

Key Points
ts-AML (arising from treated antecedent hematological disorder) is less responsive to currently applied treatment strategies.
Future trial designs should accommodate this entity as a distinct category, and patients would be best evaluated on investigational therapies.
Abstract
Secondary acute myeloid leukemia (s-AML) includes therapy-related AML and AML evolving from antecedent hematological disorder (AHD). s-AML arising after treating AHD likely represents a prognostically distinct, high-risk disease category. In this study, treated s-AML (ts-AML) was defined by: (1) prior diagnosis of myelodysplasia, myeloproliferative neoplasm, or aplastic anemia and (2) at least 1 therapy for that diagnosis. ts-AML was categorized by age (< or ≥60 years), and each cohort assessed for response rates and overall survival (OS) on various treatment regimens. Survival outcomes were compared against other high-risk prognostic subsets. Results showed that complete response and 8-week mortality rates were 32% and 27% in the younger, and 24% and 19% in the older age groups, respectively. There was a significant OS difference within s-AML based on prior treatment of AHD (ie, ts-AML vs s-AML with untreated AHD, 4.2 vs 9.2 months; P < .001). Survival in ts-AML was poor across both cohorts (younger and older, 5 and 4.7 months, respectively). In younger AML, survival was significantly inferior in ts-AML when compared with deletion 5/7, TP53, 3q abnormality, and therapy-related AML groups (median, 5 vs 7.9, 7.8, 7.9, and 11.2 months, respectively; P < .01). Additional adverse karyotype within ts-AML was associated with even worse outcomes (OS range, 1.6-2.8 months). ts-AML represents a very high-risk category, even in younger AML patients. s-AML should be further classified to describe ts-AML, an entity less responsive to currently applied treatment approaches. Future AML trial designs should accommodate ts-AML as a distinct subgroup.
Visual Abstract
Introduction
Secondary acute myeloid leukemia (s-AML) refers to a leukemic process: (1) evolving from prior myelodysplasia (MDS), myeloproliferative disorder (MPN), or aplastic anemia (AA) with or without treatment or (2) as a product of previous exposure to a proven leukemogenic chemotherapeutic agent (therapy-related AML [t-AML]).1,2 This term, although implying a process relating to the nature of its neoplastic origins, also bears negative connotations in that it conveys a poor prognosis.2,3 s-AML, although used interchangeably with t-AML, is a more inclusive diagnosis that additionally incorporates AML preceded by a hematological disorder irrespective of treatment of the disorder.2
In the 2008 revision, the World Health Organization refined its AML classification schema by incorporating “AML with myelodysplasia-related changes,” which includes: AML from prior MDS/MPN, AML with MDS-related cytogenetic abnormality, and AML with morphologic multilineage dysplasia.4,5 In the most recent 2016 update, AML-myelodysplasia-related changes and “therapy related myeloid neoplasms” have been retained as distinct subcategories.6 Therapy-related AML defines myeloid neoplasms arising consequent to attempts at curing a variety of malignancies with certain cytotoxic chemotherapeutics.7-10 Lindsley and colleagues were able to categorize clinically defined t-AML into various genetic ontogeny subgroups by their mutational pattern profiles.11 Additionally, attempts at resolving clinicopathologic heterogeneity within AML by the same group of investigators, identified a core set of mutations in selective genes to be highly specific for s-AML.11 Nevertheless, the current umbrella diagnosis of clinically defined s-AML encompasses a group of heterogeneous entities, each tied to different underlying biologic processes.
