

Abstract
Several hereditary neurological and neuromuscular diseases are caused by an abnormal expansion of trinucleotide repeats. To date, there have been ten of these trinucleotide repeat disorders associated with an expansion of the codon CAG encoding glutamine (Q). For these polyglutamine (polyQ) diseases, there is a critical threshold length of the CAG repeat required for disease, and further expansion beyond this threshold is correlated with age of onset and symptom severity. PolyQ expansion in the translated proteins promotes their self-assembly into a variety of oligomeric and fibrillar aggregate species that accumulate into the hallmark proteinaceous inclusion bodies associated with each disease. Here, we review aggregation mechanisms of proteins with expanded polyQ-tracts, structural consequences of expanded polyQ ranging from monomers to fibrillar aggregates, the impact of protein context and post translational modifications on aggregation, and a potential role for lipids membranes in aggregation. As the pathogenic mechanisms that underlie these disorders are often classified as either a gain of toxic function or loss of normal protein function, some toxic mechanisms associated with mutant polyQ tracts will also be discussed.
Introduction
A major genetic cause of a variety of neurological and neuromuscular diseases is an expansion of trinucleotide repeats to a number much larger than what occurs in the normal, stable gene.1–3 Diseases caused by these expansions are often termed trinucleotide repeat disorders (TRDs). In the majority of these disorders, the codon CAG, which encodes for glutamine (Q), is repeated, resulting in an expanded polyglutamine (polyQ) tract in the translated protein. This subset of TRDs is commonly referred to as CAG repeat disorders or polyQ diseases. To date, ten CAG repeat disorders have been identified (Table 1). The nature of the proteins containing an expanded polyQ domain and the associated pathologies are varied (Figure 1).4, 5 That is, each mutant polyQ protein underlies a distinct neurodegenerative disease that targets different populations of neurons. These polyQ diseases include Huntington’s disease (HD), the Spinocerebellar ataxias, type 1 (SCA-1), type 2 (SCA-2), type 3 (SCA-3, also known as Machado-Joseph disease), type 6 (SCA-6), type 7 (SCA-7), Dentatorubropallidoluysian atrophy (DRPLA) and Spinobulbar muscular atrophy (SBMA, also known as Kennedy’s disease).6–18 In addition CTG/CAG expansion in the JPH3 gene is associated with Huntington Disease-like 2; although, it has not formally been classified as a polyQ disorder.19
Table 1.
CAG-repeat disorders
| Disease | Common Name | Protein | PolyQ* |
|---|---|---|---|
| Huntington Disease (HD) | Huntingtin | 36–100 | |
| Spinobulbar Muscular Atrophy (SBMA) | Kennedy’s Disease | Androgen Receptor | 38–65 |
| Dentatorubral-pallidoluysian atrophy (DRPLA) | Haw River Syndrome | Atrophin-1 | 49–88 |
| Spinocerebellar Ataxia Type 1 (SCA1) | Ataxin-1 | 39–88 | |
| Spinocerebellar Ataxia Type 2 (SCA2) | Ataxin-2 | 33–77 | |
| Spinocerebellar Ataxia Type 3 (SCA3) | Machado-Joseph | Ataxin-3 | 55–86 |
| Spinocerebellar Ataxia Type 6 (SCA6) | CACNA1A | 21–33 | |
| Spinocerebellar Ataxia Type 7 (SCA7) | Ataxin-7 | 38–120 | |
| Spinocerebellar Ataxia Type 12 (SCA12) | PPP2R2B | 66–78 | |
| Spinocerebellar Ataxia Type 17 (SCA17) | TATA-box Binding Protein | 47–63 |
Indicates the typical pathogenic polyQ range associated with each disease.
Figure 1. Schematic representation of various proteins associated with CAG-disorders.
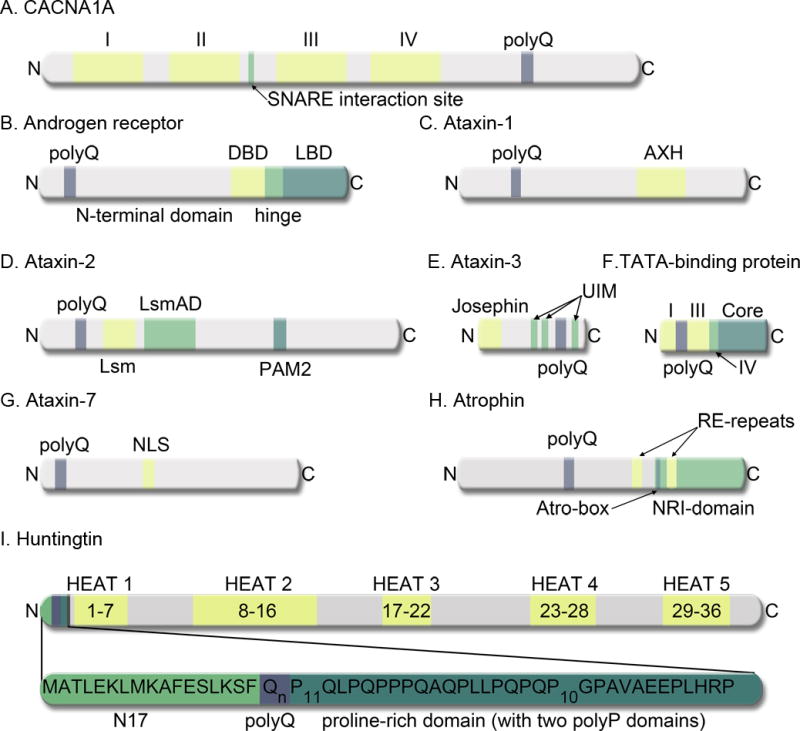
A, CACNA1A, in addition to the polyQ tract, contains four homologous domains (I–IV), each with six transmembrane segments. There is a SNARE interacting site between domains II and III. B, Androgen receptor, in addition to the polyQ tract, contains an N-terminal domain (NTD), a DNA-binding domain (DBD), a hinge region, and a ligand-binding domain (LBD). C, Ataxin-1 contains a polyQ tract and an AXH domain. D, Ataxin-2, contains the polyQ domain, a Like RNA splicing domain Sm1 and Sm2 (Lsm), a Like-Sm-associated domain (LsmAD), and a poly (A)-binding protein interacting motif 2 (PAM2). E, Ataxin-3 contains an N-terminal Josephin domain, three Ub-interacting motifs (UIM), and the polyQ domain. F, The N-terminal region of the Tata-box binding protein consists of four domains (I-IV) followed by a core region. The polyQ region is domain II. G, Ataxin-7 contains a polyQ domain and a putative nuclear localization signal (NLS). H, Atrophin-1, in addition to the polyQ tract, contains two arginine-glutamic acid dipeptide repeats (RE repeats), a nuclear receptor interacting domain (NRI domain), and a highly conserved Atro-box domain. I, The full-length htt protein contains several HEAT repeats. The inset indicates the location of htt exon1, with the N17, polyQ, and proline-rich domains indicated.
While the genes associated with the different polyQ diseases are structurally and functionally distinct, there are commonalities across all of these diseases. With SBMA being a notable exception due to it being X-linked, polyQ diseases are inherited in an autosomal dominant manner. PolyQ diseases are progressive, usually fatal disorders. For each disease, there is a critical threshold of polyQ expansion that must be exceeded for disease onset, and both age of onset and symptom severity are strongly correlated to increasing polyQ expansion beyond this critical threshold (Table 1). For example in HD, polyQ lengths shorter than 35 are not associated with disease, 35–39 repeats may induce disease, 40–60 repeats result in adult onset, and repeats longer than 60 elicit juvenile forms of HD.20, 21 This correlation with polyQ length, the dominant inheritance pattern of these diseases, the late onset of symptoms, and indistinguishable clinical presentation in homozygote and heterozygote patients suggest a toxic gain of function associated with proteins containing expanded polyQ tracts. However, polyQ expansion may also inhibit the normal functions of these proteins, i.e. a toxic loss of function.22
Disease-related proteins containing expanded polyQ tracts, as well as synthetic polyQ peptides, aggregate into detergent-insoluble, amyloid-like fibrils.23–28 There is also a direct correlation between the rate of fibril formation and polyQ length, with longer polyQ tracts resulting in increased rates of fibril formation.24, 29–31 This correlation between aggregation rate and polyQ length is recapitulated in a variety of cell culture models.32–34 Beyond the formation of fibrillar aggregates, proteins containing expanded polyQ tracts also accumulate into a variety of other aggregates, including small and large oligomers, annular structures, and amorphous aggregates.25, 30, 35–38 Precise characterization of aggregation mechanisms and aggregate structure may prove vital in understanding how proteins with expanded polyQ tracts are toxic. Throughout the literature, the terminology used to describe these different aggregate species can be used inconsistently and ambiguously. For this reason, we present a short definitions of some of these terms for clarification of how they will be used in this review (Table 2).
Table 2.
Definitions of different aggregate species associated with polyQ aggregation
| Aggregate Species | Definition |
|---|---|
| amorphous aggregate | Protein aggregates that do not have a fibrillar morphology but are larger than what is typically classified as an oligomer. Amorphous aggregates often have a granular appearance as assessed by EM or AFM. |
| amyloid fibril | Filamentous peptide or protein aggregates characterized by thermodynamic stability, high insolubility, and highly ordered cross-β rich structure. They are often comprised of intertwined protofilaments. |
| annular aggregate | Ring-shaped aggregates that are sometimes further classified as annular protofibrils or annular oligomers in the literature. Ring diameter can vary greatly. Smaller annular aggregates are hypothesized to potentially form pathogenic pores in membranes. |
| critical nucleus | The specie that directly initiates fibril formation. The critical nucleus may be monomeric or multimeric. |
| inclusion body | A large (on the order of microns) accumulation of aggregated material (including fibrils and other aggregate species) within a cell. Their large size makes them easily visible by light microscopy. |
| off pathway aggregate | An aggregate specie that is associated with an aggregation mechanism that does not lead to fibril formation. |
| oligomer | Small (on the order of 1-15 nm) protein aggregates with a globular morphology. The term oligomer has been used to describe a variety of aggregates ranging from small multimeric species containing 3-10 proteins to larger structures comprised of over 100 proteins. Oligomers are also often sub-categorized. Sub-categories include reference to their composition (i.e. tetramers, dodecamers, etc.) and being on or off pathway to fibril formation. |
| on pathway aggregate | An aggregate specie that is associated with an aggregation mechanism that leads directly to fibrils. |
| protofibril | A soluble, short filamentous aggregate that is smaller than a mature fibril and is often considered an intermediate aggregation structure. |
| protofilament | A single strand that is intertwined to from a mature fibril. |
One major hallmark of polyQ diseases is the accumulation of proteins with expanded polyQ tracts into proteinaceous inclusions that are microns in size. Abnormal nuclear structures were observed in HD brains over 40 years ago,39 and the first mouse model of HD clarified that these inclusions were comprised of fibrillar aggregates of the protein huntingtin (htt) with expanded polyQ tracts.40, 41 Similar observations have been made with the spinocerebellar ataxias.42 While the formation of intranuclear and cytoplasmic inclusion bodies of aggregated proteins is a prominent feature of polyQ diseases,43 controversy remains concerning their exact role in the etiology of these diseases. Several lines of evidence support the notion that inclusion body formation, by removing soluble forms of toxic protein species, may represents a protective cellular response.33, 44–46 These soluble forms that can be removed by the formation of large inclusions can include small oligomeric and fibrillar aggregates that are significantly smaller than the micron sized inclusions. As such, some polyQ aggregate may be benign and represent epiphenomena associated with these diseases.
Here, the underlying biochemical and biophysical aspects of the aggregation of proteins containing expanded polyQ tracts will be reviewed. We will begin by reviewing aggregation schemes associated with polyQ expansion and defining the array of aggregate species that can result. Properties of monomeric polyQ species, oligomer formation, and fibril structure will be explored in detail. Then, the influence of protein context and post-translational modifications to influence aggregation will be discussed. Finally, a brief review of potential toxic mechanisms associated with proteins containing expanded polyQ will be provided. Several other extensive reviews are available that cover other topics related to polyQ diseases such as the physical chemical and aggregation properties of polyQ,28, 47 mouse models of polyQ diseases,48–50 the impact of post-translational modifications on polyQ proteins,51 mechanisms of toxicity related to polyQ,4 RNA-mediated pathogenic mechanisms associated with polyQ diseases,52 gene and stem cell therapeutic strategies for polyQ diseases,53 the interaction of polyQ with other proteins,54 and the biology of the htt protein.55
PolyQ-mediated protein aggregation is a complex process
A popular notion is that the aggregation of proteins with expanded polyQ tracts mediate neurodegeneration, as both protein aggregation and pathogenesis are strongly correlated with polyQ length.26 This is consistent with the amyloid hypothesis associated with other common neurodegenerative diseases like Alzheimer’s disease and Parkinson diseases that also feature the accumulation of disease-related proteins into large inclusions. The aggregation process of pure polyQ peptides and polyQ-containing proteins is a complex process involving a number of distinct aggregate species (Figure 2), making it difficult to gain consensus on the prominent aggregate species involved in toxicity.
Figure 2. A schematic model for misfolding and aggregation of proteins containing expanded polyQ tracts.
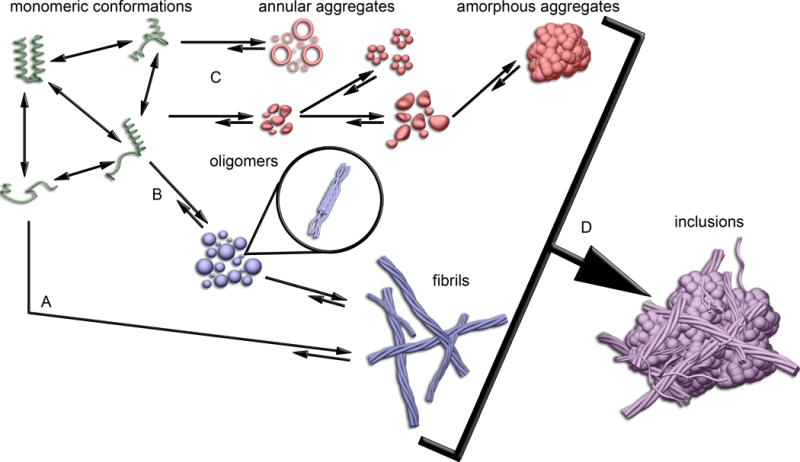
Monomeric proteins (shown in green) can sample a variety of distinct conformations, with the relative number and stability of these conformers potentially being polyQ length-dependent. Some of these monomeric conformation are aggregation prone and may lead to distinct aggregation pathway, some on pathway (shown in blue) to fibril formation and some off pathway (shown in red) to fibril formation. There appear to be two generic aggregation mechanisms toward fibril formation: (A) a monomeric critical nucleus that leads directly to fibrils and (B) fibril formation via oligomeric intermediates. Protein context of the polyQ domain can influence which pathway is dominant, and flanking sequences may facilitate the formation of some oligomeric intermediates. These two mechanisms are not necessarily mutually exclusive. (C) There are also several off-pathway aggregation routes that can lead to distinct oligomers of various sizes, a variety of annular aggregates, and large amorphous structures. (D) All of these higher order aggregates may accumulate together to form the large inclusions that are hallmarks of polyQ diseases.
Amyloids typically form via a nucleus-dependent growth polymerization mechanism.56–58 With this mechanism, the rate of aggregation is dependent on a slow nucleation phase (often called the lag phase) that involves a thermodynamically unfavorable transition from a native to non-native protein conformation that initiates the formation and elongation of fibrils. This lag phase is followed by an elongation or growth phase characterized by a relatively rapid extension of fibril aggregates. The critical nucleus that forms during the lag phase can be either monomeric or multimeric in nature. Several aggregation pathways have been proposed for the formation of fibrils of polyQ-containing proteins, ranging from simple to complex (Figure 2). While the specific details associated with the aggregation of distinct proteins can be varied, there are two generic, prominent aggregation schemes associated with polyQ-containing proteins. These are: 1) re-arrangement of polyQ monomers into a structure that directly nucleates fibril formation23, 24, 58 and 2) the formation of soluble oligomeric intermediates that undergo a structural re-arrangement into a multimeric nucleus that is on pathway to fibril formation.30, 59–61 In the first scenario, there can be multimeric critical nuclei that can directly nucleate fibrilization without a required structural rearrangement. The nature of the oligomeric aggregates associated with the second scheme can be quite heterogeneous and dependent on the specific polyQ-containing protein.30, 62, 63 It should be noted that oligomers that are off pathway to fibril formation may also form.
