

Abstract
Background
Macrophages stand at the forefront of both innate and adapted immunity through their capacities to recognize, engulf, and eliminate foreign particles, and to stimulate adapted immune cells. They are also involved in controlling pro- and anti-inflammatory pathways. Macrophage activity against Mycobacterium tuberculosis (M. tuberculosis) has been shown to involve Toll-like receptor (TLR) activation and ROS production. Previous studies have shown that lipopolysaccharide (LPS), through TLR4, could activate macrophages, improve their bactericidal ROS production, and facilitate anti-infective immune responses. We sought to better understand the role of the TLR4-NOX2 axis in macrophage activation during M. tuberculosis infection.
Methods
THP-1 macrophages and PMA primed THP-1 macrophages [THP-1(A)] were treated with LPS and infected by M. tuberculosis. Cells were analyzed by flow cytometry for TLR4 expression, ROS production, phagocytosis, and killing of M. tuberculosis. Western blotting was used to analyze NOX2 expression. Inhibitors of the TLR4-NOX2 pathway were used to assess this pathway’s role in these processes, and their role in LPS activation of macrophages.
Results
We found that THP1-derived macrophages or PMA primed THP-1 macrophages exhibit higher surface TLR4 levels and increased NOX2 expression levels following LPS treatment. M. tuberculosis infection reduced these levels, but LPS was able to limit the negative effects of M.tb. Additionally, LPS increases THP-1(A) cells’ bactericidal activities including phagocytosis, ROS production, and destruction of M. tuberculosis. Significantly, all of these activities are impaired when TLR4 or NOX2 are inhibited.
Conclusion
These studies demonstrate the importance of the TLR4-NOX2 axis in M. tuberculosis elimination by macrophages and may lead to novel therapies for tuberculosis and other bacterial infections.
Electronic supplementary material
The online version of this article (10.1186/s12890-017-0517-0) contains supplementary material, which is available to authorized users.
Keywords: TB, TLR, NOX2, Phagocytosis
Background
Macrophages, a key host-defense cell type, recognize invading pathogens via pathogen-associated pattern recognition receptors to initiate anti-infection innate immune responses [1]. During Mycobacterium tuberculosis (M. tuberculosis) infection, Toll-like receptors (TLRs) on the surface of macrophages are shown to recognize pathogens, trigger endocytosis to form phagosomes, produce cytokines, improve cell apoptosis, and thus exert bactericidal activities [2]. The activation of macrophages is the first step of acquiring specific anti-TB immunity, which inhibits the growth or even kills M. tuberculosis, and thus is an important defense mechanism in controlling the infection and spread of M. tuberculosis [3]. TLR4, similar to some other TLR family members [4], is a critical molecule in the anti-TB immune response, through its ability to recognize M. tuberculosis and its components to trigger further innate immune responses [5].
Upon recognition and phagocytosis of M. tuberculosis, macrophages produce excessive reactive oxygen species (ROS) and other bactericidal substances to kill and inactivate M. tuberculosis [6]. Previous studies have shown that lipopolysaccharide (LPS) could activate macrophages through TLR4 to improve the production of ROS, thus directly exerting bactericidal activities, and facilitating the anti-infective immune response [7]. NADPH oxidase (NOX) may produce superoxide ions and ROS through the aggregation and activation of oligomeric enzymes to participate in the host immune defense [8], and reduced NOX activation may increase the risk of infection in patients [9]. In addition, TLR signaling pathways could affect the synthesis and activation of NOX (mainly NOX2), and thus regulate the level of inflammation [10–12]. These findings strongly suggest a possible central role for the TLR-NOX-ROS signaling axis in host defense against M. tuberculosis by macrophages.
In this study, we aim to confirm the role of the TLR4-NOX2 axis in the phagocytic and bactericidal functions of macrophages in M. tuberculosis infection. The characterization of this key pathway in macrophage-mediated innate immunity against M. tuberculosis will reveal details relating to the molecular mechanism of M. tuberculosis infection, and facilitate the identification of novel therapies.
Methods
Cells and bacteria
The human acute monocytic leukemia cell line THP-1 was purchased from American Type Culture Collection (ATCC, Manassas, USA), suspended in Roswell Park Memorial Institute (RPMI) medium 1640 (Sigma-Aldrich, Shanghai, China) to achieve a density of 5 × 105/mL, and cultured in a six-well culture plate. Then, 2 mL of complete culture medium was added to each well. Phorbol-12-myristate-13-acetate (PMA, 120 ng/mL, Sigma-Aldrich, Shanghai, China) was added to stimulate the cells for 24 h, which induced the macrophage-like change in the cells [THP-1(A)] [13]. An attenuated strain of Mycobacterium tuberculosis (H37Ra) was purchased from the Type Culture Collection of Chinese National Institute (Beijing, China) for the Control of Pharmaceutical and Biological Products (batch number: 9,302,025). The H37Ra strain was seeded in solid medium and incubated at 37 °C in a constant-temperature incubator. When the cells reached the logarithmic phase (about 1 month), an aseptic inoculating loop was used to transfer a small amount of bacterial inoculum into a 1-mL homogenizer containing some normal saline. The mixture was homogenized repeatedly, and normal saline was used to wash the concentrate twice (10,000 rpm, 2 min each). Then, a 1-mL syringe was used to add 0.9% NaCl to the bacterial suspension, which was pipetted repeatedly to obtain a single bacterial suspension. The concentration of the bacteria was measured with a spectrophotometer at 600 nm. The concentration of the bacteria was adjusted to 5 × 108/mL, and the suspension was stored in an Eppendorf (EP) tube at 4 °C till use.