Older age, adverse risk cytogenetics, and antecedent hematological disorder (AHD) are well-established, poor risk prognostic factors in AML.12,13 There is expected to be a complex interplay between these competing risk factors on disease outcomes. MDS/MPN and AA are premalignant hematological diseases with the risk of evolving to AML.14,15 Leukemic transformations are believed to occur stably over time, likely propelled by stochastic processes involving random genetic events.16 However, some studies suggest that treatment of a preleukemic disorder has an influence on the incidence of subsequent AML.17,18 Clinical emergence of covert leukemic clones that emerge through the selective pressures of cytotoxic therapy, while treating for AHD, would affect leukemia biology and disease aggressiveness.19 An ontological conundrum is whether the secondary leukemia and its subsequent behavior are purely a manifestation along the natural course of, and determined by, its AHD, influenced by potentially genotoxic exposure in treating these AHDs, or a culminating effect of pathophysiologic interactions between the two. Irrespective, patients who develop s-AML evolving from an antecedent hematological disorder represent a high-risk AML subset with bleak long-term survival outcomes.17,18
In an effort to further refine the prognosis of s-AML (arising from AHD), we sought to specifically describe outcomes of patients who developed AML consequent to a previously treated s-AHD (ts-AML). The objective of this study is to outline disease characteristics of this patient subset with ts-AML, in whom there exists a critical unmet need for new treatments. Results of our study will also establish baseline parameters from which to assess the effectiveness of emerging therapies in future clinical trials.
Materials and methods
Study design
A retrospective chart review was done to evaluate patients with newly diagnosed AML treated at our institution from January 2000 to December 2015. Baseline patient and disease characteristics of ts-AML were analyzed and compared with other subsets of AML in terms of therapy responses, response duration, and survival outcomes. All patients on protocols were approved by the institutional review board, and written informed consent obtained before enrollment in accordance with the Declaration of Helsinki.
Patient population and study definitions
ts-AML for this study analysis was defined strictly by meeting the following requisite criteria: (1) history of prior MDS, MPN, or AA and (2) at least 1 chemo- or immunomodulator therapy for that AHD. t-AML was defined by AML arising after exposure to cytotoxic therapy for conditions other than the previously mentioned disease entities such as solid tumor malignancies and lymphomas.6 Patients’ karyotypes were divided into 3 categories: adverse (including complex defined by ≥3 chromosomal abnormalities),20 diploid/intermediate, and favorable.21 Survival estimates were compared with those of other high-risk AML subsets (ie, del5 and/or 7 chromosomal abnormalities, TP53/chr.17p abnormality [TP53 mutated], and t-AML). 3q abnormalities were defined by involvement of 3q21 or 3q26 abnormalities including addition/deletion of chromosome 3q, t(3;3), and inv(3).22 Subgroup analyses were performed within ts-AML comparing overall survival (OS) based on the presence or absence of concurrent high-risk features such as 3q abnormality, 5/7 chromosomal abnormalities, and t-AML within ts-AML. Response rates and survival outcomes within ts-AML were also assessed by type of therapy received. Therapies were grouped by the intensity of treatment regimens into 3 cohorts: (1) high-dose cytarabine (HiDAC)/intensive chemotherapy; (2) intermediate: twice daily fludarabine and cytarabine,23 7+3, CPX-351; and (3) low: hypomethylating agent (HMA)/HMA combinations, low-dose cytarabine/low-dose cytarabine (Ara-C) combinations, and investigational therapies. Regimen type, karyotype, prior AHD type, t-AML, white blood cells (WBCs), platelets, bone marrow blasts, and prior AHD therapy were included in the analysis.
Study end points
Study end points included complete response (CR) rate/CR with low platelets (CRp),20 OS, duration of response and 8-week mortality rates. OS was calculated from time of AML treatment to date of death and censored at last follow-up date if the patient was alive. CR duration was calculated from the time at CR/CRp to date of relapse or censored at last follow-up/death if no relapse.
Statistical analysis
Descriptive statistics were presented as medians and ranges for continuous data, and as numbers and percentages for categorical data. The χ2 and Mann-Whitney U tests were used to compare categorical and continuous data, respectively. Predictive factors for response rates were analyzed using the χ2 test for univariate comparisons. Covariates were included for multivariate comparisons if P < .2 and chosen by backward selection model. For time to event analyses (OS), survival curves were established by the Kaplan-Meier approach. Comparison of survival curves was done using the log-rank test. Variables with P ≤ .05 were chosen for the multivariate Cox regression model. Hazards ratios (HRs) are given with 95% confidence intervals (CIs). Statistical significance was determined at a P value of .05.