These two generic mechanisms are not necessarily mutually exclusive as both scenarios could occur simultaneously to varying extents. Longer pure polyQ peptides (longer than 26 repeats) typically aggregate into fibrils via the monomeric nucleation-elongation model.23, 24, 58, 64, 65 Shorter pure polyQ peptides appear to aggregate into fibrils via a tetrameric critical nucleus.58, 65 However, some studies have suggested some smaller oligomers of pure polyQ proteins may form prior to the nucleation of fibril formation.30, 60, 66, 67,67–70 The appearance of these oligomers may be due to differences in peptide preparation protocols as commonly used solvents such as hexafluoroisopropanol promote oligomer formation,71 and there is no formal kinetic evidence of how these oligomers contribute to fibril formation. However, individual oligomers of a pure polyQ peptide with 32 repeats were tracked on a mica surface, were shown to be stable for up to 200 min (but typically shorter than 50 min), and directly transition into extended fibril species.30 In this instance, the presence of the surface may have promoted or stabilized these oligomeric aggregates. It should be also noted that the dominant pathway to fibril formation on the mica surface appeared to not be oligomer mediated. The potential of these environmental factors, i.e., the presence of specific solvents or surfaces, to directly alter or promote different pathways toward fibril formation may provide valuable insight into the polyQ aggregation process. It also points to the importance of designing experiments to mechanistically investigate aggregation in relevant cellular environments.
Further complicating polyQ aggregation is the observation that heterogeneous mixtures of distinct oligomers and other aggregates can form in a polyQ length- and concentration-dependent manner (Figure 3A).30 These other aggregate types can range from large amorphous protein conglomerates to annular structures of various size (Figure 3). In addition, a variety of these other aggregates species, including some small oligomers, are off pathway to fibril formation, and these on and off pathway aggregates can perhaps form simultaneously via competing processes (Figure 2).66, 72 PolyQ length and concentration can also push aggregation toward specific species. For example, htt exon1 with 35 repeat glutamines formed large amorphous aggregates at low concentrations in vitro, but aggregated into fibrils at higher concentrations.30 In addition, the androgen receptor has been shown to aggregate into annular aggregates with short, non-pathogenic length polyQ domains, but with increasing polyQ length fibrillar species are observed.73
Figure 3. Atomic force microscopy images of a variety of aggregates formed by htt-exon1 proteins.

(A) Htt exon1 can form a variety of globular, oligomeric species (purple arrows indicate oligomers ~5 nm in height; green arrows indicate oligomers ~10 nm in height). (B) Two morphologically distinct fibrils structures formed by exon1, (orange circles indicates thinner, smooth fibril structures; pink circles indicate thicker fibrils with a beaded morphology). (C) Blue circles indicate large bundles of htt exon1 fibrils. (D) Large, amorphous aggregates of htt exon1 are indicated by red arrows (note the color scale goes up to 50 nm). (E) When htt exon1 aggregates on a lipid bilayer, a variety of oligomeric aggregates (blue arrows) associated with regions of increased membrane roughness (outlined with the green dashes lines) are observed. (F) When adding monomeric htt exon1 to pre-formed fibrils, the monomer can accumulate around the fibrils and form a variety of branching points (numbers indicated the same fibril at 0 minutes and 120 minutes after exposure to monomeric htt exon1).
Understanding the variety and mixture of aggregates formed by polyQ-containing proteins is important because these different species could play varying roles in an assortment of pathological mechanisms. Some aggregate species could represent toxic entities; while others may actually impart a protective effect. In HD for example, it is true that the formation of inclusions bodies on the order of several microns in size preceded behavioral deficits in mouse models;40 however, several observations suggest that the formation of inclusions is not involved in pathology. While it is true that large scale cellular degeneration is observed in the striatum coinciding with the presence of intracellular aggregates comprised of mutant htt, there is only moderate degeneration within the cerebral cortex despite a typically larger inclusion load.74 Specifically within the striatum, large interneurons typically contain more inclusions compared to medium spiny neurons, yet medium spiny neurons are selectively lost.75 With that being said, larger perinuclear inclusions of mutant htt can directly disrupt the nuclear envelope, leading to cell death.76 The poor correlation between inclusion body formation and toxicity is also observed in cellular models of HD.33 By tracking the fate of individual neurons using robotic live cell imaging, the formation of inclusion bodies of mutant htt correlated with cell survival compared with cells in which htt remained diffuse, suggesting an actual protective role of sequestering htt into large inclusions.77 Similar protective effects of inclusion formation have also been observed for the accumulation of AR in SBMA.46
The diffuse htt population implicated in toxicity is still a complex mixture that includes a variety of nanoscale aggregate species such as oligomers and small fibrils (Figure 3), making it difficult to pinpoint toxic species. Recently, it was estimated that the large inclusions can contain ~1 billion molecules of htt exon1; whereas, smaller diffuse aggregates can contain only up to 1,000-3,000 molecules.78 As aggregate species as small as tetramers have been observed,65 the potential heterogeneity of possible aggregate species containing anywhere from a few to thousands of molecules can be quite complex. Recently, a variety of super-resolution fluorescent microscopic techniques have been applied to study the aggregation of fluorescently-labeled htt.79–81 Initially, it was verified that sub-diffraction imaging could resolve morphological details of htt fibrils by direct comparison with AFM images on the same aggregates.79 Subsequent studies defined the spatial distributions of fluorescently-labeled htt exon1 species in a PC12m model of HD with a particular focus on small aggregate species.81 To accomplish this, methods to make these small aggregates visible in the presence of highly fluorescent inclusion bodies were developed, and small fibrillar aggregate species were detected in cells that were reminiscent of those observed in vitro. While some of these small aggregates formed a perinuclear shell-like arrangement, they were also observed dispersed throughout the entire cell.81 Interestingly, this study demonstrates that small aggregate species of htt are still present in the cell after inclusion body formation. Finally, temporal studies indicated that these fibrils only appeared in cells at late aggregation stages when inclusion bodies were already present.80 This suggests that inclusion bodies initially form from monomeric or small oligomeric htt species. An important caveat to these studies is the addition of large fluorescent tags to htt exon1 protein in these models, as fluorescent fusion partners have been shown to alter htt aggregations and toxicity in yeast.82 Still, taken in light of the potential protective effect of inclusion body formation,44, 77 this suggests these non-fibrillar aggregates may play a prominent role in toxicity.
Another aspect of polyQ aggregation (as well as amyloid formation in general) is the ability of specific pre-formed aggregates to seed aggregation via a prion-like mechanism.83, 84 In the case of polyQ proteins, this can occur with seeds inducing aggregation in the same polyQ-containing protein (self-seeding)23 or different polyQ-containing proteins (cross-seeding).37, 85, 86 Seeding can also occur across different polyQ lengths,23 causing proteins containing typically non-pathogenic polyQ lengths to aggregate. Fibrillar polyQ peptide aggregates can be internalized by mammalian cells in culture and seed aggregation, even if the polyQ length expressed in the cell is below the pathogenic threshold.87 Furthermore, aggregates isolated from several HD transgenic mouse models seed the aggregation of pure polyQ peptides in vitro,88 and seeding of mutant htt aggregation is also induced by cerebrospinal fluid from transgenic rats and human HD patients.89 Aggregates of synthetic htt exon1-like peptides grown under conditions that promote different aggregation pathways have varying abilities to seed further aggregation.38 This observation is interesting considering that, in a Drosophila model, polyQ amyloids formed in older flies more efficiently seeded in vitro aggregation than those derived from younger flies,90 suggesting that aggregates formed in flies vary in their biophysical properties and perhaps aggregation mechanism as a function of fly age. The ability to seed aggregation has several key implications for polyQ diseases. For example, the seeding phenomenon plays a critical role in non-cell autonomous effects associated with polyQ diseases based on a prion-like mechanism of transmission.83, 87, 91 These prion-like effects may cause the degeneration of neural transplants in HD patients.92
Structural features of monomeric polyQ peptides and polyQ-containing proteins
While aggregation is a major feature of polyQ-related diseases, the biochemical and structural properties of polyQ-containing proteins as monomers play a fundamental role in initiating the aggregation process. In general, the functional activity of a protein is dependent on its ability to correctly fold into and maintain its native state.57 Due to their dynamic nature, it is possible that a protein can sample a variety of conformers. Typically, there is a large kinetic barrier associated with the formation of nonnative conformations.93–96 However, there are intrinsically disordered proteins (IDPs), which lack a fixed three-dimensional structure and can range from being completely unstructured or contain unstructured regions.97, 98 Pure polyQ tracts are archetypal IDPs due to their homopolymeric nature and polar side groups.99 While many factors may lead to destabilization of native proteins, the expansion of the polyQ tract in CAG-disease related proteins promotes the formation of aberrant protein structures that promote aggregation into fibrils.
Many of the initial efforts to study the basic properties of polyQ were performed on pure polyQ peptides, i.e. in the absence of any other protein context. Due to polyQ’s poor solubility in aqueous buffers, often charged residues, most commonly lysine, are added to the ends of pure polyQ peptides to aid in solubility. This has led to a debate concerning the potential influence of these additional residues and if this limits the applicability of results obtained using these peptides. The addition of flanking lysine residues most certainly alters the biochemical and aggregation properties of polyQ peptides as they increase solubility; however, these are often experimentally necessary additions. Furthermore, the results from such studies have provided valuable insights into polyQ biochemistry and provide a valuable baseline for comparison with polyQ tracts placed in the context of disease proteins, providing insight into the role protein context may play.
PolyQ peptides behave as a polymer in a poor solvent under aqueous conditions as polyQ peptides collapse into compact conformations that exclude water, as demonstrated by a variety of experimental techniques and computational methods.59, 99–103,104 Based on a series of circular dichroism (CD) and NMR experiments, pure polyQ monomers, both below and above the typical pathological threshold in length, are predominately disordered with little difference in secondary structure as a function of polyQ length.23, 64, 67, 105–108 With these pure polyQ peptides, NMR studies did not detect distinct monomeric intermediates prior to aggregation into fibrils.107 A number of computational analyses, while producing slight variations in monomeric structure between the different studies, imply that pure polyQ peptides transiently sample a variety of secondary elements, e.g. disordered structure, α-helix, β-sheet, and β-turn.99, 102, 109–113 Some of these computational results back the experimentally observed preference of polyQ to form collapsed structures stabilized by intrapeptide H-bonding.99, 102, 113 A clear dependence of transient structural features of monomers on polyQ length has not been unambiguously established.
An extenuating circumstance is that polyQ tracts associated with disease are incorporated into the context of larger proteins, that is, there are a variety of protein sequences directly adjacent to the polyQ tract. Flanking sequences have a profound impact on aggregation kinetics and mechanism associated with polyQ aggregation (as will be discussed later), indicating that these sequences influence structural properties associated with monomeric polyQ tracts. For example, the polyQ tract in htt is flanked on the N-terminal side by the first 17 amino acids of the protein (N17) and by a polyproline (polyP) region on its C-terminal side (Figure 1I). The addition of a polyP region to the C-terminal side of a 40-repeat polyQ stretch reduces the rate of aggregation and aggregate stability.106 Interestingly, the addition of this polyP domain also removes α-helical content of the peptide as measured by CD spectroscopy.106 Whereas pure polyQ peptides are intrinsically disordered, solved X-ray diffraction crystal structures of a model htt N-terminal fragment revealed a variety of clear structural elements in the polyQ tract.114 In these structures, the N17 domain was predominately α-helical and the polyP domain formed a poly-l-proline type II (PPII) helix. Conformational variability of the polyQ domain was also recently observed in crystal structures of the carboxy‐terminal region of ataxin‐3.115 There are potential caveats, however, in that these structural elements may be induced via the crystallization process or the inclusion of fusion partners to htt that can also induce secondary structure. An α-helical structure has been observed in monomeric polyQ tracts within the androgen receptor by NMR,116 and CD spectroscopy suggests that this α-helical content increases with polyQ length.117 Structural predictions of ataxin-1, however, suggest that its polyQ domain remains predominately intrinsically disordered.118
The possibility that polyQ tracts in the context of the htt proteins can obtain a variety of interchangeable, transient conformations suggests that polyQ specific antibodies may recognize a variety of different conformers of htt.45, 119–123 The ability of polyQ-specific monoclonal antibodies specific for polyQ to alter the aggregation of a mutant htt fragment to varying degrees appears to support this notion.124 Considerable effort has been given to determine the epitopes that different polyQ specific antibodies recognize.32, 92–96 A variety of polyQ specific antibodies have been shown to have higher affinities for longer polyQ tracts.119, 121, 125 Specifically, the 1C2 monoclonal antibody has a higher affinity for longer polyQ tracts within the context of htt, SCA1, and SCA3.125 Another antibody, MW1, preferentially binds longer polyQ tracts as well; however, this occurs via a “linear lattice” mechanism in which several antibodies can bind to a single polyQ domain as available binding epitopes increase with polyQ length.119, 121 It was postulated that a third polyQ-specific antibody, 3B5H10, recognizes a β-hairpin structure of polyQ;120 however, subsequent studies have revealed that 3B5H10 recognizes a similar epitope as MW1 and 1C2.126, 127 Interestingly, the higher affinity of several polyQ antibodies for expanded polyQ tracts does not appear to be based on a unique conformational epitope associated with expansion; rather, this higher affinity is likely due to avidity effects associated with the “linear lattice” mechanism.126, 127 In addition, all of these antibodies bind to nonpathogenic and expanded lengths of polyQ tracts.127 The similarities in binding between polyQ specific antibodies argue against the notion that expansion of polyQ beyond a critical threshold results in a structurally unique toxic conformation.
The exact role monomeric species play in disease is a complex issue based on the ability of monomeric pure polyQ and polyQ-containing proteins to sample a variety of thermodynamically accessible transient conformations similar to a freely jointed homopolymeric chain.99, 102, 113, 128 A subset of these conformers might lead to distinct higher order aggregation pathways. That is, at least in principle, different polyQ monomeric conformations could promote specific oligomeric or fibrillar aggregates that are mediated by distinct hydrogen-bonding networks between polyQ residues.45, 129 Some of the monomeric forms of proteins with expanded polyQ tracts may prove toxic themselves, as a thioredoxin-polyQ fusion protein exhibits toxic properties in a meta-stable, β-sheet-rich monomeric conformation.130 Factors modulating the transient structural properties of polyQ proteins (i.e., protein context, modifications, and environmental factors) could have downstream effects on the aggregation process.