PBMC collection
All protocols of the clinical study involving human participants have been approved by the ethics committee of Bengbu Medical College and signed informed consent was obtained from all participants. 20 mL acid-citrate dextrose-treated blood was obtained from healthy donors or TB patients. PBMCs were collected by centrifugation over lymphocyte separation medium, and washed three times with phosphate-buffered saline.
Antibody, blocker, and probe
PMA (#P1585) and lipopolysaccharide were obtained from Sigma-Aldrich (Shanghai, China), fluorescein-labeled antibody TLR4-PE from eBioscience (San Diego, USA), anti-human TLR4 blocking antibody HTA125 and anti-human NOX2 protein antibody from Santa Cruz Biotechnology (Santa Cruz, USA), and NOX2 inhibitor diphenyleneiodonium (DPI) from Sigma. Fluorescein isothiocyanate (FITC) and dimethyl sulfoxide were from Merck (Kenilworth, USA). Fluorescein diacetate (FDA) was from Invitrogen (New York, USA).
Grouping and treatment of the cells
The bacteria were used to infect the cells at a ratio of 1:10, and the TLR4 blocking antibody HTA125 (1 μg/mL) or the NOX2 inhibitor DPI (20 μM) was used to pretreat the infected and noninfected cells, after which LPS (100 ng/mL, Sigma-Aldrich, Shanghai, China) was used to stimulate the cells for 24 h. Cells not treated with the blocker were also used in the study as the control.
Measuring NOX2 protein expression level by western blotting
After treatment, the cells were treated with cell lysis buffer to extract the total protein. After electrophoresis, transfer, incubation with primary and secondary antibodies, and enhancement with 3,3′-diaminobenzidine development, a Tanon GIS gel documentation system was used to obtain and record the images. Imaging analysis software was used for the quantitative analysis of the grayscale of the bands.
Measuring TLR4 expression and ROS levels using flow cytometry
The cells in each group were collected into the tubes of the flow cytometer and washed, for a final volume of 30 μL/tube. Fluorescent probe DHR123 (10 μL/tube, purchased from Sigma-Aldrich) was added to the control group, while 30 μL/tube unlabeled M. tuberculosis suspension (5 × 108/mL) and 10 μL/tube DHR123 (final concentration of 1 μmol/L) were added to the experiment tubes, mixed, and incubated in a 37 °C water bath for 15 min. Then, the cells were quickly washed in ice-cold phosphate-buffered saline (PBS) twice, and flow cytometry was used to measure the ROS level in the M. tuberculosis-infected THP-1(A) cells. For each sample, the level in 10,000 cells was measured. The region of THP-1(A) was first selected in the forward scatter and side scatter two-dimensional scatter diagram, and then a FL1(DHO) histogram was used to analyze the mean fluorescence intensity (MFI) of the oxidized rhodamine123 (namely DHO), which represented the cellular ROS level. Blank control group (macrophage group) and macrophage + DHR123 15-min group were also used in the measurement.
Measuring TLR4 and NOX2 mRNA expression levels using fluorescence quantitative polymerase chain reaction
TRIzol was used to extract the total RNA from the cells. The sequences of TLR4 primers were as follows: F: ACCTGTCCCTGAACCCTATGAA, R: CTTCTAAACCAGCCAGACCTTG. The sequences of NOX2 primers were as follows: F: CAAGATGCGTGGAAACTACC, R: TTGAGAATGGATGCGAAGG. The sequences of human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers were as follows: F: TGCACCACCAACTGCTTAGC, R: GGCATGGACTGTGGTCATGAG. The amplification was performed for 40 cycles. After completion of the amplification, the amplification curve and solubility curve of the real-time polymerase chain reaction were obtained, and the 2-ΔΔCT method was used to analyze the expression of TLR4 and NOX2 mRNA in the cells.
Measuring the phagocytosis and killing activity of THP-1(A) on M. tuberculosis
THP-1(A) cells derived from THP-1 after induction with 120 ng/mL of PMA were obtained and transferred into the tubes of a flow cytometer (1 × 105/tube). Then, 10 μL/tube of FITC-labeled M. tuberculosis suspension (5 × 106/mL), or FDA-labeled M. tuberculosis suspension (5 × 108/mL) was added, mixed, incubated in a 37 °C water bath for 30 min, and then quickly washed with ice-cold PBS twice (10,000 rpm, 2 min each). The supernatant was discarded, and the cells were resuspended. Complete RPMI-1640 culture medium containing 10% fetal bovine serum (FBS) was added (100 μL/tube), the cells were incubated in a 37 °C water bath, and 200 μL/tube ice-cold tri-distilled water was added to each tube at time 0, 10, 30, or 60 min. The tubes were incubated for 2 min to lyse the cells, and then flow cytometry was used to obtain the time-phagocytosis kinetic curve of THP-1(A) against FITC-labeled M. tuberculosis, and time-killing kinetic curve of THP-1(A) against FDA-labeled M. tuberculosis.
