improve on these results using other strategies that might incorporate surgery, combination medical therapy (additional signaling pathway inhibitors or immune modulation agents), or some form of intermittent therapy. Alternatively, enhanced imaging and biomarker analysis to detect residual GIST cells in long-term responding patients may identify those patients who might benefit from less intense medical therapy.
Supplementary Material
Acknowledgments
Conflict of Interest Disclosures: Dr. Baker has consulting/advisory role with Tevan and Morphotek; Dr. Borden has stock/other ownership with Alios BioPharma; he has patient/intellectual property through Medical College of Wisconsin, University of Wisconsin, Cleveland Clinic; Dr. Benjamin has consulting/advisory role with Novartis; Dr. Corless has consulting/advisory role with Roche and Asuragen; he receives research funding through his institution from Roche; he receives travel, accommodations, or expenses from Roche; Dr. Heinrich has stock/other ownership with MolecularMD; he has honoraria to report from Novartis and Pfizer ; he has consulting/advisory role with Novartis, Ariad, Blueprint; he has research funding from Blueprint, Inhibikase, Ariad and Deciphera; he has patent/intellectual property with Novartis (via his institution); he has provided expert testimony for Novartis; Dr. Maki reports honoraria from Novartis andhad a consulting/advisory role with Novartis; he receive research funding through his institution from Novartis; Dr. Owzar has patent/intellectual property through Duke University; Ms. Rankin reports employment with Pathology Associates Medical Laboratories. Dr. Tap has a consulting/advisory role with Novartis.
No conflicts to report: Drs. Blackstein, Blanke, Crowley, Demetri, Fletcher, Priebat, Ryan, Von Mehren.
Funding/Support: Supported in part by the following PHS/DHHS grant numbers awarded by the National Cancer Institute (NCI), National Clinical Trials Network (NCTN): CA180888, CA180819, CA180801, CA180846, CA180835, CA180818, CA180834, CA180828, CA180830, CA180820, CA180821, CA180863; NCI Community Oncology Research Program (NCORP): CA189953, CA189952, CA189954, CA189860, CA189804, CA189822, CA189971, CA189830, CA189858, CA189853, CA189957, CA189854, CA189856; NIH/NCI legacy grants: CA13612, CA 46282, CA63850, CA35119, CA74811, CA45450, CA58686, CA35262, CA46368, CA04919, CA68183, CA58348, CA16385, CA76447, CA46113, CA58416, CA12644, CA37981, CA22433; NCI SPORE Program: 5P50CA127003-08 and U54CA168512-04; Canadian Cancer Society grant 021039; and in part by Novartis. Also sponsored in part by VA Merit Review Grants to MCH (1I01BX000338-01, 2I01BX000338-05) and funding from the GIST Cancer Research Fund (MCH) and the Life Raft Group (MCH).
Role of the Funder/Sponsor: The funders/sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Trial Registration: ClinicalTrials.gov Identifier: NCT00009906
Author Contributions: Dr. Heinrich had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Baker, Blanke, Borden, Crowley, Demetri, Fletcher, Heinrich, Ryan, von Mehren
Acquisition, analysis, or interpretation of data: Baker, Blackstein, Benjamin Blanke, Corless, Crowley, Demetri, Fletcher, Heinrich, Maki, Priebat, Owzar, Rankin, Tap, von Mehren
Drafting of the manuscript: Baker, Benjamin, Blackstein, Blanke, Borden, Corless, Crowley, Demetri, Fletcher, Heinrich, Maki, Owzar, Preibat, Rankin, Ryan, Tap, von Mehren
Critical revision of the manuscript for important intellectual content: Baker, Benjamin, Blackstein, Blanke, Borden, Corless, Crowley, Demetri, Fletcher, Heinrich, Maki, Owzar, Priebat, Rankin, Ryan, Tap, von Mehren
Statistical analysis: Owzar, Rankin
Obtained funding: Blanke, Fletcher, Heinrich
Administrative, technical, or material support: Blanke, Benjamin, Demetri, Maki, Ryan, Tap, von Mehren
Study supervision: Heinrich
References
- 1.Nilsson B, Bümming P, Meis-Kindblom J, et al. Gastrointestinal stromal tumors: The incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era—a population-based study in western Sweden. Cancer. 2005;103(4):821–829. doi: 10.1002/cncr.20862. [DOI] [PubMed] [Google Scholar]
- 2.Mucciarini C, Rossi G, Bertolini F, et al. Incidence and clinicopathologic features of gastrointestinal stromal tumors. A population-based study. BMC Cancer. 2007;7:230. doi: 10.1186/1471-2407-7-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steigen SE, Eide TJ. Trends in incidence and survival of mesenchymal neoplasm of the digestive tract within a defined population of northern Norway. APMIS. 2006;114:192–200. doi: 10.1111/j.1600-0463.2006.apm_261.x. [DOI] [PubMed] [Google Scholar]
- 4.Tryggvason G, Gislason H, Magnusson M, et al. Gastrointestinal stromal tumors in Iceland, 1990-2003: the Icelandic GIST study, a population-based incidence and pathologic risk stratification study. Intl J Cancer. 2005;117(2):289–293. doi: 10.1002/ijc.21167. [DOI] [PubMed] [Google Scholar]