Oligomers formed by expanded polyQ-containing proteins are diverse
With the observation that inclusion formation within cells may represent a protective mechanism against polyQ-related toxicity,33, 44–46 there has been a recent research focus on small soluble aggregates, often termed oligomers, due to evidence pointing to their role in initiating toxic events.30, 63, 73, 131–137 There are several difficulties associated with characterizing oligomers. First, populations of oligomers comprised of polyQ-containing proteins are often heterogenic (Figure 3A).30, 72, 73 Second, oligomeric species are transiently populated as they are intrinsically unfavorable species and often represent on-pathway intermediates to fibril formation.138, 139 That being noted, there is also the potential to form oligomeric species that are off-pathway to fibril formation.132, 140
While there have been numerous reports of oligomers comprised of polyQ containing proteins,30, 36, 63, 73, 132, 135, 137, 140–145 the role of these structures in CAG repeat disorders has not been fully elucidated. Oligomeric species of polyQ-containing proteins can range in size from small dimers, tetramers, octamers, and dodecamers28, 36, 78 to much larger oligomers containing upwards of tens and even hundreds of individual proteins.30, 146,78 PolyQ peptides containing the first 17 amino acids of htt (N17) form a variety of oligomers, including tetramers, octamers and larger oligomers,36 that appear to be direct precursors to fibril formation. That is, self-association of the N17 domain localizes expanded polyQ tracts, facilitating further aggregation into fibrils.36, 61 Supporting these findings, species ranging from a monomer to a tetramer of the N17 domain of htt peptides were detected in the gas phase by ion mobility mass spectrometry (MS) analysis.147 Oligomer formation is a rate-limiting step in fibrillization of AR148 and ataxin-3.149, 150 While this suggests that oligomers may act as an on pathway nucleation site for fibril formation,151 off pathway oligomers may serve as a reservoir that supplies monomers to elongating fibrils.36 As such, different oligomers may have distinct roles in the formation and eventual elongation of fibrils. Mutant htt exon1 can also form oligomers that are generic to amyloid formation in general, as they are recognized by the A11 antibody that is specific for a generic motif in amyloid derived oligomers.142, 152 Further complicating the situation, the formation of oligomers are mediated by the exact protein fragment involved in the process. Different N-terminal fragments of htt aggregate into distinct oligomer species. For example, htt exon1 forms a distinct subset of oligomers that were not observed in aggregation of htt shortstop, a 117 amino acid fragment that is non-toxic.63
While many of the studies detecting oligomeric aggregates were performed with purified proteins, there is increasing evidence that oligomers exist in cellular environments, animal models, and HD patients.137, 143, 144, 153, 154,78 While most studies of htt oligomerization have been performed with N-terminal fragments, full length htt detected from HD brains or HD mouse models was monomeric;155 however, dimers and other larger multimers of full-length htt purified from insect156 or mammalian cells157 have been detected. Using the Seprion ligand, oligomers were isolated from mouse brains from both R6/2 and HdhQ150 knock-in mice, and these oligomers were similar in morphology to those formed by purified htt exon1 proteins in vitro.137 The R6/2 mouse model expresses htt-exon1 with an expanded polyQ domain and exhibit a progressive neurological phenotype that recapitulates several features of HD.158 The HdhQ150 mouse is a knock-in model in which a short polyQ repeat in murine htt was replaced with a 150Q repeat, resulting in measurable abnormalities in the live mouse and neuropathological features consistent with HD.159, 160 Further studies with the HdhQ150 mouse model demonstrated that a soluble oligomer pool forms within one month of age, but this oligomer pool declines with age as insoluble aggregates form.144 Oligomers similar in size to those observed by AFM in vitro were detected in HD patient brains with EM using the immunoperoxidase method and a primary antibody to N17.30 Htt exon1 with a non-disease length polyQ domain can form dimers in cells;78, 157 however with an expanded polyQ domain, an invariant oligomer pool of htt oligomers is present in cells.154 Using a number and brightness method to analyze confocal images of cells, it was determined that mutant htt forms oligomers that are 5-15 proteins in size and on pathway to inclusion formation in living cells.161 Oligomers have also been directly linked to ER-stress.143, 162, 163 Recently, it was reported that expanded htt exon1 expressed in PC12 cells did not remain monomeric; rather, tetramers were detected.78 Small, diffusible aggregates larger than tetramers formed in PC12 within 6-9 h, and sedimentable aggregates were present within 12 h, making it clear that htt oligomers of various size can exist in the cellular environment.78 Interestingly, significant nuclear DNA damage occurred when diffusible aggregates (potentially oligomers and smaller fibrils) start to accumulate, and this damage represented an early potential toxic event.78 A novel oligomeric specie of the androgen receptor with expanded polyQ was isolated from PC12 cells via cryo-extraction, and these oligomers were less compacted compared with late stage aggregates and correlated with toxicity.62
Structural analysis of polyQ fibrils
Amyloid-like fibrils formed by a number of proteins share several biochemical properties. These include a cross-β sheet quaternary structure and detergent insolubility.164 Fibrils of polyQ-containing proteins share most of these traditional amyloid hallmarks.165 While the polyQ length associated with disease thresholds is typically on the order of 35-40 repeat glutamines, pure polyQ peptides shorter than this can form fibrils under certain conditions.24 Interestingly, ataxin-3 containing either non-pathogenic or pathogenic length polyQ tracts aggregate into fibrils in vitro.149, 166–168 Furthermore, the rate of fibril formation is positively correlated to polyQ length with longer polyQ sequences aggregating faster.23, 24, 30, 31, 64, 85, 106 Structurally, Max Perutz first postulated that the β-sheet structure of polyQ fibrils are stabilized by hydrogen bonding between side-chains, creating a polar zipper,169 and this basic motif has been supported by structural analysis of polyQ fibrils. Using magic angle spinning solid state nuclear magnetic resonance (ssNMR), the amyloid core of htt exon 1 fibrils was confirmed to be a steric zipper structure that contains β-hairpins.170, 171 The polyQ core of htt fibrils is also rigidly packed based on these SS-NMR studies170, 171 and electroparamagentic resonance spectroscopy.172
Many other amyloid-forming proteins have the ability to form polymorphic fibrils structures,173–176 and EM images of fibrils of various htt fragments appear different morphologically compared with fibrils of pure polyQ peptides.25, 26, 61, 85 Despite this apparent difference in morphology, the same polyQ core structure in htt fibrils is always observed by ssNMR, despite dramatic differences in aggregation behavior and aggregation conditions.170, 171, 177, 178 A conserved core of polyQ may underlie observations that polyQ peptides with different flanking sequences form similar extended, fibrillar aggregates on an anionic surface179 Internally, polyQ fibril cores are assembled from differently structured monomers containing β-hairpins, providing an “intrinsic” polymorphism supported by ssNMR studies.170
Flanking sequences adjacent to polyQ tracts influence aggregation and toxicity
The ability of protein context to alter aggregation of polyQ tracts has been established via a wide array of systems,5, 36, 61, 105, 110, 149, 150, 179–185 suggesting a potentially critical role of flanking sequences to polyQ structure and to potential mechanisms of aggregation. For example, α-helical structure in the polyQ domain in a fragment of the androgen receptor is promoted by a preceding leucine-rich region, inhibiting aggregation.116 Addition of a three-helical B domain from staphylococcus aureus protein A retards aggregation, and adding more of these domains further slows aggregation.186 The AXH domain and the nuclear localization signal in ataxin-1 are involved in inclusion formation.187–190 The AXH domain itself has a propensity to aggregate and may be involved in initiating fibrilization.189, 191, 192 The pathogenic threshold length associated with some polyQ diseases appears dependent on the polarity of its flanking sequence.193 The polyQ length threshold increases when flanked by extended high polarity or alternating low and high polarity amino acids when compared to a long sequence of low polarity amino acids.193
Considerable attention has been focused on the impact of flanking sequences in htt on aggregation, and both the N17 and polyP regions flanking the polyQ tract have been shown to alter aggregation (Figure 1I). The addition of a 10-residue polyP sequence to the C-terminus of a polyQ peptide impacts the underlying conformation of polyQ and aggregation rates.61, 106, 181, 194 Strikingly, addition of a polyP domain to the N-terminus of a polyQ domain does not affect aggregation.106 At least in vitro, the ability of C-terminal polyP domains to slow aggregation appears to be related to its influence on the transient conformational sampling of the polyQ tract.106, 181, 194 The polyP domain typically has a PPII-like helical structure that has been observed to propagate into the polyQ tract in crystal structures.114 However, this propagation of PPII-like helical structure into the polyQ tract is not observed in the fibril structure171, suggesting that if this propagation does occur in monomers in solution that it is lost during the aggregation process. The addition of a polyP domain to the C-terminal side of polyQ stabilizes the protein reducing the rate of aggregation and increases the required polyQ repeat length that results in fibril formation.106 Some computational studies support the ability of polyP to prevent polyQ to convert to a β-sheet rich conformation as a monomer.110 The ability of polyP to influence polyQ aggregation has been extended into the cellular environment with impacts on toxicity. In yeast cells, the absence of the 38 amino acid proline-rich region affects the shape and number of polyQ inclusions and also activated toxicity in yeast cells.182, 195 A scFv of a polyP specific antibody, MW7, promotes htt turnover in HEK293 cells and mouse models of HD, reducing htt toxicity.196, 197 This same antibody inhibits htt aggregation in vitro and destabilized pre-formed htt fibrils,124 perhaps by stabilize a conformation of the polyP domains that prevents the necessary conformational changes in the polyQ domain associated with fibril core structure.
The N17 flanking sequence also influences polyQ structure, aggregation mechanisms, aggregate stability, and other important biochemical/biophysical properties of htt. Specifically, N17 drives the initial interaction between htt exon 1 monomers by self-association to form oligomers.103, 178, 179, 185, 198 N17 attached to the N-terminal side of synthetic PolyQ increases aggregation rates exponentially.61, 195 Unlike polyP, N17’s influence on polyQ aggregation is not position-dependent as it promotes polyQ aggregation when incorporated onto the C-terminal end of polyQ peptides and even when separated from the polyQ by a solubility imparting sequence like lysines.61 It also appears that N17 preferentially shifts the aggregation mechanisms to an oligomer mediated pathway.61, 199, 200 Interestingly, N17 can interact with two regions of htt exon1, N17 and the polyQ tract.201 One proposed mechanism is that the aggregation process occurs via self-association of N17 from different htt peptides to form small, α-helix rich oligomers that bring the polyQ tracts into close proximity to facilitate fibril formation36, 61, 202 Alternatively, intermolecular interactions between N17 and polyQ may serve a role in initial steps leading to aggregation.201 Computational studies have also observed intermolecular interactions associated with N17.200 Support for both intramolecular N17/N17 and intermolecular N17/polyQ interactions playing a role in aggregation is provided by the effect of the addition of exogenous N17 peptides lacking polyQ tracts to aggregating model peptides.185, 201 When N17 peptides are incubated with pure polyQ peptides, the aggregation of the polyQ peptides is enhanced.201 When N17 peptides are incubated with htt-mimicking peptides that contain the N17 domain, aggregation is suppressed.185 Similar mechanisms of aggregation are associated with the Josephin domain bringing polyQ tracts together to aid fibril formation in ataxin-3.149, 150 In cellular models, N17 mediated oligomerization appears to be a rate-determining step in aggregation as htt exon 1 oligomer populations are relatively stable even when monomers are recruited into inclusion bodies.38, 203
At low concentrations, N17 is essentially disordered.36, 204 However, N17 can form an amphipathic α-helix, consisting of a predominately hydrophilic face and a predominately hydrophobic face, that is observed in some aggregate structures of htt to varying extents66, 146, 177 and plays a role in aggregation.201 Amphipathic α-helices tend to be disordered until interacting directly with a binding partner, as when N17 domains from different monomers interact to form α-helix rich oligomers.36 Amphipathic α-helices are also often associated with lipid membrane binding, particularly highly curved membranes, in a regulated manner.205 N17 indeed functions as a lipid binding domain179, 204, 206, 207 and demonstrates preferential binding to curved membranes.208 Understanding how binding partners induce α-helical structure in N17 could be important, as the ability of N17 to form an amphipathic α-helix has been implicated in the formation of a variety of helix bundles associated with the initial phases of htt aggregation.
Since the flanking amino acids on either side of polyglutamine affect toxicity,182 the existence of possible interactions between the two regions are possible.209, 210 Elegant FRET based studies in cells suggest that the polyQ domain in htt acts as a flexible hinge facilitating an interaction between N17 the polyP region creating a molecular switch that regulates htt activity.210 This action was correlated with polyQ length with a critical threshold in line with HD where the flexibility of the polyQ domain is sufficiently lost, leading to dysfunction of the switch and perhaps toxicity.210 Interplay between the two flanking sequences in htt diseases also modulates the interaction of htt with lipids, as having both N17 and the C-terminal polyP domains flanking the polyQ tract resulted in enhanced aggregation on bilayers, destabilized membrane structure, and lead directly to membrane leakage compared with peptides that contained only one of the flanking sequences.179 This observation was further supported by computational studies demonstrating that N17 regulates htt/lipid interactions and the presence of both N17 and polyP enhance the interaction of htt with bilayers.211
While it is well-appreciated that flanking sequences influence polyQ structure and aggregation, most studies with recombinantly produced htt proteins have exogenous amino acids at the N-terminus. Chemically synthesized polyQ peptides typically have flanking lysines added to one or more termini. While often necessary from a technical point of view, the addition of exogenous tags can also exert influence on the biochemical and aggregation properties of polyQ-containing proteins. In a yeast model, sequences flanking the polyQ region of huntingtin exon I heavily influenced toxicity and aggregate morphology.182 Specifically, a non-native FLAG tag promoted toxicity. The addition of a myc-tag preceding N17 in full htt exon 1 can shift the balance between aggregation from an oligomeric-mediated mechanism toward a monomer aggregation pathway, suggesting that the N17 promoted oligomer formation can be influenced by steric hindrances.146 This same myc-tag can reduce the affinity of htt exon1 for lipid membranes.212 Recently, a variety of fluorescent fusion partners were shown to differentially influence htt aggregations and toxicity in yeast.82 Such observations make it important to remember the experimental context of aggregation studies of polyQ proteins. Recently, successful efforts to synthesize and characterize full length htt exon1 lacking any non-native amino acids have been reported,78, 146,170,213 and further biochemical analysis of these proteins without exogenous tags and amino acids should provide clearer insights into polyQ structure and aggregation.