THP-1(A) cells were seeded on a six-well plate (2 mL/well). For the cells in the control group, complete RPMI-1640 culture medium was added. However, for the cells in the LPS treatment group, different concentrations of LPS (final concentrations of 10, 100, 1000, and 10,000 ng/mL) were added to stimulate the cells for 24 h. Ice-cold PBS was used to elute the cells into the tubes of the flow cytometer, and 100 μL of complete culture medium was used to resuspend the cells. Nonlabeled bacterial suspension (10 μL/tube) was added to the blank control group, while 10 μL/tube FITC-labeled or FDA-labeled M. tuberculosis (labeling rate > 90%) was added to the negative control and LPS treatment groups. After incubation in a 37 °C water bath for 30 min, the tubes were quickly washed in ice-cold PBS (10,000 rpm for 5 min) twice. The supernatant was discarded, and 100 μL/tube of complete RPMI-1640 culture medium was added to each tube. Then, the tubes were incubated in 0 °C or 37 °C water baths for 0−60 min, and ice-cold tri-distilled water (250 μL/tube) was added immediately. After incubation for 2 min, flow cytometry was used to measure the MFI of FITC or FDA in M. tuberculosis.
The TLR4 and NOX2 blocker groups were also used in this study. In brief, the cells were pretreated with HTA125 (1 μg/mL) for 30 min or DPI (10−20 μM) for 90 min. Then, LPS (100 ng/mL) was used to stimulate the cells for 24 h, and the cells were eluted into the corresponding flow cytometer tubes with ice-cold PBS. The cells were incubated with 5 × 106/mL M. tuberculosis-FITC or 5 × 108/mL M. tuberculosis-FDA for 30 min, washed in ice-cold PBS, and then washed again with ice-cold RPMI-1640 medium once. Next, complete culture medium containing 10% FBS was added, and the tubes were incubated in a 37 °C bath for 0−60 min. Ice-cold tri-distilled water (200 μL/tube) was added to lyse the cells for 2 min, and flow cytometry was used to measure the killing rate. THP-1(A) added to the unlabeled bacteria was used as the blank control.
Statistical analysis
SPSS 17.0 for Windows software (SPSS, IL, USA) was used for the statistical analysis. All data is expressed as mean and standard deviation () and compared by one-way analysis of variance or Q test. A P value less than 0.05 was considered statistically significant.
Results
LPS upregulates TLR4/NOX2 expression and ROS levels in THP-1 cells
As a surface marker of leukocytes, TLR4 was quantified with flow cytometry methods [14]. LPS challenge (24 h) significantly increased TLR4 expression on naïve THP-1 cells or PMA-activated THP-1 cells [THP-1(A) cell] (Fig. 1a, b). Interestingly, PMA treatment and LPS challenge exert a synergistic effect on TLR4 upregulation. Similarly, LPS significantly increased surface TLR4 expression on M. tuberculosis-infected THP-1(A) cells (Fig. 1a, b). We next examined the expression of NOX2 protein under similar conditions. M. tuberculosis-infected THP-1(A) cells exhibit reduced NOX2 protein expression, compared to naïve THP-1(A) cells. Since NOX2 is known to be responsible for immune-defensive ROS generation [15], M. tuberculosis infection seems to reduce NOX2 expression to suppress the immune response in macrophages. However, LPS challenge (100 ng/mL, 24 h) significantly promoted NOX2 protein expression in M. tuberculosis-infected THP-1(A) cells (Fig. 1c, d). In addition, we have confirmed that the regulation of TLR4 and NOX2 expression is on the mRNA levels (Additional file 1: Figure S1).
Fig. 1.

LPS upregulates TLR4 and NOX2 expression and ROS levels in THP-1 cells. a THP-1 cells or THP-1(A) cells were treated with or without LPS, incubated with TLR4-PE, and bound TLR4-PE levels were measured using flow cytometry as an indicator of TLR4 expression. Additionally, some THP-1(A) cells were infected with M. tuberculosis. Histograms of the results, with percentage of cells expressing high TLR4 indicated. b Bar graph of percentages from a. (*; P < 0.05 compared to its control). c Western blot of NOX2 expression in THP-1(A) cells with or without pretreatment with LPS, with or without subsequent M. tuberculosis infection. d Bar graph of band intensities from c. (*; P < 0.05). e Intracellular ROS levels were measured using flow cytometry. f Bar graph of ROS levels from e. [*; P < 0.05 compared to LPS(−)]
NOX2 exerts its immune-defensive effects via ROS generation upon pathogen stimulation [16]. Thus we next examined the intracellular ROS levels in THP-1(A) cells. The ROS level in the M. tuberculosis-infected THP-1(A) cells was significantly lower than that in the uninfected cells, consistent with our findings on NOX2 expression regulation in these cells. In addition, LPS stimulation (24 h) significantly promoted ROS generation in infected cells (Fig. 1e, f). To confirm the regulation of ROS generation is dependent on TLR4-NOX2 axis, the selective TLR4 inhibitor HTA125 and the NOX2 inhibitor DPI were used. HTA125 or DPI treatment significantly reduced the ROS level in M. tuberculosis-infected THP-1(A) cells post LPS stimulation (Fig. 1e, f). These findings confirm that generation of immune-defensive ROS is dependent on the TLR4-NOX2 axis.