Results
We examined a total of 2912 patients with newly diagnosed AML seen at MD Anderson Cancer Center from January 2000 to December 2015. Of these patients, 254 (9%) were designated ts-AML and the other 2658 (91%) patients as non–ts-AML. Included within the non–ts-AML cohort were 215 patients with s-AML evolving from an untreated AHD. Compared with the non–ts-AML population, ts-AML cohort patients were older in age and had lower baseline platelet and bone marrow blast percentage counts (Table 1). Mutational data in genes frequently mutated in AML were available in a proportion of these patients. Among patients with molecular characterization data, RAS, ASXL1, TET2, and RUNX1 mutations were observed more frequently in ts-AML, whereas NPM1 occurred at a lower frequency compared with non–ts-AML. TP53 mutations occurred in 9 patients (1 in younger AML, 9 in older AML); mutations in all cases accompanied a complex karyotype.
Table 1.
Ts- AML and non–ts-AML population disease characteristics and outcomes
| Variable | ts-AML (N = 254) | Non–ts-AML (N = 2659) | P |
|---|---|---|---|
| Median (range) or no./proportion (%) | |||
| Age, y | 68 (15-92) | 63 (12-92) | <.01 |
| WBCs, ×109/L | 4 (0-195) | 5 (0.1-479) | .13 |
| Platelets, ×109/L | 30 (1-936) | 46 (1-1 069) | .03 |
| PB blasts, % | 15 (3-98) | 15 (0-99) | .13 |
| LDH, IU/L | 747 (13-22 798) | 762 (190-42 000) | .12 |
| BM blasts, % | 32 (6-94) | 46 (0-99) | <.01 |
| t-AML, no. (%) | 54 (21) | 454 (17) | .27 |
| Prior AHD treatment, no. (%) | |||
| Hypomethylating agents | 185 (73) | — | — |
| Lenalidomide/thalidomide | 16 (6) | — | — |
| Others | 53 (21) | — | — |
| Karyotype, no. (%) | |||
| Adverse | 109 (43) | 849 (32) | .005 |
| Diploid/intermediate | 118 (46) | 1 159 (44) | .86 |
| Favorable | 3 (1) | 226 (8.5) | <.001 |
| Not done/insufficient | 24 (10) | 558 (15.5) | |
| Mutations* | |||
| FLT3-ITD | 24/220 (11) | 349/2 200 (16) | .052 |
| FLT3D835 | 9/220 (4) | 120/2 200 (5.4) | .39 |
| NPM1 | 17/157 (11) | 293/1 462 (20) | .005 |
| RAS | 42/214 (20) | 278/1 980 (14) | .03 |
| RUNX1 | 11/35 (31) | 30/350 (8.5) | <.001 |
| ASXL1 | 13/34 (38) | 33/354 (9) | <.001 |
| TET2 | 18/55 (33) | 44/359 (12) | <.001 |
| DNMT3A | 14/75 (19) | 110/835 (13) | .18 |
| IDH1 | 13/115 (11) | 113/1 100 (10) | .72 |
| IDH2 | 18/115 (16) | 141/1 100 (13) | .39 |
| TP53 | 9/64 (14) | 112/613 (18) | .40 |
—, Not applicable; BM, bone marrow; LDH, lactate dehydrogenase; PB, peripheral blood.
Molecular mutational data available in limited samples as numerated in the denominators.
Patients were stratified by age into 2 study groups: younger (<60 years) and older (≥60 years) AML. Patient and disease characteristics of the 2 ts-AML age cohorts are outlined in Table 2. The 2 age cohorts varied significantly by prior AHD type and regimens received. Included among the 254 ts-AML patients were 53 with t-AML (previously treated solid tumor or lymphoma history), including 14 of 51 (29%) and 39 of 203 (19%) in the younger and older ts-AML cohorts, respectively. Five of 16 t-AML patients, who had previously received anthracyclines for treatment of their prior malignancy, were induced with an anthracycline-based regimen for their AML.