Post-translational modifications impact aggregation and toxicity
An additional factor influencing polyQ-related disease are post translational modifications that can occur within polyQ-containing proteins. For example, phosphorylation, acetylation, and SUMOylation of AR influence its structure and stability.214–217 There are two verified phosphorylation sites within ataxin-1, one of which is located near the polyQ domain.118 There are at least 48 distinct PTMs associated with htt that have been identified.55 Several of these PTMs modulate toxicity of mutant htt, and much effort has been exerted to understand the role of PTMs in the disease state. PTMs have been implicated in htt localization, function, and aggregation. For example, acetylation at lysine 444 tags mutant htt for clearance via the autophagic-lysosomal pathway leading to neuroprotection in a C.elegans HD model,218 and phosphorylation at S434 or S536 regulates proteolysis by caspase 3 and calpain with a reduction in associated toxicity.219, 220 Phosphorylation at S421 in cellular models reduces accumulation of htt fragments in the nucleus,221 and decreases NMDA-mediated excitotoxicity.222 The ability to phosphorylate S421 is altered by polyQ expansion, which results in a smaller fraction of htt phosphorylated at this site.223, 224 Recently, mimicking S421 phosphorylation improved behavioral dysfunction and striatal neurodegeneration in mice by increasing proteasome-dependent turnover of htt.225 Several other PTMs have been demonstrated to function in axonal transport.157, 220, 226, 227 Here, we will focus on PTMs that occur within the N17 domain of htt, as several of these impact polyQ structure and aggregation.228 Several PTMs occur in this domain – i.e., phosphorylation,229–232 acetylation,233 ubiquitination,234 SUMOylation,235, 236 palmitoylation,237 oxidation,238 and transglutamination.239 Additionally, the initiator methionine of N17 can be proteolytically removed, and this may occur rapidly after or during synthesis.157, 229 Several PTMs in N17 impact htt function, translocation, aggregation, and the toxicity of mutant htt.51, 229, 235, 240, 241
The htt N17 domain has three phosphorylatable sites: threonine 3, (T3), serine 13 (S13), and serine (S16) (full length htt can also be phosphorylated at serine residues downstream from the N terminal domain), and phosphorylation in N17 has been linked to reduced toxicity.227, 229, 231, 232, 240, 242, 243 The addition of phosphomimetic mutation at S13 and S16 within full length htt with 97Q expressed in a transgenic mouse model reduced visible htt inclusions within the brain and ameliorated HD–like behavioral phenotypes.232 Phosphorylation at S13 and S16 can be triggered by ganglioside, GM1, and initiate a protective effect that restores normal motor function in transgenic mice.230 Phosphomimetic amino acid substitution at T3 promotes htt aggregation in cultured cells but reduced neurodegeneration in Drosophila models of HD.229 Based on studies using a variety of phosophomimetic combinations of mutations at serine 13 and 16, phosphorylation at these sights was linked to a significant decrease in aggregation rates and reduced structural stability of the resulting fibrils.177, 232 As such, phosphorylation, which is often viewed from a signaling point of view, has biophysical consequences that impact structural features of N17 and htt aggregation. For example, phosphorylation has a generic ability to destabilize α-helicity in proteins,244 this has been specifically demonstrated for htt peptides.177 Inhibited aggregation of htt due to phosphorylation in N17 could be partially attributed to destabilization of α-helical mediated self-association of N17 into tetramers, which appears to be an early step in fibril formation.36
Ubiquitination, SUMOylation, and acetylation can also occur within N17 of htt at lysine residues 6, 9, and 15. Ubiquitination of N17 tags htt for degradation by the ubiquitin proteasome system (UPS), reducing the toxicity of the mutant htt.245 Based on cell and animal models, SUMOylation of N17 plays a role in mediating HD pathogenesis.235, 236 SUMOylation at K6 and K9 reduced aggregation by stabilizing htt exon1 fragments in cultured cells and exacerbates toxicity in Drosophila.235 A small G protein, Rhes, binds mutant htt with a higher affinity compared with wild type and acts as a SUMO E3 ligase, resulting in higher concentration of soluble levels of htt with expanded polyQ with a concomitant enhancement of toxicity.246 Proteomic mapping by MS demonstrated that K9 is substantially acetylated in mammalian cell lysates,233 and this residue is preferentially chemically acetylated in vitro.247 Interestingly, acetylation of N17 reduces aggregation into fibrils while promoting the formation of large oligomeric species.247 Acetylation of N17 also decreases htt’s affinity for lipid membranes, and computational studies suggested that K9 may play a key role in the initial binding of N17 to lipids.247
Interaction with lipids influence polyQ aggregation and localization
PolyQ peptides, htt exon1, and ataxin-3 all bind lipid membranes in a polyQ-length dependent manner.167, 212, 248 Both normal and mutant htt localizes to numerous subcellular compartments, i.e. endosomes, pre-synaptic, and clathrin-coated vesicles, and dendritic plasma membrane.249 Htt also associates with acidic phospholipids250 and localizes to brain membrane fractions.251 In cell lines expressing expanded n-terminal htt fragments, multi-vesicular membranes, autophagosomes, and mitochondria are incorporated into inclusion bodies.252, 253 Collectively, this suggests a strong htt/lipid interaction. As previously mentioned, the N17 domain of htt can adopt a highly conserved amphipathic α-helix, which has been shown to bind membranes.198, 204, 206
With the localization of htt to membranous surfaces well established, it is appropriate to note that lipids are a common modulator of amyloid formation, as studies of α-synuclein,254, 255 islet amyloid polypeptide,256 β-amyloid,257–259 and polyQ179, 212, 260, 261 all demonstrate altered aggregation in the presence of membranes when compared to bulk solution. With regards to the amyloid formation in general, several physicochemical properties of lipid membrane, i.e. fluidity, curvature, elasticity, modulus, surface charge, and degree of hydration, influence protein aggregation.262, 263 Although lipid bilayers can directly influence aggregation, lipid membranes can also be directly disrupted by the expanded polyQ proteins, leading to permeabilization.179, 264 Htt aggregates can associate directly lipid membranes and induce local mechanical changes.68, 179, 212
High local concentrations due to subcellular localization of polyQ-containing proteins on lipid membranes may create aggregation nucleation sites or even stabilize specific conformers or aggregate species involved in toxicity that otherwise are only transiently formed in bulk solution. In this regard, the α-helical content of htt is altered in the presence of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS): POPC SUVs, based on CD spectroscopy.198 Htt/lipid interaction is further modulated by membrane composition and polyQ length.248, 265 For example, exogenous cholesterol protects lipid bilayers from htt induced disruption,264 which is particularly important provided that HD patients have altered cholesterol homeostasis.266, 267 Membrane degradation of the nuclear envelope, ER, and mitochondria has been described to the presence of mutant htt,76, 162 and expanded polyQ can generically distort ER membranes and the nuclear envelope in cell culture.163 Stabilized oligomers species of htt are associated with mitochondrial structural proteins, and can lead to mitochondrial fragmentation, abnormal mitochondrial dynamics, and oxidative DNA damage.268
Toxic Mechanisms attributed to polyQ expansion
In terms of discovering toxic mechanisms associated with expanded polyQ tracts, the impact of expressing mutant polyQ-containing proteins in cells lines, primary cultures, and animal models has been extensively pursued. Similar to other amyloid-associated diseases, expanded polyQ tracts within proteins are often associated with a toxic gain of function (Figure 4).94, 269, 270 Many common toxic mechanisms have been hypothesized to contribute to pathology across these diseases, including nuclear DNA damage,78, 232, 271 transcription dysregulation,272, 273 impairing function of other proteins via sequestration within aggregates,274,275 interference with central protein quality control and clearance mechanisms,276–278 alteration in endocytosis and microtubule-based transport,249 disruption of cellular/subcellular membranes,179, 264 and mitochondrial dysfunction.279, 280 These varied toxic mechanisms may act in a synchronistic or cooperative manner to produce pathology. Alternatively, these toxic pathways could potentially act in series with one mechanism triggering the next, resulting in a cascading effect of toxicity. Unraveling how these toxic pathways are interdependent will require careful temporal analysis of these processes with a particular focus on determining the earliest events. Some of these mechanisms will be discussed in further detail below.
Figure 4.
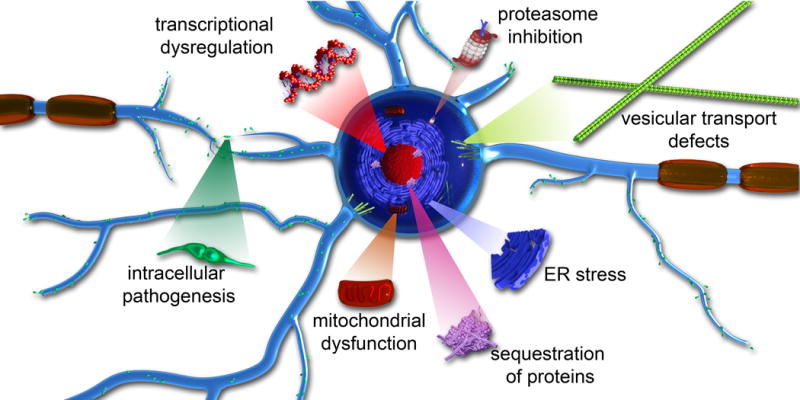
Toxic gain of function pathogenic mechanisms associated with polyQ diseases.
Aggregate species often have excessive exposure of hydrophobic residues that facilitate aberrant interactions with other proteins in the cellular milieu. For example, numerous cell regulatory proteins are co-opted into aggregate structures, effectively sequestering these proteins and disabling their functionality. Key proteins that are found to be co-localized within protein aggregates function in a variety of roles including transcription, vesicle trafficking, redox-homeostasis, and protein homeostasis. Accordingly, polyQ-expanded disease proteins can accumulate in the nucleus, and interfere with transcription factors and regulators, leading to toxicity.281 Oligomers of expanded polyQ-containing proteins directly interact with functional proteins containing polyQ tracts, i.e. the transcriptions-factors CBP, TBP, and Sp1, impairing their function.135, 272 Specifically, htt interference of CBP-mediated transcription occurs in a polyQ length-dependent manner.275, 282, 283 Nuclear inclusions of ataxin-3 can be seeded by itself or ataxin-1, and several unrelated proteins, i.e., TATA-binding protein and Eyes Absent protein, are recruited into nuclear inclusions comprised of ataxin 3.284
While observed in many age-related amyloid diseases, the sequestration of components of the degradation and protein quality control systems by aggregates of polyQ-containing proteins also occurs.276–278, 285, 286 Several components of protein quality control machinery, including the proteasome and molecular chaperones, are irreversibly pulled into protein aggregates, and the presence of proteins containing expanded polyQ tracts can eventually overwhelm the cells ability to effectively maintain protein homeostasis.152, 287 Beyond sequestration, proteins with expanded polyQ tracts can impact protein homeostasis indirectly, as a diverse set of metastable proteins that do not co-localize have been shown to lose function with expression of mutant polyQ-containing proteins.278 Protease-mediated cleaving events produce a variety of N-terminal fragments of mutant htt which can translocate to the nucleus and interfere with transcription.33, 55, 288, 289 Furthermore, HSF1, the major transcription factor for stress-inducible molecular chaperones, is dysregulated in HD patients and mouse models, reducing chaperone levels and suggesting an additional transcriptional mechanisms for impaired protein quality control.290, 291 Combined, the direct and indirect impairment of protein quality control can initiate a feedback loop that exasperates the situation with age.
Disease mechanisms discussed so far are associated with a toxic gain of function associated with expanded polyQ. Most patients with polyQ-related diseases are heterozygotes, expressing one mutant allele and one wild-type allele. As a result, both wild type and mutant forms are typically present in vivo with the concentration of the fully functional wild type forms being approximately cut in half compared to the non-disease state. Deleterious effects associated with disease can also arise from a loss of function associated with expanded polyQ. For example, htt is associated with a variety of beneficial functions including vesicle trafficking,292, 293 mediating endocytosis and endosomal trafficking,294, 295 regulating ciliogenesis,296–298 and regulating transcription.283, 299 While knocking out htt results in embryonic lethality in mice,300–302 HD-like pathology develops in mice when htt is knocked out in adulthood.303 Heterozygous htt knockout mice also have impaired motor activity, cognitive deficits, and neurodegeneration;301, 304 although, the phenotype is milder compared with mouse models overexpressing mutant htt fragments.305 Endocrine associated symptoms in SBMA are potentially due to reduce AR activity,306 and expanded polyQ disrupts the interaction between transcriptional coactivators and the amino-terminal transactivation domain of AR.307 Evidence points to ataxin-1 being involved in transcription regulation, and loss of this function due to expanded polyQ can contribute to pathology.308, 309 These findings support that loss of normal function of proteins containing expanded polyQ tracts may also contribute to disease pathology.
Conclusions
Due to the complexity in structure, dynamics, and aggregation properties of proteins containing expanded polyQ tracts, consensus on underlying toxic specie(s) and their mode of operation in disease has not been reached. As there are likely numerous bioactive aggregate species potentially within the range of monomers through oligomers and eventually fibrils, the precise characterization of aggregates and underlying structure and bio-reactivity will prove invaluable in understanding the underlying toxic mechanisms associates with these proteins. While much progress has been made since the mutations associated with these diseases were first discovered, unequivocally identifying precise toxic species has remained elusive. Biochemical and biophysical analyses of the aggregation of polyQ have revealed that a myriad of factors contribute to aggregation in vitro, i.e. expansion length, flanking sequences, membranes, and post-translational modifications. Aggregation reactions can be quite diverse, resulting in complex, heterogeneous mixtures of aggregates with varying stabilities. When placed in context of the environmental complexity within cells, potential factors modulating aggregation are even more numerous. As cellular dysfunction and toxicity likely lies in the balance between these various species of different polyQ-containing proteins, therapies designed to manipulate aggregation will need to critically evaluate the extent to which the mixture of monomeric/oligomeric/fibrillar aggregates are altered, as some aggregates may be toxic while others are protective. Ultimately to identify toxic entities in these polyQ diseases, novel experimental techniques and approaches capable of distinguishing the complex mixtures of aggregates in cells and correlating this information to toxic outcomes will need to be further developed and applied.
Acknowledgments
Funding Information: JL received funding by the National Institutes of Health grant R15NS090380.