- 5.Tran T, Davila JA, El-Serag HB. The epidemiology of malignant gastrointestinal stromal tumors: an analysis of 1458 cases from 1992 to 2000. Am J Gastroenterol. 2005;100:162–168. doi: 10.1111/j.1572-0241.2005.40709.x. [DOI] [PubMed] [Google Scholar]
- 6.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- 7.Rubin BP, Fletcher JA, Fletcher CD. Molecular Insights into the histogenesis and pathogenesis of gastrointestinal stromal tumors. Int J Surg Pathol. 2000;8(1):5–10. doi: 10.1177/106689690000800105. [DOI] [PubMed] [Google Scholar]
- 8.Platt BE, Hollema H, Molenaar WM, et al. Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: differences in clinical outcome and expression of multidrug resistance protein. J Clin Oncol. 2000;18:3211–3220. doi: 10.1200/JCO.2000.18.18.3211. [DOI] [PubMed] [Google Scholar]
- 9.DeMatteo R, Lewis J, Leung D, Satvinder M, Woodruff J, Brennan M. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231(1):51–58. doi: 10.1097/00000658-200001000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeMatteo RP, Heinrich MC, el-Rifai W, Demetri GD. Clinical management of gastrointestinal stromal tumors: Before and after STI-571. Hum Pathol. 2002;33:466–77. doi: 10.1053/hupa.2002.124122. [DOI] [PubMed] [Google Scholar]
- 11.Joensuu H, Roberts PJ, Sarlomo-Kikala M, et al. Effect of the tryrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344(14):1052–1056. doi: 10.1056/NEJM200104053441404. [DOI] [PubMed] [Google Scholar]
- 12.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 13.Verwij J, Casali P, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomized trial. Lancet. 2004;364(9440):1127–1134. doi: 10.1016/S0140-6736(04)17098-0. [DOI] [PubMed] [Google Scholar]
- 14.Blanke CD, Rankin C, Demetri GD, et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the KIT receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26(4):626–632. doi: 10.1200/JCO.2007.13.4452. [DOI] [PubMed] [Google Scholar]
- 15.Blanke C, Demetri G, Mehren M, et al. Long-term results from a randomized phase II trial of standard-versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 16.From the Gastrointestinal Stromal Tumor Meta-Analysis group (MetaGIST) Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28(7):1247–1253. doi: 10.1200/JCO.2009.24.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Mehren M, Heinrich MC, Jeonsuu H, et al. Follow-up results after 9 years (yrs) of the ongoing, phase II B2222 trial of imatinib mesylate (IM) in patients (pts) with metastatic or unresectable KIT+ gastrointestinal stromal tumors (GIST) J Clin Oncol. 2011;29(suppl) abstr 10016. [Google Scholar]
- 18.Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360–5367. doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaplan EL, Meier R. Nonparametric estimation from incomplete observation. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 20.Grasso C, Butler T, Rhodes K, et al. Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn. 2015;17(1):53–63. doi: 10.1016/j.jmoldx.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–3825. doi: 10.1200/JCO.2004.05.140. [DOI] [PubMed] [Google Scholar]
- 22.Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39(10):1411–1419. doi: 10.1016/j.humpath.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 23.Miettinen M, Lasota J. Gastrointestinal stromal tumors: Review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466–1478. doi: 10.5858/2006-130-1466-GSTROM. [DOI] [PubMed] [Google Scholar]
- 24.Rubin B, Heinrich M, Corless Gastrointestinal stromal tumour. Lancet. 2007;369(9574):1731–1741. doi: 10.1016/S0140-6736(07)60780-6. [DOI] [PubMed] [Google Scholar]
- 25.Nannini M, Astolfi A, Urbini M, et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGFRA/SDH/RAS pathway wild-type GIST) BMC Cancer. 2014;14:685. doi: 10.1186/1471-2407-14-685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urbini M, Astolfi A, Indio V, et al. SDHC methylation in gastrointestinal stromal tumors (GIST): a case report. BMC Medical Genetics. 2015;16:87. doi: 10.1186/s12881-015-0233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonvalot S, Eldweny H, Péchoux CL, et al. Impact of surgery on advanced gastrointestinal stromal tumors (GIST) in the imatinib era. Ann Surg Oncol. 2006;13:1596–1603. doi: 10.1245/s10434-006-9047-3. [DOI] [PubMed] [Google Scholar]
- 29.Bauer S, Hartmann JT, de Wit M, et al. Resection of residual disease in patients with metastatic gastrointestinal stromal tumors responding to treatment with imatinib. Int J Cancer. 2005;117:316–325. doi: 10.1002/ijc.21164. [DOI] [PubMed] [Google Scholar]
- 30.Le Cesne A, Ray-Coquard I, Bui B, et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomized phase 3 trial. Lancet Oncol. 2010;11(10):942–949. doi: 10.1016/S1470-2045(10)70222-9. [DOI] [PubMed] [Google Scholar]
- 31.Du CY, Zhou Y, Song C, et al. Is there a role of surgery in patients with recurrent or metastatic gastrointestinal stromal tumours responding to imatinib: a prospective randomised trial in China. Eur J Cancer. 2014;50(10):1772–8. doi: 10.1016/j.ejca.2014.03.280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.