Blockade of TLR4 and NOX2 inhibits phagocytosis of M. tuberculosis by THP-1 cells
After we confirmed that LPS restores the expression of TLR4 and NOX2 and generation of ROS in M. tuberculosis infected THP-1(A) cells, next we examined the effects of LPS on the phagocytic activity of THP-1(A) cells. THP-1(A) cells were incubated with FITC labeled M. tuberculosis (1:50) for different time periods (1, 2, 4, and 6 h) and the phagocytic effects of the cells were determined by flow cytometry methods. The mean fluorescence intensity (MFI) of FITC was detected and used as an indicator of phagocytic activity in THP-1(A) macrophages. M. tuberculosis infection increased phagocytosis of THP-1(A) time-dependently and peaked at 6 h (Fig. 2a). In addition, M. tuberculosis infection dose dependently activated the phagocytic effects of THP-1(A) cells (Fig. 2c). These findings also have been reported previously [17], proving a working model of phagocytosis assessment in the THP-1(A) system. As expected, LPS (100 or 1000 ng/mL 24 h pretreatment) significantly increased phagocytosis of M. tuberculosis (at the ratio of 1:50 and 1:100, 30 min) in THP-1(A) cells (Fig. 2c). We also found that LPS dose-dependently (10−10,000 ng/mL) activates phagocytosis in both THP-1 and THP-1(A) cells incubated with M. tuberculosis-FITC at the ratio of 1:50 for 30 min (Additional file 2: Figure S2).
Fig. 2.
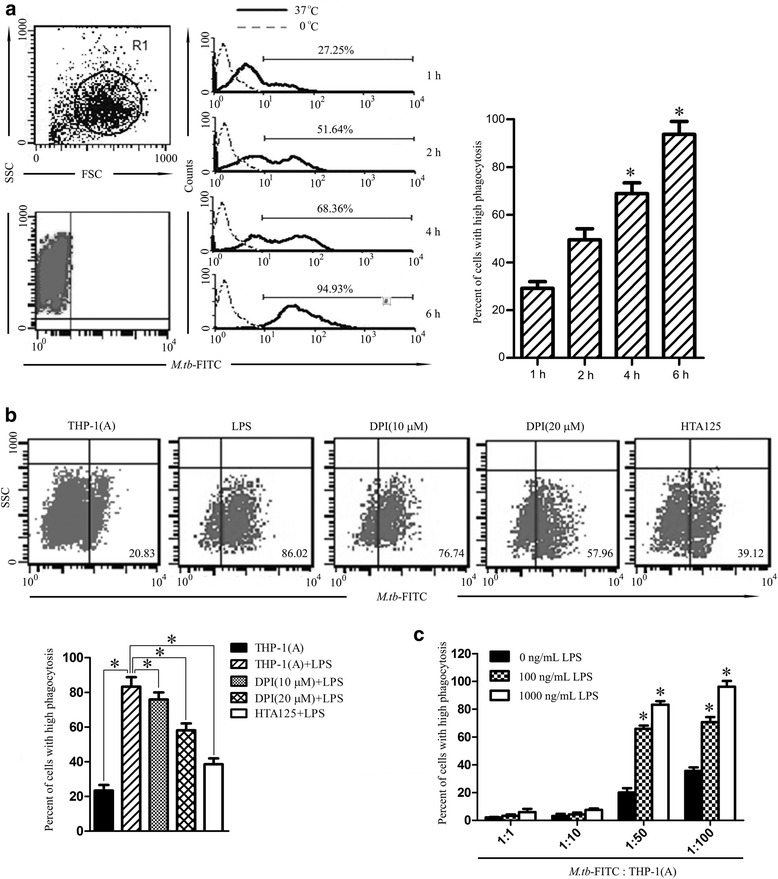
Blocking TLR4 and NOX2 inhibits the phagocytosis of M. tuberculosis by THP-1 cells. a THP-1(A) cells were incubated with M. tuberculosis-FITC for various time periods (1–6 h) at 0 °C or 37 °C. THP-1(A) cells were isolated and identified by flow cytometry (top left), and amount of M. tuberculosis-FITC in those cells was also measured by flow cytometry (bottom left). Histogram of results (middle) with percentage of cells containing high levels of FITC indicated for the 37 °C group. Bar graph of percentages (right). (*; P < 0.05 compared to t = 1 h). b THP-1(A) cells treated without LPS, with LPS, or with LPS in combination with DPI (10 μM), DPI (20 μM), or HTA125 pretreatment were infected with M. tuberculosis-FITC, isolated, and analyzed by flow cytometry. The proportion of cells containing high levels of FITC is indicated (top). Bar graph of percentages (bottom). (*; P < 0.05). c THP-1(A) cells were incubated with various concentrations of LPS (0–1000 ng/mL) and subsequently infected with various concentrations of M. tuberculosis-FITC (1:1–1:100). The proportion of cells containing high levels of FITC was measured as in b. Bar graph of the results. (*; P < 0.05 compared to lowest concentration of LPS). (FSC; forward scatter, SSC; side scatter)
We next tested the role of TLR4-NOX2 in LPS-activated phagocytosis in M. tuberculosis infected THP-1(A) cells. THP-1(A) cells were activated by LPS (1000 ng/mL) for 24 h and then incubated with M. tuberculosis at the ratio of 1:50 for 30 min. The phagocytosis rate of M. tuberculosis was significantly increased by LPS priming (Fig. 2b). However, the TLR4 receptor blocker HTA125 (1 μg/mL, 30 min pretreatment) significantly reduced phagocytosis in THP-1(A) cells treated by LPS and infected by M. tuberculosis. Similarly, NOX2 inhibitor DPI pre-treatment (10 μM, 90 min) significantly reduced the LPS stimulated M. tuberculosis phagocytosis in THP-1(A) cells, and a higher concentration of DPI (20 μM) further attenuated phagocytosis. These findings have demonstrated that LPS activates phagocytosis in M. tuberculosis infected cells via the TLR4-NOX2 axis.