Table 2.
ts-AML population disease characteristics and outcomes
| Variable | Age <60 y (N = 51) | Age ≥60 y (N = 203) | P |
|---|---|---|---|
| Median (range) or no./proportion (%) | |||
| Age, y | 52 (15-59) | 71 (60-92) | — |
| WBCs, ×109/L | 3 (1-78) | 4 (0-195) | .71 |
| Platelets, ×109/L | 26 (1-227) | 31 (2-936) | .22 |
| PB blasts, % | 21 (0-99) | 14 (0-98) | .68 |
| LDH, IU/L | 779 (287-22 798) | 688 (13-8564) | .46 |
| Total bilirubin, mg/dL | 1 (0-2) | 1 (0-2) | .62 |
| Creatinine, mg/dL | 1 (0-1) | 1 (0-3) | .002 |
| BM blasts, % | 29 (9-93) | 33 (6-94) | .62 |
| Number of prior therapies | 1 (1-5) | 1 (1-6) | .14 |
| Prior AHD, no. (%) | |||
| MDS | 43 (84) | 186 (92) | .002 |
| MPN | 5 (10) | 16 (8) | |
| AA | 3 (6) | 1 (1) | |
| Prior AHD therapy, no. (%) | |||
| Hypomethylating agent | 28 (55) | 153 (75) | .002 |
| Thalidomide/lenalidomide/cyclosporine | 8 (16) | 25 (12) | |
| Ruxolitinib/hydroxyurea | 5 (10) | 13 (18) | |
| Other chemotherapy/immunomodulator therapy* | 10 (19) | 10 (5) | |
| Karyotype | |||
| Adverse | 30/51 (58) | 79/179 (39) | .10 |
| Diploid/intermediate | 20/51 (40) | 98/179 (51) | |
| Favorable | 1/51 (2) | 2/179 (1) | |
| 5q/7q abnormality | 23/51 (45) | 66/179 (37) | .28 |
| Complex | 21/51 (41) | 57/179 (32) | .14 |
| 3q abnormality | 8/51 (16) | 22/179 (12) | .53 |
| Mutations* | |||
| FLT3-ITD | 3/41 (7) | 21/179 (12) | .41 |
| FLT3D835 | 1/41 (3) | 8/179 (4.5) | .55 |
| NPM1 | 3/26 (4) | 14/131 (11) | .89 |
| RAS | 8/38 (21) | 34/176 (19) | .81 |
| TP53 | 1/10 (10) | 8/54 (5) | .68 |
| Regimens | |||
| N, evaluable | 47/51 | 179/203 | — |
| High intensity, no. (%) | 31 (66) | 31 (17) | .001 |
| Medium intensity, no. (%) | 7 (15) | 11 (6) | |
| Low intensity, no. (%) | 9 (19) | 137 (77) | |
Patients treated with stimulating agents such as growth factor supplements, recombinant erythropoietin and analogs, and danazol alone not considered to have ts-AML.
Younger ts-AML study cohort
A total of 1160 (40%) patients were younger than 60 years at diagnosis. Of these, 51 had ts-AML and remaining 1109 were non–ts-AML. The median age at diagnosis was 52 years (range, 15-59). Median number of prior AHD therapies received was 1 (range, 1-5). Thirty of 51 (59%) patients had an adverse karyotype. The cytogenetic landscape is illustrated in supplemental Figure 1. On comparing frequency distribution of karyotypes between ts-AML, t-AML, and de novo AML groups, ts-AML and t-AML groups were associated with a high frequency of adverse karyotype (68% vs 79%, respectively; P = .52), which as statistically different from the de novo AML group (P = .001) (supplemental Figure 2A).
Response and early mortality rates.