References
- 1.Budworth H, McMurray CT. A brief history of triplet repeat diseases. Methods Mol Biol. 2013;1010:3–17. doi: 10.1007/978-1-62703-411-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hegde MV, Saraph AA. Unstable genes unstable mind: beyond the central dogma of molecular biology. Med Hypotheses. 2011;77:165–170. doi: 10.1016/j.mehy.2011.03.051. [DOI] [PubMed] [Google Scholar]
- 3.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber JJ, Sowa AS, Binder T, Hubener J. From Pathways to Targets: Understanding the Mechanisms behind Polyglutamine Disease. BioMed Res Int. 2014;22:701758. doi: 10.1155/2014/701758. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saunders HM, Bottomley SP. Multi-domain misfolding: understanding the aggregation pathway of polyglutamine proteins. Protein Eng Des Sel. 2009;22:447–451. doi: 10.1093/protein/gzp033. [DOI] [PubMed] [Google Scholar]
- 6.Orr HT, Chung M-y, Banfi S, Kwiatkowski TJ, Servadio A, Beaudet AL, McCall AE, Duvick LA, Ranum LPW, Zoghbi HY. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet. 1993;4:221–226. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 7.The Huntington’s Disease Collaborative Research Group. A Novel Gene Containing a Trinucleotide Repeat That Is Expanded and Unstable on Huntington’s Disease Chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 8.Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S, Kawakami H, Nakamura S, Nishimura M, Akiguchi I, Kimura J, Narumiya S, Kakizuka A. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14Q32.1. Nat Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- 9.Benton CS, de Silva R, Rutledge SL, Bohlega S, Ashizawa T, Zoghbi HY. Molecular and clinical studies in SCA-7 define a broad clinical spectrum and the infantile phenotype. Neurology. 1998;51:1081–1086. doi: 10.1212/wnl.51.4.1081. [DOI] [PubMed] [Google Scholar]
- 10.David G, Abbas N, Stevanin G, Durr A, Yvert G, Cancel G, Weber C, Imbert G, Saudou F, Antoniou E, Drabkin H, Gemmill R, Giunti P, Benomar A, Wood N, Ruberg M, Agid Y, Mandel JL, Brice A. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet. 1997;17:65–70. doi: 10.1038/ng0997-65. [DOI] [PubMed] [Google Scholar]
- 11.Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, Yamada M, Takahashi H, Tsuji S. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet. 1999;8:2047–2053. doi: 10.1093/hmg/8.11.2047. [DOI] [PubMed] [Google Scholar]
- 12.Komure O, Sano A, Nishino N, Yamauchi N, Ueno S, Kondoh K, Sano N, Takahashi M, Murayama N, Kondo I, Nagafuchi S, Yamada M, Kanazawa I. DNA analysis in hereditary Dentatorubral-Pallidoluysian Atrophy – correlation between CAG repear length and phenotypic variation and the molecular-basis of anticipation. Neurology. 1995;45:143–149. doi: 10.1212/wnl.45.1.143. [DOI] [PubMed] [Google Scholar]
- 13.Matilla T, Volpini V, Genis D, Rosell J, Corral J, Davalos A, Molins A, Estivill X. Presympomatic analysis of Spinocerebellar Ataxia Type-1 (SCA1) via the expansion of the SCA-1 CAG-repeat in a large pedigree displaying anticipation and parental male bias. Hum Mol Genet. 1993;2:2123–2128. doi: 10.1093/hmg/2.12.2123. [DOI] [PubMed] [Google Scholar]
- 14.Nahhas FA, Garbern J, Krajewski KM, Roa BB, Feldman GL. Juvenile onset Huntington disease resulting from a very large maternal expansion. Am J Med Genet A. 2005;137A:328–331. doi: 10.1002/ajmg.a.30891. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–1448. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 16.Pulst SM, Santos N, Wang D, Yang HY, Huynh D, Velazquez L, Figueroa KP. Spinocerebellar ataxia type 2: polyQ repeat variation in the CACNA1A calcium channel modifies age of onset. Brain. 2005;128:2297–2303. doi: 10.1093/brain/awh586. [DOI] [PubMed] [Google Scholar]
- 17.Ranum LPW, Chung MY, Banfi S, Bryer A, Schut LJ, Ramesar R, Duvick LA, McCall A, Subramony SH, Goldfarb L, Gomez C, Sandkuijl LA, Orr HT, Zoghbi HY. Molecular and clinical correlations in Spinocerebellar Ataxia Type-1 – evidence for familial effects on teh age at onset. Am J Hum Genet. 1994;55:244–252. [PMC free article] [PubMed] [Google Scholar]
- 18.Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C, Dobyns WB, Subramony SH, Zoghbi HY, Lee CC. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha(1A)-voltage-dependent calcium channel. Nat Genet. 1997;15:62–69. doi: 10.1038/ng0197-62. [DOI] [PubMed] [Google Scholar]
- 19.Margolis RL, Rudnicki DD, Holmes SE. Huntington’s disease like-2: review and update. Acta Neurol Taiwan. 2005;14:1–8. [PubMed] [Google Scholar]
- 20.Penney JB, Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann Neurol. 1997;41:689–692. doi: 10.1002/ana.410410521. [DOI] [PubMed] [Google Scholar]
- 21.Snell RG, Macmillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, Macdonald ME, Gusella JF, Harper PS, Shaw DJ. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntingtons disease. Nat Genet. 1993;4:393–397. doi: 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- 22.Cisbani G, Cicchetti F. An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis. 2012;3:e382. doi: 10.1038/cddis.2012.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 24.Chen SM, Berthelier V, Hamilton JB, O’Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 25.Poirier MA, Li HL, Macosko J, Cai SW, Amzel M, Ross CA. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem. 2002;277:41032–41037. doi: 10.1074/jbc.M205809200. [DOI] [PubMed] [Google Scholar]
- 26.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 27.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: Implications for Huntington’s disease pathology. P Natl Acad Sci USA. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wetzel R. Physical Chemistry of Polyglutamine: Intriguing Tales of a Monotonous Sequence. J Mol Biol. 2012;421:466–490. doi: 10.1016/j.jmb.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lunkes A, Mandel JL. A cellular model that recapitulates major pathogenic steps of Huntington’s disease. Hum Mol Genet. 1998;7:1355–1361. doi: 10.1093/hmg/7.9.1355. [DOI] [PubMed] [Google Scholar]
- 30.Legleiter J, Mitchell E, Lotz GP, Sapp E, Ng C, DiFiglia M, Thompson LM, Muchowski PJ. Mutant Huntingtin Fragments Form Oligomers in a Polyglutamine Length-dependent Manner in Vitro and in Vivo. J Biol Chem. 2010;285:14777–14790. doi: 10.1074/jbc.M109.093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhattacharyya AM, Thakur AK, Wetzel R. Polyglutamine aggregation nucleation: Thermodynamics of a highly unfavorable protein folding reaction. P Natl Acad Sci USA. 2005;102:15400–15405. doi: 10.1073/pnas.0501651102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang TQ, Kalchman M, Hayden MR. The influence of Huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka M, Morishima I, Akagi T, Hashikawa T, Nukina N. Intra- and intermolecular beta-pleated sheet formation in glutamine-repeat inserted myoglobin as a model for polyglutamine diseases. J Biol Chem. 2001;276:45470–45475. doi: 10.1074/jbc.M107502200. [DOI] [PubMed] [Google Scholar]
- 35.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 36.Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R. Slow Amyloid Nucleation via alpha-Helix-Rich Oligomeric Intermediates in Short Polyglutamine-Containing Huntingtin Fragments. J Mol Biol. 2012;415:881–899. doi: 10.1016/j.jmb.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jayaraman M, Kodali R, Wetzel R. The impact of ataxin-1-like histidine insertions on polyglutamine aggregation. Protein Eng Des Sel. 2009;22:469–478. doi: 10.1093/protein/gzp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jayaraman M, Mishra R, Kodali R, Thakur AK, Koharudin LMI, Gronenborn AM, Wetzel R. Kinetically Competing Huntingtin Aggregation Pathways Control Amyloid Polymorphism and Properties. Biochemistry. 2012;51:2706–2716. doi: 10.1021/bi3000929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roizin L, Stellar S, Willson N, Whittier J, Liu JC. Electron microscope and enzyme studies in cerebral biopsies of Huntington’s chorea. Trans Am Neurol Assoc. 1974;99:240–243. [PubMed] [Google Scholar]
- 40.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 41.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 42.Paulson HL, Perez MK, Trottier Y, Trojanowski JQ, Subramony SH, Das SS, Vig P, Mandel JL, Fischbeck KH, Pittman RN. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. doi: 10.1016/s0896-6273(00)80943-5. [DOI] [PubMed] [Google Scholar]
- 43.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 44.Arrasate M, Finkbeiner S. Protein aggregates in Huntington’s disease. Exp Neurol. 2012;238:1–11. doi: 10.1016/j.expneurol.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, Curtis J, Cheung K, Krishnan P, Mitra S, Widjaja K, Shaby BA, Lotz GP, Newhouse Y, Mitchell EJ, Osmand A, Gray M, Thulasiramin V, Saudou F, Segal M, Yang XW, Masliah E, Thompson LM, Muchowski PJ, Weisgraber KH, Finkbeiner S. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7:925–934. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 47.Hoffner G, Djian P. Monomeric, oligomeric and polymeric proteins in huntington disease and other diseases of polyglutamine expansion. Brain Sci. 2014;4:91–122. doi: 10.3390/brainsci4010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ehrnhoefer DE, Butland SL, Pouladi MA, Hayden MR. Mouse models of Huntington disease: variations on a theme. Dis Model Mech. 2009;2:123–129. doi: 10.1242/dmm.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Figiel M, Szlachcic WJ, Switonski PM, Gabka A, Krzyzosiak WJ. Mouse models of polyglutamine diseases: review and data table. Part I. Mol Neurobiol. 2012;46:393–429. doi: 10.1007/s12035-012-8315-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Switonski PM, Szlachcic WJ, Gabka A, Krzyzosiak WJ, Figiel M. Mouse Models of Polyglutamine Diseases in Therapeutic Approaches: Review and Data Table. Part II. Mol Neurobiol. 2012;46:430–466. doi: 10.1007/s12035-012-8316-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pennuto M, Palazzolo I, Poletti A. Post-translational modifications of expanded polyglutamine proteins: impact on neurotoxicity. Hum Mol Genet. 2009;18:R40–R47. doi: 10.1093/hmg/ddn412. [DOI] [PubMed] [Google Scholar]
- 52.Chan HYE. RNA-mediated pathogenic mechanisms in polyglutamine diseases and amyotrophic lateral sclerosis. Front Cell Neurosci. 2014;8:431. doi: 10.3389/fncel.2014.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fan HC, Ho LI, Chi CS, Chen SJ, Peng GS, Chan TM, Lin SZ, Harn HJ. Polyglutamine (PolyQ) diseases: genetics to treatments. Cell Transplant. 2014;23:441–458. doi: 10.3727/096368914X678454. [DOI] [PubMed] [Google Scholar]
- 54.Margulis BA, Vigont V, Lazarev VF, Kaznacheyeva EV, Guzhova IV. Pharmacological protein targets in polyglutamine diseases: Mutant polypeptides and their interactors. FEBS Lett. 2013;587:1997–2007. doi: 10.1016/j.febslet.2013.05.022. [DOI] [PubMed] [Google Scholar]
- 55.Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Xue WF, Homans SW, Radford SE. Systematic analysis of nucleation-dependent polymerization reveals new insights into the mechanism of amyloid self-assembly. P Natl Acad Sci USA. 2008;105:8926–8931. doi: 10.1073/pnas.0711664105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 58.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18:328–336. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walters RH, Murphy RM. Examining Polyglutamine Peptide Length: A Connection between Collapsed Conformations and Increased Aggregation. J Mol Biol. 2009;393:978–992. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walters RH, Murphy RM. Aggregation Kinetics of Interrupted Polyglutamine Peptides. J Mol Biol. 2011;412:505–519. doi: 10.1016/j.jmb.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJL, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berger TR, Montie HL, Jain P, Legleiter J, Merry DE. Identification of novel polyglutamine-expanded aggregation species in spinal and bulbar muscular atrophy. Brain Res. 2015;1628:254–264. doi: 10.1016/j.brainres.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nucifora LG, Burke KA, Feng X, Arbez N, Zhu SS, Miller J, Yang GC, Ratovitski T, Delannoy M, Muchowski PJ, Finkbeiner S, Legleiter J, Ross CA, Poirier MA. Identification of Novel Potentially Toxic Oligomers Formed in Vitro from Mammalian-derived Expanded huntingtin Exon-1 Protein. J Biol Chem. 2012;287:16017–16028. doi: 10.1074/jbc.M111.252577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen SM, Ferrone FA, Wetzel R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. P Natl Acad Sci USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Landrum E, Wetzel R. Biophysical Underpinnings of the Repeat Length Dependence of Polyglutamine Amyloid Formation. J Biol Chem. 2014;289:10254–10260. doi: 10.1074/jbc.C114.552943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Burke KA, Godbey J, Legleiter J. Assessing mutant huntingtin fragment and polyglutamine aggregation by atomic force microscopy. Methods. 2011;53:275–284. doi: 10.1016/j.ymeth.2010.12.028. [DOI] [PubMed] [Google Scholar]
- 67.Inayathullah M, Tan A, Jeyaraj R, Lam J, Cho NJ, Liu CW, Manoukian MAC, Ashkan K, Mahmoudi M, Rajadas J. Self-assembly and sequence length dependence on nanofibrils of polyglutamine peptides. Neuropeptides. 2016;57:71–83. doi: 10.1016/j.npep.2016.01.011. [DOI] [PubMed] [Google Scholar]
- 68.Ho CS, Khadka NK, She F, Cai J, Pan J. Polyglutamine aggregates impair lipid membrane integrity and enhance lipid membrane rigidity. BBA-Biomembranes. 2016;1858:661–670. doi: 10.1016/j.bbamem.2016.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi Y, Okamoto Y, Popiel HA, Fujikake N, Toda T, Kinjo M, Nagai Y. Detection of polyglutamine protein oligomers in cells by fluorescence correlation spectroscopy. J Biol Chem. 2007;282:24039–24048. doi: 10.1074/jbc.M704789200. [DOI] [PubMed] [Google Scholar]
- 70.Lee CC, Walters RH, Murphy RM. Reconsidering the mechanism of polyglutamine peptide aggregation. Biochemistry. 2007;46:12810–12820. doi: 10.1021/bi700806c. [DOI] [PubMed] [Google Scholar]
- 71.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 72.Ruggeri FS, Vieweg S, Cendrowska U, Longo G, Chiki A, Lashuel HA, Dietler G. Nanoscale studies link amyloid maturity with polyglutamine diseases onset. Sci Rep. 2016;6:31155. doi: 10.1038/srep31155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jochum T, Ritz ME, Schuster C, Funderburk SF, Jehle K, Schmitz K, Brinkmann F, Hirtz M, Moss D, Cato ACB. Toxic and non-toxic aggregates from the SBMA and normal forms of androgen receptor have distinct oligomeric structures. BBA-Mol Basis Dis. 2012;1822:1070–1078. doi: 10.1016/j.bbadis.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 74.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntingtin aggregates may not predict neuronal death in Huntington’s disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 76.Liu KY, Shyu YC, Barbaro BA, Lin YT, Chern Y, Thompson LM, James Shen CK, Marsh JL. Disruption of the nuclear membrane by perinuclear inclusions of mutant huntingtin causes cell-cycle re-entry and striatal cell death in mouse and cell models of Huntington’s disease. Hum Mol Genet. 2015;24:1602–1616. doi: 10.1093/hmg/ddu574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 78.Sahoo B, Arduini I, Drombosky KW, Kodali R, Sanders LH, Greenamyre JT, Wetzel R. Folding Landscape of Mutant Huntingtin Exon1: Diffusible Multimers, Oligomers and Fibrils, and No Detectable Monomer. PLoS One. 2016;11:e0155747. doi: 10.1371/journal.pone.0155747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Duim WC, Chen B, Frydman J, Moerner WE. Sub-diffraction imaging of huntingtin protein aggregates by fluorescence blink-microscopy and atomic force microscopy. Chemphyschem. 2011;12:2387–2390. doi: 10.1002/cphc.201100392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sahl SJ, Lau L, Vonk WI, Weiss LE, Frydman J, Moerner WE. Delayed emergence of subdiffraction-sized mutant huntingtin fibrils following inclusion body formation. Q Rev Biophys. 2016;49:e2. doi: 10.1017/S0033583515000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sahl SJ, Weiss LE, Duim WC, Frydman J, Moerner WE. Cellular inclusion bodies of mutant huntingtin exon 1 obscure small fibrillar aggregate species. Sci Rep. 2012;2:895. doi: 10.1038/srep00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang Y, Di Gregorio SE, Duennwald ML, Lajoie P. Polyglutamine toxicity in yeast uncovers phenotypic variations between different fluorescent protein fusions. Traffic. 2016;18:58–70. doi: 10.1111/tra.12453. [DOI] [PubMed] [Google Scholar]