Blockade of TLR4-NOX2 inhibits the killing of M. tuberculosis by THP-1 cells
We next examined the effect of LPS on bactericidal activity of THP-1(A) cells. We successfully labeled M. tuberculosis with FDA (0.1–1.0 μg/mL) and analyzed it by flow cytometry (Fig. 3a). The maximum labeling efficiency was achieved at an FDA concentration of 1.0 μg/mL (98.27% ± 0.92). The bactericidal effects of THP-1(A) on M. tuberculosis-FDA were found to be time dependent, with the proportion of FDA removal peaking at 60 min (53.54% ± 1.85) (Fig. 3b). The time point 30 min was chosen to evaluate the role of TLR4 and NOX2 on bactericidal activity of THP-1(A) cells. After pretreatment with the anti-TLR4 receptor antibody HTA125 (1 μg/mL), the number of viable M. tuberculosis-FDA was increased significantly at both 0 °C and 37 °C compared to LPS treatment alone (P < 0.05) (Fig. 3c). Similarly, the NOX2 inhibitor DPI significantly reduced bactericidal activity at both 0 °C and 37 °C compared to LPS treatment alone (P < 0.05). LPS was also found to increase bactericidal activity in THP-1(A) cells in a dose dependent manner, with significant effects observed at concentrations above 100 ng/mL at both 0 °C and 37 °C and continuously increasing until the maximum testing dose at 10,000 ng/mL (P < 0.01) (Fig. 3d).
Fig. 3.
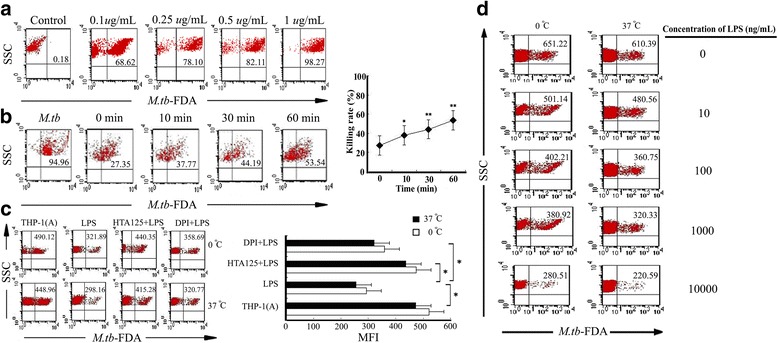
Blocking TLR4 and NOX2 inhibits the killing of M. tuberculosis by THP-1 cells. a M. tuberculosis was incubated with various concentrations of FDA to achieve labeling. Percentage of bacteria labeled is indicated. b FDA labeled M. tuberculosis (left most scatter plot) was incubated with THP-1(A) cells for various time periods (0–60 min). THP-1(A) cells were lysed and total phagocytized M. tuberculosis and M. tuberculosis in serum were isolated. FDA levels in M. tuberculosis were measured by flow cytometry with percentage of M. tuberculosis with FDA removed indicated (left). Graph of FDA removal levels (right). (*; P < 0.05 compared to t = 0, **; P < 0.01 compared to t = 0). c THP-1(A) cells treated without LPS, with LPS, or with LPS in combination with DPI or HTA125 pretreatment were infected with M. tuberculosis-FDA at 0 °C or 37 °C. THP-1(A) cells were lysed and total phagocytized M. tuberculosis and M. tuberculosis in serum were analyzed by flow cytometry for FDA levels. The number of viable M. tuberculosis retaining FDA is indicated (left). Graph of MFI from left (right). (*; P < 0.05). d THP-1(A) cells were treated with various concentrations of LPS (0–10,000 ng/mL), then infected with M. tuberculosis-FDA at 0 °C or 37 °C. THP-1(A) cells were lysed and total phagocytized M. tuberculosis and M. tuberculosis in serum were analyzed by flow cytometry for FDA levels. The number of viable M. tuberculosis retaining FDA is indicated. (SSC; side scatter, MFI; median fluorescence intensity)
Discussion
Macrophages are one of the first lines of defense against bacteria invasion through their ability to directly recognize and eliminate microbes. LPS, a component of Gram-negative bacteria including M. tuberculosis, induces pro-inflammatory activities in various cell types [18]. Here, we demonstrate the role of LPS in stimulating bactericidal activities against M. tuberculosis by macrophages. We found that LPS increased surface levels of TLR4 and induced expression of NOX2. Consistently, we found that LPS enhanced ROS generation, phagocytosis, and bactericide activities in THP-1(A) macrophages. Significantly, all these activities induced by LPS were dependent on the TLR4-NOX2 axis. As an interesting validation, we used PBMCs isolated from TB patients or healthy volunteers, and examined the levels of TLR4 expression (Additional file 3: Figure S3), as well as intracellular ROS levels (Additional file 4: Figure S4). We confirmed that LPS restored reduced surface expression of TLR4, as well as intracellular ROS levels in the TB-infected PBMCs.
M. tuberculosis is mainly recognized by macrophages via TLR2/4 signaling pathways, but the TLR2 signal can be inhibited by the antigens secreted by bacteria [19, 20]. In addition, bacterial components such as phosphatidylinositol mannosides strongly inhibit TLR4 signaling. The combined effects of these factors effectively inhibit autophagy and allow the bacteria to survive for prolonged periods of time in macrophages [21, 22]. M. tuberculosis contains multiple TLR agonists (for instance, the TLR2 agonists LpqH, LprA, and LprG). However, such agonists cannot effectively activate TLR signaling and dowstream phagocytic and bactericidal functions [23]. The possible reasons are: (1) the TLR2 signal is inhibited by some antigens secreted by M. tuberculosis and (2) the bacterial components such as phosphatidylinositol mannosides strongly inhibit the TLR4 signal. The combined effects of these factors ultimately inhibit the effective activation of macrophages [24].