Forty-seven of 51 patients were evaluable for response after receiving chemotherapy (Table 3). CR/CRp rate was 32% (CR, 12/47; CRp = 3/47). The 4- and 8-week mortality rates were 5/47 (10%) and 14/47 (27%), respectively (causes of death: sepsis/multiorgan failure, 11; progressive leukemia, 1; intracranial hemorrhage, 1; undetermined, 1). The CR/CRp, 4-week, and 8-week mortality rates by high, intermediate, and investigational/low therapy are outlined in Table 3. Response rates were far lower compared with the t-AML (67.6%) and de novo AML (79.5%) cohorts (P < .001) (supplemental Figure 2B). Similarly, 8-week mortality rates were significantly higher in the ts-AML cohort compared with t-AML (9.2%) and de novo AML (6.1%) (P < .001).
Table 3.
Response rates by treatments received in the ts-AML cohort (grouped by age)
| Parameter | High intensity | Intermediate intensity | Low intensity | P |
|---|---|---|---|---|
| Age <60 y | ||||
| N, evaluable | 31 | 7 | 9 | |
| CR | 6 (19) | 2 (29) | 1 (11) | .67 |
| CRp | 2 (6) | 1 (14) | 0 (0) | .51 |
| CR/CRp | 8 (26) | 3 (43) | 1 (11) | .2 |
| 4-wk mortality | 3 (10) | 1 (14) | 1 (11) | .93 |
| 8-wk mortality | 11 (35) | 1 (14) | 2 (22) | .46 |
| Age ≥60 y | ||||
| N, evaluable | 31 | 11 | 134 | |
| CR | 6 (19) | 3 (27) | 20 (15) | .47 |
| CRp | 4 (13) | 0 (0) | 10 (7) | .35 |
| CR/CRp | 10 (32) | 3 (27) | 30 (22) | .46 |
| 4-wk mortality | 1 (3) | 1 (9) | 10 (7) | .67 |
| 8-wk mortality | 10 (32) | 4 (36) | 25 (19) | .11 |
Data presented as no. (%).
Survival outcomes.
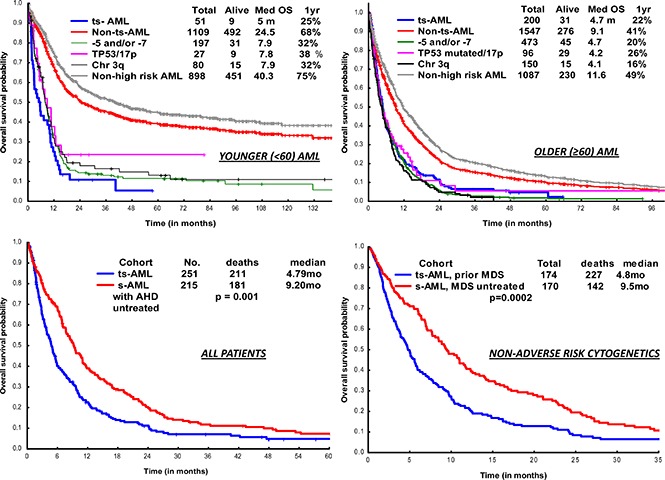
OS in ts-AML patients was significantly inferior compared with non–ts-AML (5 vs 24 months, P < .001; Figure 1A). Estimated 1-year survival probability was 25%. Also, there was a significant difference in OS among the high-risk AML subsets, ts-AML included (P < .01 Figure 1B). A dense clustering of survival curves with virtually identical outcomes was observed across the non–ts-AML high-risk subsets (Figure 1B). Median survival in ts-AML was statistically lower when compared with other high-risk cohorts: 5/7 chromosome abnormality, TP53 mutated, and chromosome 3q (5 vs 7.9, 7.8, and 7.9 months, respectively; P < .01). Additionally, t-AML was associated with superior survival compared with ts-AML (11.2 vs 5 months, P = .002; Figure 1C).
Figure 1.

Younger AML. OS comparisons between the (A) ts-AML and non–ts-AML population, (B) various prognostic risk groups including ts-AML, (C) ts-AML and t-AML, and (D) ts-AML based on whether patients had undergone transplant. chr, chromosome; SCT, allogeneic stem cell transplant; tot, total.