- 83.Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010;11:301–307. doi: 10.1038/nrm2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holmes BB, Diamond MI. Cellular mechanisms of protein aggregate propagation. Curr Opin Neurol. 2012;25:721–726. doi: 10.1097/WCO.0b013e32835a3ee0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thakur AK, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. P Natl Acad Sci USA. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Arango M, Holbert S, Zala D, Brouillet E, Pearson J, Regulier E, Thakur AK, Aebischer P, Wetzel R, Deglon N, Neri C. CA150 expression delays striatal cell death in overexpression and knock-in conditions for mutant huntingtin neurotoxicity. J Neurosci. 2006;26:4649–4659. doi: 10.1523/JNEUROSCI.5409-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gupta S, Jie S, Colby DW. Protein misfolding detected early in pathogenesis of transgenic mouse model of Huntington disease using amyloid seeding assay. J Biol Chem. 2012;287:9982–9989. doi: 10.1074/jbc.M111.305417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tan Z, Dai W, van Erp TG, Overman J, Demuro A, Digman MA, Hatami A, Albay R, Sontag EM, Potkin KT, Ling S, Macciardi F, Bunney WE, Long JD, Paulsen JS, Ringman JM, Parker I, Glabe C, Thompson LM, Chiu W, Potkin SG. Huntington’s disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol Psychiatry. 2015;20:1286–1293. doi: 10.1038/mp.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tonoki A, Kuranaga E, Ito N, Nekooki-Machida Y, Tanaka M, Miura M. Aging causes distinct characteristics of polyglutamine amyloids in vivo. Genes to Cells. 2011;16:557–564. doi: 10.1111/j.1365-2443.2011.01505.x. [DOI] [PubMed] [Google Scholar]
- 91.Pearce MM, Spartz EJ, Hong W, Luo L, Kopito RR. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat Commun. 2015;6:6768. doi: 10.1038/ncomms7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cicchetti F, Saporta S, Hauser RA, Parent M, Saint-Pierre M, Sanberg PR, Li XJ, Parker JR, Chu Y, Mufson EJ, Kordower JH, Freeman TB. Neural transplants in patients with Huntington’s disease undergo disease-like neuronal degeneration. P Natl Acad Sci USA. 2009;106:12483–12488. doi: 10.1073/pnas.0904239106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jahn TR, Radford SE. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch Biochem Biophys. 2008;469:100–117. doi: 10.1016/j.abb.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med -JMM. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 95.Gillam JE, MacPhee CE. Modelling amyloid fibril formation kinetics: mechanisms of nucleation and growth. J Phys Condens Matter. 2013;25:373101. doi: 10.1088/0953-8984/25/37/373101. [DOI] [PubMed] [Google Scholar]
- 96.Knowles TPJ, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 97.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat Rev Mol Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van der Lee R, Buljan M, Lang B, Weatheritt RJ, Daughdrill GW, Dunker AK, Fuxreiter M, Gough J, Gsponer J, Jones DT, Kim PM, Kriwacki RW, Oldfield CJ, Pappu RV, Tompa P, Uversky VN, Wright PE, Babu MM. Classification of Intrinsically Disordered Regions and Proteins. Chem Rev. 2014;114:6589–6631. doi: 10.1021/cr400525m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vitalis A, Wang X, Pappu RV. Quantitative characterization of intrinsic disorder in polyglutamine: Insights from analysis based on polymer theories. Biophys J. 2007;93:1923–1937. doi: 10.1529/biophysj.107.110080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. P Natl Acad Sci USA. 2006;103:16764–16769. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dougan L, Li J, Badilla CL, Berne BJ, Fernandez JM. Single homopolypeptide chains collapse into mechanically rigid conformations. P Natl Acad Sci USA. 2009;106:12605–12610. doi: 10.1073/pnas.0900678106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vitalis A, Wang X, Pappu RV. Atomistic Simulations of the Effects of Polyglutamine Chain Length and Solvent Quality on Conformational Equilibria and Spontaneous Homodimerization. J Mol Biol. 2008;384:279–297. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Crick SL, Ruff KM, Garai K, Frieden C, Pappu RV. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. P Natl Acad Sci USA. 2013;110:20075–20080. doi: 10.1073/pnas.1320626110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Digambaranath JL, Campbell TV, Chung A, McPhail MJ, Stevenson KE, Zohdy MA, Finke JM. An accurate model of polyglutamine. Proteins. 2011;79:1427–1440. doi: 10.1002/prot.22970. [DOI] [PubMed] [Google Scholar]
- 105.Altschuler EL, Hud NV, Mazrimas JA, Rupp B. Random coil conformation for extended polyglutamine stretches in aqueous soluble monomeric peptides. J Pept Res. 1997;50:73–75. doi: 10.1111/j.1399-3011.1997.tb00622.x. [DOI] [PubMed] [Google Scholar]
- 106.Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, Creamer TP, Wetzel R. Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol. 2006;355:524–535. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 107.Klein FAC, Pastore A, Masino L, Zeder-Lutz G, Nierengarten H, Oulad-Abdeighani M, Altschuh D, Mandel JL, Trottier Y. Pathogenic and non-pathogenic polyglutamine tracts have similar structural properties: Towards a length-dependent toxicity gradient. J Mol Biol. 2007;371:235–244. doi: 10.1016/j.jmb.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 108.Masino L, Kelly G, Leonard K, Trottier Y, Pastore A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002;513:267–272. doi: 10.1016/s0014-5793(02)02335-9. [DOI] [PubMed] [Google Scholar]
- 109.Khare SD, Ding F, Gwanmesia KN, Dokholyan NV. Molecular origin of polyglutamine aggregation in neurodegenerative diseases. PLoS Comput Biol. 2005;1:230–235. doi: 10.1371/journal.pcbi.0010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lakhani VV, Ding F, Dokholyan NV. Polyglutamine Induced Misfolding of Huntingtin Exon1 is Modulated by the Flanking Sequences. PLoS Comput Biol. 2010;6 doi: 10.1371/journal.pcbi.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leitgeb B, Kerenyi A, Bogar F, Paragi G, Penke B, Rakhely G. Studying the structural properties of polyalanine and polyglutamine peptides. J Mol Model. 2007;13:1141–1150. doi: 10.1007/s00894-007-0241-4. [DOI] [PubMed] [Google Scholar]
- 112.Nakano M, Watanabe H, Rothstein SM, Tanaka S. Comparative Characterization of Short Monomeric Polyglutamine Peptides by Replica Exchange Molecular Dynamics Simulation. J Phys Chem B. 2010;114:7056–7061. doi: 10.1021/jp9122024. [DOI] [PubMed] [Google Scholar]
- 113.Wang XL, Vitalis A, Wyczalkowski MA, Pappu RV. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 114.Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary Structure of Huntingtin Amino-Terminal Region. Structure. 2009;17:1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhemkov VA, Kulminskaya AA, Bezprozvanny IB, Kim M. The 2.2-Angstrom resolution crystal structure of the carboxy-terminal region of ataxin-3. FEBS Open Bio. 2016;6:168–178. doi: 10.1002/2211-5463.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eftekharzadeh B, Piai A, Chiesa G, Mungianu D, Garcia J, Pierattelli R, Felli IC, Salvatella X. Sequence Context Influences the Structure and Aggregation Behavior of a PolyQ Tract. Biophys J. 2016;110:2361–2366. doi: 10.1016/j.bpj.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Davies P, Watt K, Kelly SM, Clark C, Price NC, McEwan IJ. Consequences of poly-glutamine repeat length for the conformation and folding of the androgen receptor amino-terminal domain. J Mol Endocrinol. 2008;41:301–314. doi: 10.1677/JME-08-0042. [DOI] [PubMed] [Google Scholar]
- 118.de Chiara C, Pastore A. Prediction and Experimental Detection of Structural and Functional Motifs in Intrinsically Unfolded Proteins. Selected Works in Bioinformatics 2011 [Google Scholar]
- 119.Bennett MJ, Huey-Tubman KE, Herr AB, West AP, Ross SA, Bjorkman PJ. A linear lattice model for polyglutamine in CAG-expansion diseases. P Natl Acad Sci USA. 2002;99:11634–11639. doi: 10.1073/pnas.182393899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Peters-Libeu C, Newhouse Y, Krishnan P, Cheung K, Brooks E, Weisgraber K, Finkbeiner S. Crystallization and diffraction properties of the Fab fragment of 3B5H10, an antibody specific for disease-causing polyglutamine stretches. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:1065–1068. doi: 10.1107/S1744309105036547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li P, Huey-Tubman KE, Gao T, Li X, West AP, Jr, Bennett MJ, Bjorkman PJ. The structure of a polyQ-anti-polyQ complex reveals binding according to a linear lattice model. Nat Struct Mol Biol. 2007;14:381–387. doi: 10.1038/nsmb1234. [DOI] [PubMed] [Google Scholar]
- 122.Ko J, Ou S, Patterson PH. New anti-huntingtin monoclonal antibodies: Implications for huntingtin conformation and its binding proteins. Brain Res Bull. 2001;56:319–329. doi: 10.1016/s0361-9230(01)00599-8. [DOI] [PubMed] [Google Scholar]
- 123.Heiser V, Scherzinger E, Boeddrich A, Nordhoff E, Lurz R, Schugardt N, Lehrach H, Wanker EE. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: Implications for Huntington’s disease therapy. P Natl Acad Sci USA. 2000;97:6739–6744. doi: 10.1073/pnas.110138997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Legleiter J, Lotz GP, Miller J, Ko J, Ng C, Williams GL, Finkbeiner S, Patterson PH, Muchowski PJ. Monoclonal Antibodies Recognize Distinct Conformational Epitopes Formed by Polyglutamine in a Mutant Huntingtin Fragment. J Biol Chem. 2009;284:21647–21658. doi: 10.1074/jbc.M109.016923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L, Agid Y, Brice A, Mandel JL. Polyglutamine expansion as a pathological eptiote in Huntingtons-disease and 4 dominant Cerebellar Ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 126.Klein FA, Zeder-Lutz G, Cousido-Siah A, Mitschler A, Katz A, Eberling P, Mandel JL, Podjarny A, Trottier Y. Linear and extended: a common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Hum Mol Genet. 2013;22:4215–4223. doi: 10.1093/hmg/ddt273. [DOI] [PubMed] [Google Scholar]
- 127.Owens GE, New DM, West AP, Jr, Bjorkman PJ. Anti-PolyQ Antibodies Recognize a Short PolyQ Stretch in Both Normal and Mutant Huntingtin Exon 1. J Mol Biol. 2015;427:2507–2519. doi: 10.1016/j.jmb.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vitalis A, Lyle N, Pappu RV. Thermodynamics of beta-Sheet Formation in Polyglutamine. Biophys J. 2009;97:303–311. doi: 10.1016/j.bpj.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Miller J, Rutenber E, Muchowski PJ. Polyglutamine dances the conformational cha-cha-cha. Structure. 2009;17:1151–1153. doi: 10.1016/j.str.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, Goto Y, Naiki H, Toda T. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–340. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 131.Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, Cecchi C, Chiti F. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat Chem Biol. 2010;6:140–147. doi: 10.1038/nchembio.283. [DOI] [PubMed] [Google Scholar]
- 132.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–566. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 133.Faendrich M. Oligomeric Intermediates in Amyloid Formation: Structure Determination and Mechanisms of Toxicity. J Mol Biol. 2012;421:427–440. doi: 10.1016/j.jmb.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 134.Glabe CG. Structural Classification of Toxic Amyloid Oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hands SL, Wyttenbach A. Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol. 2010;120:419–437. doi: 10.1007/s00401-010-0703-0. [DOI] [PubMed] [Google Scholar]
- 136.Lotz GP, Legleiter J. The role of amyloidogenic protein oligomerization in neurodegenerative disease. J Mol Med -JMM. 2013;91:653–664. doi: 10.1007/s00109-013-1025-1. [DOI] [PubMed] [Google Scholar]
- 137.Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, Bates GP. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet. 2010;19:65–78. doi: 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Porat Y, Kolusheva S, Jelinek R, Gazit E. The human islet amyloid polypeptide forms transient membrane-active prefibrillar assemblies. Biochemistry. 2003;42:10971–10977. doi: 10.1021/bi034889i. [DOI] [PubMed] [Google Scholar]
- 139.Sandberg A, Luheshi LM, Sollvander S, de Barros TP, Macao B, Knowles TPJ, Biverstal H, Lendel C, Ekholm-Petterson F, Dubnovitsky A, Lannfelt L, Dobson CM, Hard T. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. P Natl Acad Sci USA. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Behrends C, Langer CA, Boteva R, Bottcher UM, Stemp MJ, Schaffar G, Rao BV, Giese A, Kretzschmar H, Siegers K, Hartl FU. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 141.Beitel LK, Alvarado C, Mokhtar S, Paliouras M, Trifiro M. Mechanisms mediating spinal and bulbar muscular atrophy: investigations into polyglutamine-expanded androgen receptor function and dysfunction. Front Neurol. 2013;4:53. doi: 10.3389/fneur.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 143.Leitman J, Ulrich Hartl F, Lederkremer GZ. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;4:2753. doi: 10.1038/ncomms3753. [DOI] [PubMed] [Google Scholar]
- 144.Marcellin D, Abramowski D, Young D, Richter J, Weiss A, Marcel A, Maassen J, Kauffmann M, Bibel M, Shimshek DR, Faull RLM, Bates GP, Kuhn RR, Van der Putten PH, Schmid P, Lotz GP. Fragments of HdhQ150 Mutant Huntingtin Form a Soluble Oligomer Pool That Declines with Aggregate Deposition upon Aging. PLoS One. 2012;7:e44457. doi: 10.1371/journal.pone.0044457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gales L, Cortes L, Almeida C, Melo CV, Costa MD, Maciel P, Clarke DT, Damas AM, Macedo-Ribeiro S. Towards a structural understanding of the fibrillization pathway in Machado-Joseph’s disease: Trapping early oligomers of non-expanded ataxin-3. J Mol Biol. 2005;353:642–654. doi: 10.1016/j.jmb.2005.08.061. [DOI] [PubMed] [Google Scholar]
- 146.Sahoo B, Singer D, Kodali R, Zuchner T, Wetzel R. Aggregation Behavior of Chemically Synthesized, Full-Length Huntingtin Exon1. Biochemistry. 2014;53:3897–3907. doi: 10.1021/bi500300c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Arndt JR, Kondalaji SG, Maurer MM, Parker A, Legleiter J, Valentine SJ. Huntingtin N-Terminal Monomeric and Multimeric Structures Destabilized by Covalent Modification of Heteroatomic Residues. Biochemistry. 2015;54:4285–4296. doi: 10.1021/acs.biochem.5b00478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wyttenbach A. Role of heat shock proteins during polyglutamine neurodegeneration – Mechanisms and hypothesis. J Mol Neurosci. 2004;23:69–95. doi: 10.1385/JMN:23:1-2:069. [DOI] [PubMed] [Google Scholar]
- 149.Ellisdon AM, Pearce MC, Bottomley SP. Mechanisms of ataxin-3 misfolding and fibril formation: Kinetic analysis of a disease-associated polyglutamine protein. J Mol Biol. 2007;368:595–605. doi: 10.1016/j.jmb.2007.02.058. [DOI] [PubMed] [Google Scholar]
- 150.Ellisdon AM, Thomas B, Bottomley SP. The two-stage pathway of ataxin-3 fibrillogenesis involves a polyglutamine-independent step. J Biol Chem. 2006;281:16888–16896. doi: 10.1074/jbc.M601470200. [DOI] [PubMed] [Google Scholar]
- 151.Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, Serpell L, Arnsdorf MF, Lindquist SL. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–1321. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 152.Lotz GP, Legleiter J, Aron R, Mitchell EJ, Huang SY, Ng CP, Glabe C, Thompson LM, Muchowski PJ. Hsp70 and Hsp40 Functionally Interact with Soluble Mutant Huntingtin Oligomers in a Classic ATP-dependent Reaction Cycle. J Biol Chem. 2010;285:38183–38193. doi: 10.1074/jbc.M110.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Weiss A, Klein C, Woodman B, Sathasivam K, Bibel M, Regulier E, Bates GP, Paganetti P. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J Neurochem. 2008;104:846–858. doi: 10.1111/j.1471-4159.2007.05032.x. [DOI] [PubMed] [Google Scholar]
- 154.Olshina MA, Angley LM, Ramdzan YM, Tang J, Bailey MF, Hill AF, Hatters DM. Tracking Mutant Huntingtin Aggregation Kinetics in Cells Reveals Three Major Populations That Include an Invariant Oligomer Pool. J Biol Chem. 2010;285:21807–21816. doi: 10.1074/jbc.M109.084434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sapp E, Valencia A, Li XY, Aronin N, Kegel KB, Vonsattel JP, Young AB, Wexler N, DiFiglia M. Native Mutant Huntingtin in Human Brain EVIDENCE FOR PREVALENCE OF FULL-LENGTH MONOMER. J Biol Chem. 2012;287:13487–13499. doi: 10.1074/jbc.M111.286609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Li W, Serpell LC, Carter WJ, Rubinsztein DC, Huntington JA. Expression and characterization of full-length human Huntingtin, an elongated HEAT repeat protein. J Biol Chem. 2006;281:15916–15922. doi: 10.1074/jbc.M511007200. [DOI] [PubMed] [Google Scholar]