In this study, we showed that TLR4 expression in M. tuberculosis-infected macrophages was significantly decreased, suggesting that M. tuberculosis could downregulate TLR4 expression to inhibit and evade cognition by macrophages. Biglycans were shown to induce NOX2 expression in mouse macrophages through a TLR4/MyD88-dependent pathway, which also led to NOX2 activation [25]. The present study showed that, in addition to upregulating TLR4 expression, LPS could also enhance the bactericidal activity of macrophages by activating the TLR4-NOX2 axis to produce ROS. Previous studies have shown that TLR signaling on activated macrophages could enhance NOX2 activity and stimulate ROS, which not only directly damaged M. tuberculosis but also initiated the activation of autophagy. All these effects contribute to anti-M. tuberculosis responses [16, 26].
A previous study has shown that TLR’s could affect NOX2 expression, activate NOX2 to produce ROS, and regulate inflammatory responses [10]. This study showed that LPS could upregulate NOX2 expression and ROS production in M. tuberculosis-infected THP-1 cells through TLR4-dependent activation. This indicates that regulating ROS through TLR’s could be a potential target for anti-M. tuberculosis treatment.
As noted, we used M.tb-FITC as a tool to reflect the relative level of intracellular M.tb. Measurement was made through an immunofluorescence microtip sensor for specific detection of Mycobacterium cells in sputum samples by the combination of electric field, streaming flow, and immuno-affinity binding [27–29]. This assay is widely used to determine relative levels of M.tb levels. This method is relatively limited due to its low reliability, especially for those assays using heterogeneous cell samples. For the current study, this method is convenient, and the results are conclusive, especially combined with all other supportive data.
In summary, our work has merged several pathways related to bactericidal activities discovered in other immune system cells and we extended these ideas to THP-1 macrophages. We demonstrated M. tuberculosis elimination’s reliance on the TLR4-NOX2 axis. These findings add to our understanding of macrophage immune action, and may contribute to novel therapies for tuberculosis and other bacterial infections.
Conclusions
LPS treatment of THP-1 macrophages leads to increased TLR4 and NOX2 expression, and enhances their ability to produce ROS, phagocytize, and kill bacteria such as M. tuberculosis. Each of these processes is dependent on the TLR4-NOX2 axis. This work demonstrates the ability for LPS to activate macrophages through the TLR4-NOX2 axis, the reliance of macrophages on the TLR4-NOX2 axis for its functions, and may contribute to developing therapies for bacterial infections.
Additional files
LPS upregulates TLR4 and NOX2 mRNA levels in M. tuberculosis-infected THP-1 cells. THP-1(A) cells were treated as in Fig. 1c. mRNA was isolated and the levels of TLR4 and NOX2 mRNA were measured, compared to GAPDH, and normalized to untreated. (*; P < 0.05). (TIFF 252 kb)
LPS dose-dependently (10−10,000 ng/mL) activates phagocytosis in both THP-1 and THP-1(A) cells incubated with M. tuberculosis-FITC at the ratio of 1:50 for 30 min. THP-1 and THP-1(A) cells were incubated with various concentrations of LPS (0–10,000 ng/mL) and subsequently infected with M. tuberculosis-FITC, isolated (top scatter plots), and analyzed by flow cytometry (bottom scatter plots). The proportion of cells containing high levels of FITC is indicated. (TIFF 82 kb)
LPS upregulates TLR4 expression in peripheral blood mononuclear cells (PBMCs) from TB patients. PBMCs were isolated from patients with active tuberculosis and healthy volunteers. Age and gender factors were considered to match. In the supplementary experiments, 5 × 105/mL PBMCs from patient or health control were treated with or without 100 ng/mL LPS for 6 h, and TLR4 levels were measured using flow cytometry. The bar graph showed statistical difference of TLR4 levels (*; P < 0.05 compared to health control). (TIFF 265 kb)
LPS upregulates ROS levels in peripheral blood mononuclear cells (PBMCs) from TB patients. PBMCs were isolated from patients with active tuberculosis and healthy volunteers. Age and gender factors were considered to match. In the supplementary experiments, 5 × 105/mL PBMCs from patient or health control were treated with or without 100 ng/mL LPS for 6 h, and ROS levels were measured using flow cytometry. The bar graph showed statistical difference of ROS levels (*; P < 0.05 compared to health control). (TIFF 203 kb)
Acknowledgements
Not applicable.