Response duration in the CR group was significantly shorter in ts-AML compared with non–ts-AML (median, 5.3 vs 31 months; P < .01). Nine of the 12 CR group patients proceeded to transplant. Patients transplanted had a superior survival when compared with patients continued on chemotherapy alone (12.4 vs 2.9 months, P = .002; Figure 1D)
Survival by presence of associated high-risk chromosomal abnormalities and therapy regimen type.
Additional presence of 5/7 chromosomal abnormality within ts-AML conferred a statistically significant inferior survival over ts-AML without the high-risk karyotypic feature (2.8 vs 5.9 months, P = .03; Figure 2A). Similarly, there was a trend toward inferior survival associated with the presence of 3q abnormality (vs absent, 1.6 vs 5.6 months, P = .17; Figure 2B). Very few ts-AML patients had a TP53 mutated or complex karyotype occurring in isolation; hence, these karyotype relationships were not examined.
Figure 2.

Younger AML cohort. OS with ts-AML based on associated presence of (A) monosomy 5/7 and (B) 3q abnormality.
Survival comparisons when analyzed by type of therapeutic approach (high, intermediate, low intensity) did not show survival difference across regimens, although there was a trend toward superior survival with intermediate-intensity regimens (3 vs 10 vs 2.9 months, respectively; Figure 3).
Figure 3.

OS within younger ts-AML by different induction regimens. INT, intermediate intensity regimen; INV, low-intensity/investigational regimens; m, months.
Factors predicting response and survival.
In a univariate analysis, none of the analyzed factors predicted for response (Table 4). Univariably, only cytogenetics were associated with differences in OS (Table 4) with nonadverse karyotype predicting for superior survival; this was retained in the multivariate analysis (HR, 0.25 [95% CI, 0.11-0.59]; P = .001).
Table 4.
Response rates and OS by patient and disease characteristics in younger AML
| Response rates (N = 47 evaluable patients) | Univariate K-M survival analysis of OS (N = 51) | ||||
|---|---|---|---|---|---|
| Variable | CR/CRp rates (%) | P | No. deaths | Median OS (mo) | P |
| Overall | — | 42/51 | 5 | — | |
| Treatment group | |||||
| High intensity | 8/31 (26) | .25 | 30/35 | 5.45 | .914 |
| Intermediate | 3/7 (43) | 6/7 | 10.05 | ||
| Inv/low intensity | 1/11 (11) | 6/9 | 3.67 | ||
| Cytogenetics group | |||||
| Adverse | 5/27 (18.5) | .13 | 27/30 | 2.89 | .003 |
| Nonadverse | 11/18 (61) | 15/21 | 10.4 | ||
| Age, y | |||||
| <50 | 3/14 (21.4) | .67 | 13/15 | 2.92 | .618 |
| 50-60 | 9/33 (27.2) | 29/36 | 5.65 | ||
| AHD type | |||||
| MDS | 9/39 (23) | .39 | 35/43 | 5.6 | .75 |
| Non-MDS | 3/8 (37.5) | 7/8 | 5.45 | ||
| t-AML | |||||
| Yes | 3/12 (25) | .96 | 11/14 | 5.02 | .88 |
| No | 9/35 (26) | 31/37 | 5.58 | ||
| WBC | |||||
| ≥10 | 2/15 (13) | .27 | 15/17 | 3.67 | .87 |
| <10 | 10/32 (31) | 27/34 | 5.45 | ||
| Platelet | |||||
| <50 | 8/36 (25) | .25 | 30/38 | 4.23 | .37 |
| ≥50 | 4/11 (36) | 12/13 | 9 | ||
| BM-blast | |||||
| <30% | 8/31 (26) | .57 | 23/29 | 9.79 | .28 |
| ≥30% | 4/21 (19) | 19/22 | 3.67 | ||
| AHD therapy | |||||
| HMA/lenalidomide/ thalidomide | 6/29 (21) | .58 | 24/29 | 5.6 | .6 |
| Other | 6/22 (27) | 18/22 | 5.02 | ||
K-M, Kaplan-Meier.