- 157.Huang B, Lucas T, Kueppers C, Dong X, Krause M, Bepperling A, Buchner J, Voshol H, Weiss A, Gerrits B, Kochanek S. Scalable Production in Human Cells and Biochemical Characterization of Full-Length Normal and Mutant Huntingtin. PLoS One. 2015;10:e0121055. doi: 10.1371/journal.pone.0121055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 159.Heng MY, Tallaksen-Greene SJ, Detloff PJ, Albin RL. Longitudinal evaluation of the Hdh(CAG)150 knock-in murine model of Huntington’s disease. J Neurosci. 2007;27:8989–8998. doi: 10.1523/JNEUROSCI.1830-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 161.Ossato G, Digman MA, Aiken C, Lukacsovich T, Marsh JL, Gratton E. A Two-Step Path to Inclusion Formation of Huntingtin Peptides Revealed by Number and Brightness Analysis. Biophys J. 2010;98:3078–3085. doi: 10.1016/j.bpj.2010.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Jiang Y, Chadwick SR, Lajoie P. Endoplasmic reticulum stress: The cause and solution to Huntington’s disease? Brain Res. 2016;1648(Pt B):650–657. doi: 10.1016/j.brainres.2016.03.034. [DOI] [PubMed] [Google Scholar]
- 163.Ueda M, Li S, Itoh M, Wang MX, Hayakawa M, Islam S, Tana, Nakagawa K, Chen H, Nakagawa T. Expanded polyglutamine embedded in the endoplasmic reticulum causes membrane distortion and coincides with Bax insertion. Biochem Biophys Res Commun. 2016;474:259–263. doi: 10.1016/j.bbrc.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 164.Rambaran RN, Serpell LC. Amyloid fibrils Abnormal protein assembly. Prion. 2008;2:112–117. doi: 10.4161/pri.2.3.7488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Amyloid, Prions, and Other Protein Aggregates. 1999;309:375–386. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- 166.Chow MKM, Mackay JP, Whisstock JC, Scanlon MJ, Bottomley SP. Structural and functional analysis of the Josephin domain of the polyglutamine protein ataxin-3. Biochem Biophys Res Commun. 2004;322:387–394. doi: 10.1016/j.bbrc.2004.07.131. [DOI] [PubMed] [Google Scholar]
- 167.Saunders HM, Hughes VA, Cappai R, Bottomley SP. Conformational Behavior and Aggregation of Ataxin-3 in SDS. PLoS One. 2013;8:e69416. doi: 10.1371/journal.pone.0069416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Masino L, Nicastro G, Menon RP, Dal Piaz F, Calder L, Pastore A. Characterization of the structure and the amyloidogenic properties of the Josephin domain of the polyglutamine-containing protein ataxin-3. J Mol Biol. 2004;344:1021–1035. doi: 10.1016/j.jmb.2004.09.065. [DOI] [PubMed] [Google Scholar]
- 169.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers – their possible role in inherited neurodegenerative diseases. P Natl Acad Sci USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hoop CL, Lin HK, Kar K, Magyarfalvi G, Lamley JM, Boatz JC, Mandal A, Lewandowski JR, Wetzel R, van der Wel PCA. Huntingtin exon 1 fibrils feature an interdigitated beta-hairpin-based polyglutamine core. P Natl Acad Sci USA. 2016;113:1546–1551. doi: 10.1073/pnas.1521933113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hoop CL, Lin HK, Kar K, Hou Z, Poirier MA, Wetzel R, van der Wel PCA. Polyglutamine Amyloid Core Boundaries and Flanking Domain Dynamics in Huntingtin Fragment Fibrils Determined by Solid-State Nuclear Magnetic Resonance. Biochemistry. 2014;53:6653–6666. doi: 10.1021/bi501010q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bugg CW, Isas JM, Fischer T, Patterson PH, Langen R. Structural Features and Domain Organization of Huntingtin Fibrils. J Biol Chem. 2012;287:31739–31746. doi: 10.1074/jbc.M112.353839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Goldsbury CS, Cooper GJS, Goldie KN, Muller SA, Saafi EL, Gruijters WTM, Misur MP. Polymorphic fibrillar assembly of human amylin. J Struct Biol. 1997;119:17–27. doi: 10.1006/jsbi.1997.3858. [DOI] [PubMed] [Google Scholar]
- 174.Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struc Biol. 2007;17:48–57. doi: 10.1016/j.sbi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 175.Kodali R, Williams AD, Chemuru S, Wetzel R. A beta(1-40) Forms Five Distinct Amyloid Structures whose beta-Sheet Contents and Fibril Stabilities Are Correlated. J Mol Biol. 2010;401:503–517. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yates EA, Cucco EM, Legleiter J. Point Mutations in Aβ Induce Polymorphic Aggregates at Liquid/Solid Interfaces. ACS Chem Neuro. 2011;2:294–307. doi: 10.1021/cn200001k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mishra R, Hoop CL, Kodali R, Sahoo B, van der Wel PCA, Wetzel R. Serine Phosphorylation Suppresses Huntingtin Amyloid Accumulation by Altering Protein Aggregation Properties. J Mol Biol. 2012;424:1–14. doi: 10.1016/j.jmb.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PCA. The Aggregation-Enhancing Huntingtin N-Terminus Is Helical in Amyloid Fibrils. J Am Chem Soc. 2011;133:4558–4566. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Burke KA, Kauffman KJ, Umbaugh CS, Frey SL, Legleiter J. The Interaction of Polyglutamine Peptides with Lipid Membranes Is Regulated by Flanking Sequences Associated with Huntingtin. J Biol Chem. 2013;288:14993–15005. doi: 10.1074/jbc.M112.446237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Arndt JR, Brown RJ, Burke KA, Legleiter J, Valentine SJ. Lysine residues in the N-terminal huntingtin amphipathic alpha-helix play a key role in peptide aggregation. J Mass Spectrom. 2015;50:117–126. doi: 10.1002/jms.3504. [DOI] [PubMed] [Google Scholar]
- 181.Darnell G, Orgel JPRO, Pahl R, Meredith SC. Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J Mol Biol. 2007;374:688–704. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 182.Duennwald ML, Jagadish S, Muchowski PJ, Lindquist S. Flanking sequences profoundly alter polyglutamine toxicity in yeast. P Natl Acad Sci USA. 2006;103:11045–11050. doi: 10.1073/pnas.0604547103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Robertson AL, Horne J, Ellisdon AM, Thomas B, Scanlon MJ, Bottomley SP. The Structural Impact of a Polyglutamine Tract Is Location-Dependent. Biophys J. 2008;95:5922–5930. doi: 10.1529/biophysj.108.138487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Saunders HM, Gilis D, Rooman M, Dehouck Y, Robertson AL, Bottomley SP. Flanking domain stability modulates the aggregation kinetics of a polyglutamine disease protein. Protein Sci. 2011;20:1675–1681. doi: 10.1002/pro.698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mishra R, Jayaraman M, Roland BP, Landrum E, Fullam T, Kodali R, Thakur AK, Arduini I, Wetzel R. Inhibiting the Nucleation of Amyloid Structure in a Huntingtin Fragment by Targeting alpha-Helix-Rich Oligomeric Intermediates. J Mol Biol. 2012;415:900–917. doi: 10.1016/j.jmb.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Robertson AL, Bate MA, Buckle AM, Bottomley SP. The Rate of PolyQ-Mediated Aggregation Is Dramatically Affected by the Number and Location of Surrounding Domains. J Mol Biol. 2011;413:879–887. doi: 10.1016/j.jmb.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 187.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 188.Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, Clark HB, Orr HT. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 189.de Chiara C, Menon RP, Adinolfi S, de Boer J, Ktistaki E, Kelly G, Calder L, Kioussls D, Pastore A. The AXH domain adopts alternative folds: The solution structure of HBP1 AXH. Structure. 2005;13:743–753. doi: 10.1016/j.str.2005.02.016. [DOI] [PubMed] [Google Scholar]
- 190.Welzel F, Kaehler C, Isau M, Hallen L, Lehrach H, Krobitsch S. FOX-2 Dependent Splicing of Ataxin-2 Transcript Is Affected by Ataxin-1 Overexpression. PLoS One. 2012;7:e37985. doi: 10.1371/journal.pone.0037985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, McCall A, Canal I, Orr HT, Zoghbi HY, Botas J. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature. 2000;408:101–106. doi: 10.1038/35040584. [DOI] [PubMed] [Google Scholar]
- 192.Tsuda H, Jafar-Nejad H, Patel AJ, Sun Y, Chen HK, Rose MF, Venken KJT, Botas J, Orr HT, Bellen HJ, Zoghbi HY. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 193.Kim M. Pathogenic polyglutamine expansion length correlates with polarity of the flanking sequences. Mol Neurodegener. 2014;9:45. doi: 10.1186/1750-1326-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Darnell GD, Derryberry J, Kurutz JW, Meredith SC. Mechanism of Cis-Inhibition of PolyQ Fibrillation by PolyP: PPII Oligomers and the Hydrophobic Effect. Biophys J. 2009;97:2295–2305. doi: 10.1016/j.bpj.2009.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dehay B, Bertolotti A. Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast. J Biol Chem. 2006;281:35608–35615. doi: 10.1074/jbc.M605558200. [DOI] [PubMed] [Google Scholar]
- 196.Southwell AL, Khoshnan A, Dunn DE, Bugg CW, Lo DC, Patterson PH. Intrabodies binding the proline-rich domains of mutant huntingtin increase its turnover and reduce neurotoxicity. J Neurosci. 2008;28:9013–9020. doi: 10.1523/JNEUROSCI.2747-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Southwell AL, Ko J, Patterson PH. Intrabody Gene Therapy Ameliorates Motor, Cognitive, and Neuropathological Symptoms in Multiple Mouse Models of Huntington’s Disease. J Neurosci. 2009;29:13589–13602. doi: 10.1523/JNEUROSCI.4286-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16:2600–2615. doi: 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- 199.Kelley NW, Huang X, Tam S, Spiess C, Frydman J, Pande VS. The Predicted Structure of the Headpiece of the Huntingtin Protein and Its Implications on Huntingtin Aggregation. J Mol Biol. 2009;388:919–927. doi: 10.1016/j.jmb.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Williamson TE, Vitalis A, Crick SL, Pappu RV. Modulation of Polyglutamine Conformations and Dimer Formation by the N-Terminus of Huntingtin. J Mol Biol. 2010;396:1295–1309. doi: 10.1016/j.jmb.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat Struct Mol Biol. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Monsellier E, Redeker V, Ruiz-Arlandis G, Bousset L, Melki R. Molecular Interaction between the Chaperone Hsc70 and the N-terminal Flank of Huntingtin Exon 1 Modulates Aggregation. J Biol Chem. 2015;290:2560–2576. doi: 10.1074/jbc.M114.603332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Vitalis A, Pappu RV. Assessing the contribution of heterogeneous distributions of oligomers to aggregation mechanisms of polyglutamine peptides. Biophys Chem. 2011;159:14–23. doi: 10.1016/j.bpc.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Michalek M, Salnikov ES, Werten S, Bechinger B. Membrane Interactions of the Amphipathic Amino Terminus of Huntingtin. Biochemistry. 2013;52:847–858. doi: 10.1021/bi301325q. [DOI] [PubMed] [Google Scholar]
- 205.Drin G, Antonny B. Amphipathic helices and membrane curvature. FEBS Lett. 2010;584:1840–1847. doi: 10.1016/j.febslet.2009.10.022. [DOI] [PubMed] [Google Scholar]
- 206.Michalek M, Salnikov ES, Bechinger B. Structure and Topology of the Huntingtin 1-17 Membrane Anchor by a Combined Solution and Solid-State NMR Approach. Biophys J. 2013;105:699–710. doi: 10.1016/j.bpj.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Côté S, Binette V, Salnikov Evgeniy S, Bechinger B, Mousseau N. Probing the Huntingtin 1-17 Membrane Anchor on a Phospholipid Bilayer by Using All-Atom Simulations. Biophys J. 2015;108:1187–1198. doi: 10.1016/j.bpj.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chaibva M, Burke KA, Legleiter J. Curvature Enhances Binding and Aggregation of Huntingtin at Lipid Membranes. Biochemistry. 2014;53:2355–2365. doi: 10.1021/bi401619q. [DOI] [PubMed] [Google Scholar]
- 209.Duennwald ML, Jagadish S, Giorgini F, Muchowski PJ, Lindquist S. A network of protein interactions determines polyglutamine toxicity. P Natl Acad Sci USA. 2006;103:11051–11056. doi: 10.1073/pnas.0604548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Caron NS, Desmond CR, Xia J, Truant R. Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. P Natl Acad Sci USA. 2013;110:14610–14615. doi: 10.1073/pnas.1301342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Nagarajan A, Jawahery S, Matysiak S. The Effects of Flanking Sequences in the Interaction of Polyglutamine Peptides with a Membrane Bilayer. J Phys Chem B. 2013;118:6368–6379. doi: 10.1021/jp407900c. [DOI] [PubMed] [Google Scholar]
- 212.Burke KA, Hensal KM, Umbaugh CS, Chaibva M, Legleiter J. Huntingtin disrupts lipid bilayers in a polyQ-length dependent manner. BBA-Biomembranes. 2013;1828:1953–1961. doi: 10.1016/j.bbamem.2013.04.025. [DOI] [PubMed] [Google Scholar]
- 213.Vieweg S, Ansaloni A, Wang ZM, Warner JB, Lashuel HA. An Intein-based Strategy for the Production of Tag-free Huntingtin Exon 1 Proteins Enables New Insights into the Polyglutamine Dependence of Httex1 Aggregation and Fibril Formation. J Biol Chem. 2016;291:12074–12086. doi: 10.1074/jbc.M116.713982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Palazzolo I, Burnett BG, Young JE, Brenne PL, La Spada AR, Fischbeck KH, Howell BW, Pennuto M. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum Mol Genet. 2007;16:1593–1603. doi: 10.1093/hmg/ddm109. [DOI] [PubMed] [Google Scholar]
- 215.Palazzolo I, Stack C, Kong LL, Musaro A, Adachi H, Katsuno M, Sobue G, Taylor JP, Sumner CJ, Fischbeck KH, Pennuto M. Overexpression of IGF-1 in Muscle Attenuates Disease in a Mouse Model of Spinal and Bulbar Muscular Atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Montie HL, Pestell RG, Merry DE. SIRT1 Modulates Aggregation and Toxicity through Deacetylation of the Androgen Receptor in Cell Models of SBMA. J Neurosci. 2011;31:17425–17436. doi: 10.1523/JNEUROSCI.3958-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mukherjee S, Thomas M, Dadgar N, Lieberman AP, Iniguez-Lluhi JA. Small Ubiquitin-like Modifier (SUMO) Modification of the Androgen Receptor Attenuates Polyglutamine-mediated Aggregation. J Biol Chem. 2009;284:21296–21306. doi: 10.1074/jbc.M109.011494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Jeong H, Then F, Melia TJ, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation Targets Mutant Huntingtin to Autophagosomes for Degradation. Cell. 2009;137:60–72. doi: 10.1016/j.cell.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Luo SQ, Vacher C, Davies JE, Rubinsztein DC. Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: implications for mutant huntingtin toxicity. J Cell Biol. 2005;169:647–656. doi: 10.1083/jcb.200412071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Schilling B, Gafni J, Torcassi C, Cong X, Row RH, LaFevre-Bernt MA, Cusack MP, Ratovitski T, Hirschhorn R, Ross CA, Gibson BW, Ellerby LM. Huntingtin phosphorylation sites mapped by mass spectrometry – Modulation of cleavage and toxicity. J Biol Chem. 2006;281:23686–23697. doi: 10.1074/jbc.M513507200. [DOI] [PubMed] [Google Scholar]
- 221.Warby SC, Doty CN, Graham RK, Shively J, Singaraja RR, Hayden MR. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol Cell Neurosci. 2009;40:121–127. doi: 10.1016/j.mcn.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 222.Metzler M, Gan L, Mazarei G, Graham RK, Liu L, Bissada N, Lu G, Leavitt BR, Hayden MR. Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J Neurosci. 2010;30:14318–14329. doi: 10.1523/JNEUROSCI.1589-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, Robertson HA, Hayden MR. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–1577. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- 224.Pardo R, Colin E, Regulier E, Aebischer P, Deglon N, Humbert S, Saudou F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci. 2006;26:1635–1645. doi: 10.1523/JNEUROSCI.3706-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kratter IH, Zahed H, Lau A, Tsvetkov AS, Daub AC, Weiberth KF, Gu X, Saudou F, Humbert S, Yang XW, Osmand A, Steffan JS, Masliah E, Finkbeiner S. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J Clin Invest. 2016;126:3585–3597. doi: 10.1172/JCI80339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- 227.Watkin EE, Arbez N, Waldron-Roby E, O’Meally R, Ratovitski T, Cole RN, Ross CA. Phosphorylation of Mutant Huntingtin at Serine 116 Modulates Neuronal Toxicity. PLoS One. 2014;9:e88284. doi: 10.1371/journal.pone.0088284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Arndt JR, Chaibva M, Legleiter J. The emerging role of the first 17 amino acids of huntingtin in Huntington’s disease. Biomol Concepts. 2015;6:33–46. doi: 10.1515/bmc-2015-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Aiken CT, Steffan JS, Guerrero CM, Khashwji H, Lukacsovich T, Simmons D, Purcell JM, Menhaji K, Zhu YZ, Green K, LaFerla F, Huang L, Thompson LM, Marsh JL. Phosphorylation of Threonine 3 IMPLICATIONS FOR HUNTINGTIN AGGREGATION AND NEUROTOXICITY. J Biol Chem. 2009;284:29427–29436. doi: 10.1074/jbc.M109.013193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Di Pardo A, Maglione V, Alpaugh M, Horkey M, Atwal RS, Sassone J, Ciammola A, Steffan JS, Fouad K, Truant R, Sipione S. Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. P Natl Acad Sci USA. 2012;109:3528–3533. doi: 10.1073/pnas.1114502109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, O’Rourke JG, Khashwji H, Lukacsovich T, Zhu YZ, Lau AL, Massey A, Hayden MR, Zeitlin SO, Finkbeiner S, Green KN, LaFerla FM, Bates G, Huang L, Patterson PH, Lo DC, Cuervo AM, Marsh JL, Steffan JS. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, Yang XW. Serines 13 and 16 Are Critical Determinants of Full-Length Human Mutant Huntingtin Induced Disease Pathogenesis in HD Mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cong X, Held JM, DeGiacomo F, Bonner A, Chen JM, Schilling B, Czerwieniec GA, Gibson BW, Ellerby LM. Mass spectrometric identification of novel lysine acetylation sites in huntingtin. Mol Cell Proteomics. 2011;10:M111. 009829. doi: 10.1074/mcp.M111.009829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Kalchman MA, Graham RK, Xia G, Koide HB, Hodgson JG, Graham KC, Goldberg YP, Gietz RD, Pickart CM, Hayden MR. Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J Biol Chem. 1996;271:19385–19394. doi: 10.1074/jbc.271.32.19385. [DOI] [PubMed] [Google Scholar]