Funding
This study was funded by Anhui Provincial Natural Science Research Project of University KJ2013A188 to ZQ (to support the collection and analysis of the data) and KJ2014A164 to JL (to support the collection and analysis of the data), International Science and Technology Cooperation Project-Key Research and Development Program of Anhui Province 1604b0602026 to ZQ (to support the analysis and interpretation of the data), Key Program of Natural Science Research of Higher Education of Anhui Province KJ2017A241 to XW (to support the analysis and interpretation of the data), Key program for excellent young talents in college and university of Anhui province gxyqZD2016168 to XW (to support the analysis and interpretation of the data), and National Natural Science Foundation of China 81,570,011 to TW (to support the analysis and interpretation of the data, and preparation of the manuscript).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DPI
Diphenyleneiodonium
- FDA
Fluorescein diacetate
- FITC
Fluorescein isothiocyanate
- FSC
Forward scatter
- LPS
Lipopolysaccharide
- MFI
Median fluorescence intensity
- NOX
NADPH oxidase
- PBS
Phosphate-buffered saline
- PMA
Phorbol-12-myristate-13-acetate
- ROS
Reactive oxygen species
- RPMI
Roswell Park Memorial Institute medium 1640
- SSC
Side scatter
- THP-1(A)
PMA-activated THP-1
- TLR
Toll-like receptor
Authors’ contributions
TW and ZQ designed the study. JL, XH, HW, and ZW collected the data. XH, HW, ZW, GTK, XW, TW, and ZQ analyzed the data. GTK, XW, YC, TW, and ZQ wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All protocols of the clinical study involving human participants have been approved by the ethics committee of Bengbu Medical College and signed informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12890-017-0517-0) contains supplementary material, which is available to authorized users.
Contributor Information
Jingzhu Lv, Email: bamboolv7778@126.com.
Xiaoyan He, Email: 541455838@qq.com.
Hongtao Wang, Email: w_hongtao@sohu.com.
Zhaohua Wang, Email: 251033026@qq.com.
Gabriel T. Kelly, gabrielkelly@email.arizona.edu
Xiaojing Wang, Email: wangxiaojing8888@163.com.
Yin Chen, Email: ychen@pharmacy.arizona.edu.
Ting Wang, Phone: (520) 626-4472, twang@email.arizona.edu.
Zhongqing Qian, Phone: (86) 0552-3175025, Email: qzq7778@hotmail.com.
References
- 1.Zhou Z, Ding M, Huang L, Gilkeson G, Lang R, Jiang W. Toll-like receptor-mediated immune responses in intestinal macrophages; implications for mucosal immunity and autoimmune diseases. Clin Immunol. 2016;173:81–86. doi: 10.1016/j.clim.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shiratsuchi A, Watanabe I, Takeuchi O, Akira S, Nakanishi Y. Inhibitory effect of toll-like receptor 4 on fusion between phagosomes and endosomes/lysosomes in macrophages. J Immunol. 2004;172(4):2039–2047. doi: 10.4049/jimmunol.172.4.2039. [DOI] [PubMed] [Google Scholar]
- 3.Wang J, Wakeham J, Harkness R, Xing Z. Macrophages are a significant source of type 1 cytokines during mycobacterial infection. J Clin Invest. 1999;103(7):1023–1029. doi: 10.1172/JCI6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jo EK, Yang CS, Choi CH, Harding CV. Intracellular signalling cascades regulating innate immune responses to Mycobacteria: branching out from toll-like receptors. Cell Microbiol. 2007;9(5):1087–1098. doi: 10.1111/j.1462-5822.2007.00914.x. [DOI] [PubMed] [Google Scholar]
- 5.Chang JS, Huggett JF, Dheda K, Kim LU, Zumla A, Rook GA. Myobacterium tuberculosis induces selective up-regulation of TLRs in the mononuclear leukocytes of patients with active pulmonary tuberculosis. J Immunol. 2006;176(5):3010–3018. doi: 10.4049/jimmunol.176.5.3010. [DOI] [PubMed] [Google Scholar]
- 6.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4(3):181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Brumell JH. NADPH oxidases contribute to autophagy regulation. Autophagy. 2009;5(6):887–889. doi: 10.4161/auto.9125. [DOI] [PubMed] [Google Scholar]
- 8.Segal BH, Grimm MJ, Khan AN, Han W, Blackwell TS. Regulation of innate immunity by NADPH oxidase. Free Radic Biol Med. 2012;53(1):72–80. doi: 10.1016/j.freeradbiomed.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh A, Zarember KA, Kuhns DB, Gallin JI. Impaired priming and activation of the neutrophil NADPH oxidase in patients with IRAK4 or NEMO deficiency. J Immunol. 2009;182(10):6410–6417. doi: 10.4049/jimmunol.0802512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsieh LT, Frey H, Nastase MV, et al. Bimodal role of NADPH oxidases in the regulation of biglycan-triggered IL-1beta synthesis. Matrix Biol. 2016;49:61–81. doi: 10.1016/j.matbio.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menden HL, Xia S, Mabry SM, Navarro A, Nyp MF, Sampath V. Nicotinamide adenine Dinucleotide phosphate Oxidase 2 regulates LPS-induced inflammation and alveolar remodeling in the developing lung. Am J Respir Cell Mol Biol. 2016;55(6):767–778. doi: 10.1165/rcmb.2016-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Chen F, Chen J, et al. Soyasaponin bb inhibits the recruitment of toll-like receptor 4 (TLR4) into lipid rafts and its signaling pathway by suppressing the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-dependent generation of reactive oxygen species. Mol Nutr Food Res. 2016;60(7):1532–1543. doi: 10.1002/mnfr.201600015. [DOI] [PubMed] [Google Scholar]
- 13.Cao H, Huang Y, Wang L, et al. Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 2016;7(40):65441–53. [DOI] [PMC free article] [PubMed]