- 235.Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 236.O’Rourke JG, Gareau JR, Ochaba J, Song W, Rasko T, Reverter D, Lee J, Monteys AM, Pallos J, Mee L, Vashishtha M, Apostol BL, Nicholson TP, Illes K, Zhu YZ, Dasso M, Bates GP, Difiglia M, Davidson B, Wanker EE, Marsh JL, Lima CD, Steffan JS, Thompson LM. SUMO-2 and PIAS1 Modulate Insoluble Mutant Huntingtin Protein Accumulation. Cell Reports. 2013;4:362–375. doi: 10.1016/j.celrep.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yanai A, Huang K, Kang R, Singaraja RR, Arstikaitis P, Gan L, Orban PC, Mullard A, Cowan CM, Raymond LA, Drisdel RC, Green WN, Ravikumar B, Rubinsztein DC, El-Husseini A, Hayden MR. Palmitoylation of huntingtin by HIP14is essential for its trafficking and function. Nat Neurosci. 2006;9:824–831. doi: 10.1038/nn1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.DiGiovanni LF, Mocle AJ, Xia J, Truant R. Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization. Hum Mol Genet. 2016 doi: 10.1093/hmg/ddw234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kahlem P, Green H, Djian P. Transglutaminase action imitates Huntington’s disease: Selective polymerization of huntingtin containing expanded polyglutamine. Mol Cell. 1998;1:595–601. doi: 10.1016/s1097-2765(00)80059-3. [DOI] [PubMed] [Google Scholar]
- 240.Ehrnhoefer DE, Sutton L, Hayden MR. Small Changes, Big Impact: Posttranslational Modifications and Function of Huntingtin in Huntington Disease. Neuroscientist. 2011;17:475–492. doi: 10.1177/1073858410390378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Díaz‐Hernández M, Valera AG, Morán MA, Gómez‐Ramos P, Alvarez‐Castelao B, Castaño JG, Hernández F, Lucas JJ. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J Neurochem. 2006;98:1585–1596. doi: 10.1111/j.1471-4159.2006.03968.x. [DOI] [PubMed] [Google Scholar]
- 242.Warby SC, Doty CN, Graham RK, Carroll JB, Yang YZ, Singaraja RR, Overall CM, Hayden MR. Activated caspase-6 and caspase-6-cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum Mol Genet. 2008;17:2390–2404. doi: 10.1093/hmg/ddn139. [DOI] [PubMed] [Google Scholar]
- 243.Humbert S, Saudou F. Huntingtin phosphorylation and signaling pathways that regulate toxicity in Huntington’s disease. Clin Neurosci Res. 2003;3:149–155. [Google Scholar]
- 244.Elbaum MB, Zondlo NJ. OGlcNAcylation and Phosphorylation Have Similar Structural Effects in α-Helices: Post-Translational Modifications as Inducible Start and Stop Signals in α-Helices, with Greater Structural Effects on Threonine Modification. Biochemistry. 2014;53:2242–2260. doi: 10.1021/bi500117c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Jana NR, Dikshit P, Goswami A, Kotliarova S, Murata S, Tanaka K, Nukina N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J Biol Chem. 2005;280:11635–11640. doi: 10.1074/jbc.M412042200. [DOI] [PubMed] [Google Scholar]
- 246.Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a Striatal Specific Protein, Mediates Mutant-Huntingtin Cytotoxicity. Science. 2009;324:1327–1330. doi: 10.1126/science.1172871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Chaibva M, Jawahery S, Pilkington AWT, Arndt JR, Sarver O, Valentine S, Matysiak S, Legleiter J. Acetylation within the First 17 Residues of Huntingtin Exon 1 Alters Aggregation and Lipid Binding. Biophys J. 2016;111:349–362. doi: 10.1016/j.bpj.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kegel KB, Sapp E, Alexander J, Valencia A, Reeves P, Li X, Masso N, Sobin L, Aronin N, DiFiglia M. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J Neurochem. 2009;110:1585–1597. doi: 10.1111/j.1471-4159.2009.06255.x. [DOI] [PubMed] [Google Scholar]
- 249.Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:425–433. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- 250.Kegel KB, Sapp E, Yoder J, Cuiffo B, Sobin L, Kim YJ, Qin ZH, Hayden MR, Aronin N, Scott DL, Isenberg F, Goldmann WH, DiFiglia M. Huntingtin associates with acidic phospholipids at the plasma membrane. J Biol Chem. 2005;280:36464–36473. doi: 10.1074/jbc.M503672200. [DOI] [PubMed] [Google Scholar]
- 251.Suopanki J, Gotz C, Lutsch G, Schiller J, Harjes P, Herrmann A, Wanker EE. Interaction of huntingtin fragments with brain membranes – clues to early dysfunction in Huntington’s disease. J Neurochem. 2006;96:870–884. doi: 10.1111/j.1471-4159.2005.03620.x. [DOI] [PubMed] [Google Scholar]
- 252.Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Qin ZH, Wang YM, Sapp E, Cuiffo B, Wanker E, Hayden MR, Kegel KB, Aronin N, DiFiglia M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci. 2004;24:269–281. doi: 10.1523/JNEUROSCI.1409-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Jo E, Darabie AA, Han K, Tandon A, Fraser PE, McLaurin J. alpha-synuclein-synaptosomal membrane interactions – Implications for fibrillogenesis. Eur J Biochem. 2004;271:3180–3189. doi: 10.1111/j.1432-1033.2004.04250.x. [DOI] [PubMed] [Google Scholar]
- 255.Jo EJ, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. alpha-synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 256.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 257.Yip CM, Darabie AA, McLaurin J. A beta 42-peptide assembly on lipid Bilayers. J Mol Biol. 2002;318:97–107. doi: 10.1016/S0022-2836(02)00028-1. [DOI] [PubMed] [Google Scholar]
- 258.Yip CM, McLaurin J. Amyloid-beta peptide assembly: A critical step in fibrillogenesis and membrane disruption. Biophys J. 2001;80:1359–1371. doi: 10.1016/S0006-3495(01)76109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Yates EA, Owens SL, Lynch MF, Cucco EM, Umbaugh CS, Legleiter J. Specific Domains of A beta Facilitate Aggregation on and Association with Lipid Bilayers. J Mol Biol. 2013;425:1915–1933. doi: 10.1016/j.jmb.2013.03.022. [DOI] [PubMed] [Google Scholar]
- 260.Monsellier E, Bousset L, Melki R. alpha-Synuclein and huntingtin exon 1 amyloid fibrils bind laterally to the cellular membrane. Sci Rep. 2016;6:19180. doi: 10.1038/srep19180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Burke KA, Yates EA, Legleiter J. Amyloid-Forming Proteins Alter the Local Mechanical Properties of Lipid Membranes. Biochemistry. 2013;52:808–817. doi: 10.1021/bi301070v. [DOI] [PubMed] [Google Scholar]
- 262.Gorbenko GP, Kinnunen PKJ. The role of lipid-protein interactions in amyloid-type protein fibril formation. Chem Phys Lipids. 2006;141:72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 263.Burke KA, Yates EA, Legleiter J. Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front Neurol. 2013;4:17. doi: 10.3389/fneur.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Gao X, Campbell WA, Chaibva M, Jain P, Leslie AE, Frey SL, Legleiter J. Cholesterol Modifies Huntingtin Binding to, Disruption of, and Aggregation on Lipid Membranes. Biochemistry. 2016;55:92–102. doi: 10.1021/acs.biochem.5b00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kegel-Gleason KB. Huntingtin Interactions with Membrane Phospholipids: Strategic Targets for Therapeutic Intervention? J Huntingtons Dis. 2013;2:239–250. doi: 10.3233/JHD-130068. [DOI] [PubMed] [Google Scholar]
- 266.Leoni V, Caccia C. The impairment of cholesterol metabolism in Huntington disease. BBA-Mol Cell Biol L. 2015;1851:1095–1105. doi: 10.1016/j.bbalip.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 267.Valenza M, Cattaneo E. Emerging roles for cholesterol in Huntington’s disease. Trends Neurosci. 2011;34:474–486. doi: 10.1016/j.tins.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 268.Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20:1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kratter IH, Finkbeiner S. PolyQ Disease: Too Many Qs, Too Much Function? Neuron. 2010;67:897–899. doi: 10.1016/j.neuron.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Illuzzi J, Yerkes S, Parekh-Olmedo H, Kmiec EB. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res. 2009;87:733–747. doi: 10.1002/jnr.21881. [DOI] [PubMed] [Google Scholar]
- 272.Chen-Plotkin AS, Sadri-Vakill G, Yohrling GJ, Bravernan MW, Berin CL, Glajch KE, DiRocco DP, Farrella LA, Krainc D, Gines S, MacDonald ME, Cha JHJ. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington’s disease. Neurobiol Dis. 2006;22:233–241. doi: 10.1016/j.nbd.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 273.Benn CL, Sun T, Sadri-Vakili G, McFarland KN, DiRocco DP, Yohrling GJ, Clark TW, Bouzou B, Cha JHJ. Huntingtin Modulates Transcription, Occupies Gene Promoters In Vivo, and Binds Directly to DNA in a Polyglutamine-Dependent Manner. J Neurosci. 2008;28:10720–10733. doi: 10.1523/JNEUROSCI.2126-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Bolognesi B, Kumita JR, Barros TP, Esbjorner EK, Luheshi LM, Crowther DC, Wilson MR, Dobson CM, Favrin G, Yerbury JJ. ANS Binding Reveals Common Features of Cytotoxic Amyloid Species. ACS Chem Biol. 2010;5:735–740. doi: 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- 275.Nucifora FC, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by Huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 276.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 277.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 278.Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- 279.Costa V, Giacomello M, Hudec R, Lopreiato R, Ermak G, Lim D, Malorni W, Davies KJA, Carafoli E, Scorrano L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol Med. 2010;2:490–503. doi: 10.1002/emmm.201000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Orr AL, Li S, Wang CE, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, Li XJ. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.La Spada AR, Taylor JP. Polyglutamines placed into context. Neuron. 2003;38:681–684. doi: 10.1016/s0896-6273(03)00328-3. [DOI] [PubMed] [Google Scholar]
- 282.Cong SY, Pepers BA, Evert BO, Rubinsztein DC, Roos RA, van Ommen GJ, Dorsman JC. Mutant huntingtin represses CBP, but not p300, by binding and protein degradation. Mol Cell Neurosci. 2005;30:560–571. [PubMed] [Google Scholar]
- 283.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. P Natl Acad Sci USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the Role of Nuclear Localization in Polyglutamine-mediated Aggregation. J Cell Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Calderwood SK, Murshid A, Prince T. The Shock of Aging: Molecular Chaperones and the Heat Shock Response in Longevity and Aging – A Mini-Review. Gerontology. 2009;55:550–558. doi: 10.1159/000225957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Koga H, Kaushik S, Cuervo AM. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res Rev. 2011;10:205–215. doi: 10.1016/j.arr.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. P Natl Acad Sci USA. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurology. 2011;10:83–98. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 289.Valor LM. Transcription, Epigenetics and Ameliorative Strategies in Huntington’s Disease: a Genome-Wide Perspective. Mol Neurobiol. 2015;51:406–423. doi: 10.1007/s12035-014-8715-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Chafekar SM, Duennwald ML. Impaired Heat Shock Response in Cells Expressing Full-Length Polyglutamine-Expanded Huntingtin. PLoS One. 2012;7:e37929. doi: 10.1371/journal.pone.0037929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi-Carter R, Paganetti P, Bates GP. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J Clin Invest. 2011;121:3306–3319. doi: 10.1172/JCI57413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Strehlow ANT, Li JZ, Myers RM. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum Mol Genet. 2007;16:391–409. doi: 10.1093/hmg/ddl467. [DOI] [PubMed] [Google Scholar]
- 293.Twelvetrees AE, Yuen EY, Arancibia-Carcamo IL, MacAskill AF, Rostaing P, Lumb MJ, Humbert S, Triller A, Saudou F, Yan Z, Kittler JT. Delivery of GABA(A)Rs to Synapses Is Mediated by HAP1-KIF5 and Disrupted by Mutant Huntingtin. Neuron. 2010;65:53–65. doi: 10.1016/j.neuron.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.El-Daher MT, Hangen E, Bruyere J, Poizat G, Al-Ramahi I, Pardo R, Bourg N, Souquere S, Mayet C, Pierron G, Leveque-Fort S, Botas J, Humbert S, Saudou F. Huntingtin proteolysis releases non-polyQ fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 2015;34:2255–2271. doi: 10.15252/embj.201490808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Engqvist-Goldstein AEY, Warren RA, Kessels MM, Keen JH, Heuser J, Drubin DG. The actin-binding protein Hip1R associates with clathrin during early stages of endocytosis and promotes clathrin assembly in vitro. J Cell Biol. 2001;154:1209–1223. doi: 10.1083/jcb.200106089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Keryer G, Pineda JR, Liot G, Kim J, Dietrich P, Benstaali C, Smith K, Cordelieres FP, Spassky N, Ferrante RJ, Dragatsis I, Saudou F. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J Clin Invest. 2011;121:4372–4382. doi: 10.1172/JCI57552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Badano JL, Teslovich TM, Katsanis N. The centrosome in human genetic disease. Nat Rev Genet. 2005;6:194–205. doi: 10.1038/nrg1557. [DOI] [PubMed] [Google Scholar]
- 298.Haremaki T, Deglincerti A, Brivanlou AH. Huntingtin is required for ciliogenesis and neurogenesis during early Xenopus development. Dev Biol. 2015;408:305–315. doi: 10.1016/j.ydbio.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 299.Marcora E, Gowan K, Lee JE. Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2. P Natl Acad Sci USA. 2003;100:9578–9583. doi: 10.1073/pnas.1133382100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL, Macdonald ME. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 301.Nasir J, Floresco SB, Okusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 302.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 303.Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- 304.O’Kusky JR, Nasir J, Cicchetti F, Parent A, Hayden MR. Neuronal degeneration in the basal ganglia and loss of pallido-subthalamic synapses in mice with targeted disruption of the Huntington’s disease gene. Brain Res. 1999;818:468–479. doi: 10.1016/s0006-8993(98)01312-2. [DOI] [PubMed] [Google Scholar]
- 305.Menalled L, El-Khodor BF, Patry M, Suarez-Farinas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol Dis. 2009;35:319–336. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Yu ZG, Dadgar N, Albertelli M, Scheller A, Albin RL, Robins DM, Lieberman AP. Abnormalities of germ cell maturation and sertoli cell cytoskeleton in androgen receptor 113 CAG knock-in mice reveal toxic effects of the mutant protein. Am J Pathol. 2006;168:195–204. doi: 10.2353/ajpath.2006.050619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Poletti A. The polyglutamine tract of androgen receptor: from functions to dysfunctions in motor neurons. Front Neuroendocrin. 2004;25:1–26. doi: 10.1016/j.yfrne.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 308.Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- 309.Serra HG, Byam CE, Lande JD, Tousey SK, Zoghbi HY, Orr HT. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum Mol Genet. 2004;13:2535–2543. doi: 10.1093/hmg/ddh268. [DOI] [PubMed] [Google Scholar]