- 14.Galdino H, Jr, Saar Gomes R, Dos Santos JC, et al. Leishmania (Viannia) braziliensis amastigotes induces the expression of TNFalpha and IL-10 by human peripheral blood mononuclear cells in vitro in a TLR4-dependent manner. Cytokine. 2016;88:184–192. doi: 10.1016/j.cyto.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Pelletier M, Lepow TS, Billingham LK, Murphy MP, Siegel RM. New tricks from an old dog: mitochondrial redox signaling in cellular inflammation. Semin Immunol. 2012;24(6):384–392. doi: 10.1016/j.smim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J, Canadien V, Lam GY, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106(15):6226–6231. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie JC, Gougerot-Pocidalo MA, Dang PM. Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev. 2016;273(1):180–193. doi: 10.1111/imr.12447. [DOI] [PubMed] [Google Scholar]
- 18.Plociennikowska A, Hromada-Judycka A, Borzecka K, Kwiatkowska K. Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2015;72(3):557–581. doi: 10.1007/s00018-014-1762-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fortune SM, Solache A, Jaeger A, et al. Mycobacterium tuberculosis inhibits macrophage responses to IFN-gamma through myeloid differentiation factor 88-dependent and -independent mechanisms. J Immunol. 2004;172(10):6272–6280. doi: 10.4049/jimmunol.172.10.6272. [DOI] [PubMed] [Google Scholar]
- 20.Pai RK, Convery M, Hamilton TA, Boom WH, Harding CV. Inhibition of IFN-gamma-induced class II transactivator expression by a 19-kDa lipoprotein from mycobacterium tuberculosis: a potential mechanism for immune evasion. J Immunol. 2003;171(1):175–184. doi: 10.4049/jimmunol.171.1.175. [DOI] [PubMed] [Google Scholar]
- 21.Doz E, Rose S, Court N, et al. Mycobacterial phosphatidylinositol mannosides negatively regulate host toll-like receptor 4, MyD88-dependent proinflammatory cytokines, and TRIF-dependent co-stimulatory molecule expression. J Biol Chem. 2009;284(35):23187–23196. doi: 10.1074/jbc.M109.037846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell DG, Mwandumba HC, Rhoades EE. Mycobacterium and the coat of many lipids. J Cell Biol. 2002;158(3):421–426. doi: 10.1083/jcb.200205034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orvedahl A, Levine B. Eating the enemy within: autophagy in infectious diseases. Cell Death Differ. 2009;16(1):57–69. doi: 10.1038/cdd.2008.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27(1):135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller-Calleja N, Kohler A, Siebald B, et al. Cofactor-independent antiphospholipid antibodies activate the NLRP3-inflammasome via endosomal NADPH-oxidase: implications for the antiphospholipid syndrome. Thromb Haemost. 2015;113(5):1071–1083. doi: 10.1160/TH14-07-0628. [DOI] [PubMed] [Google Scholar]
- 26.Sanjuan MA, Dillon CP, Tait SW, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450(7173):1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 27.Adcock IM, Mortaz E, Alipoor SD, Garssen J, Akbar VA. In vitro effects of water-pipe smoke condensate on the endocytic activity of type II alveolar epithelial cells (A549) with bacillus Calmette-Guerin. Int J Mycobacteriol. 2016;5(Suppl 1):S157–S158. doi: 10.1016/j.ijmyco.2016.09.036. [DOI] [PubMed] [Google Scholar]
- 28.Kim JH, Lee KH, Cangelosi GA, Chung JH. Immunofluorescence microtip sensor for point-of-care tuberculosis (TB) diagnosis. Methods Mol Biol. 2015;1256:57–69. doi: 10.1007/978-1-4939-2172-0_4. [DOI] [PubMed] [Google Scholar]
- 29.Sanjurjo L, Amezaga N, Vilaplana C, et al. The scavenger protein apoptosis inhibitor of macrophages (AIM) potentiates the antimicrobial response against mycobacterium tuberculosis by enhancing autophagy. PLoS One. 2013;8(11):e79670. doi: 10.1371/journal.pone.0079670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
LPS upregulates TLR4 and NOX2 mRNA levels in M. tuberculosis-infected THP-1 cells. THP-1(A) cells were treated as in Fig. 1c. mRNA was isolated and the levels of TLR4 and NOX2 mRNA were measured, compared to GAPDH, and normalized to untreated. (*; P < 0.05). (TIFF 252 kb)
LPS dose-dependently (10−10,000 ng/mL) activates phagocytosis in both THP-1 and THP-1(A) cells incubated with M. tuberculosis-FITC at the ratio of 1:50 for 30 min. THP-1 and THP-1(A) cells were incubated with various concentrations of LPS (0–10,000 ng/mL) and subsequently infected with M. tuberculosis-FITC, isolated (top scatter plots), and analyzed by flow cytometry (bottom scatter plots). The proportion of cells containing high levels of FITC is indicated. (TIFF 82 kb)
LPS upregulates TLR4 expression in peripheral blood mononuclear cells (PBMCs) from TB patients. PBMCs were isolated from patients with active tuberculosis and healthy volunteers. Age and gender factors were considered to match. In the supplementary experiments, 5 × 105/mL PBMCs from patient or health control were treated with or without 100 ng/mL LPS for 6 h, and TLR4 levels were measured using flow cytometry. The bar graph showed statistical difference of TLR4 levels (*; P < 0.05 compared to health control). (TIFF 265 kb)
LPS upregulates ROS levels in peripheral blood mononuclear cells (PBMCs) from TB patients. PBMCs were isolated from patients with active tuberculosis and healthy volunteers. Age and gender factors were considered to match. In the supplementary experiments, 5 × 105/mL PBMCs from patient or health control were treated with or without 100 ng/mL LPS for 6 h, and ROS levels were measured using flow cytometry. The bar graph showed statistical difference of ROS levels (*; P < 0.05 compared to health control). (TIFF 203 kb)
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.