Older ts-AML study cohort
Of the overall study population, 1752 patients were ≥60 years of age at AML diagnosis; 203 had ts-AML. The median age at diagnosis was 71 (60-92) and median number of prior therapies received was 1 (1-6). A total of 179/203 (88%) patients had a determinable karyotype. Cytogenetic landscape is illustrated in supplemental Figure 3.
Response and early mortality rates.
Among 203 patients, 176 were evaluable for response after receiving chemotherapy (Table 3); CR/CRp rate was 24%. The 4- and 8-week mortality rates were 12/176 (7%) and 39/176 (22%), respectively. The CR/CRp, 4-week, and 8- week mortality rates by different treatment intensity regimens are shown in Tables 2 and 5
Table 5.
Response rates by type of low intensity treatment (age ≥60 y)
| Parameter | CLAD/CLOF + LDAC | HMA | Other | P |
|---|---|---|---|---|
| N, evaluable | 36 | 21 | 77 | |
| CR | 14 (39) | 3 (14) | 3 (4) | <.001 |
| CRp | 3 (8) | 1 (5) | 6 (8) | .87 |
| CR/CRp | 17 (47) | 4 (19) | 9 (12) | <.001 |
| 4-wk mortality | 1 (3) | 1 (5) | 8 (10) | .31 |
| 8-wk mortality | 6 (17) | 5 (24) | 14 (18) | .29 |
Data presented as no. (%).
CLAD, cladribine; CLOF, clofarabine; HMA, hypomethylating agent.
Survival outcomes.
Two hundred of 203 patients were evaluable for survival analysis. OS in ts-AML was significantly lower compared with OS in non–ts-AML (4.7 vs 9.1 months, P < .001) (Figure 4A). Estimated 1-year survival probability in ts-AML was only 22%. Unlike in the younger AML cohort, there was no significant difference in OS between the high-risk AML groups and ts-AML resulting from drop in survival rates across the non–ts-AML high-risk AML age groups. There was no statistically significant difference in median survival in ts-AML compared with t-AML without ts-AML (4.7 vs 5.4 months, P = .24; Figure 4C). Similar to the younger age AML, patients who received transplant had a superior survival compared with patients not transplanted (16.2 vs 4.6 months, P < .001; Figure 4D).
Figure 4.

Older AML. OS comparisons between (A) ts-AML and non–ts-AML population, (B) various prognostic risk groups including ts-AML, (C) ts-AML and t-AML, and (D) ts-AML based on whether patients had undergone transplant.
Survival outcomes by associated high-risk chromosomal abnormalities and therapy type.
Additional presence of 5/7 chromosomal abnormality conferred a statistically significant inferior survival over ts-AML in the absence of this high-risk feature (2.8 vs 5.2 months, P = .03) (Figure 5A). Similarly, inferior survival was conferred by the additional presence of 3q abnormality (2.4 vs 5.1 months, P = .03) (Figure 5B).
Figure 5.

Younger AML cohort OS. ts-AML OS based on associated presence of (A) monosomy 5/7 and (B) 3q abnormality.
Survival comparisons when analyzed by type of therapeutic approach (ie, HiDAC vs intermediate intensity vs low intensity) did not show statistically significant survival differences (median, 3.4 vs 5.7 vs 4.8 months; Figure 6).
Figure 6.

OS within older ts-AML by different induction regimens.
Factors predicting response and survival.
On a univariate analysis, none of the analyzed factors predicted for response, except for non-adverse karyotype, which was associated with a trend toward higher response rates (Table 6). Univariably, adverse cytogenetics, white blood cell counts and platelet counts were associated with statistically significant differences in OS (Table 7). By multivariate analysis, only WBC (≥10 K/µL) and adverse karyotype were associated with significantly worse survival (Table 7).
Table 6.
Response rates by patient and disease characteristics in the older AML population (N = 176)
| Variable | CR rates (%) | P |
|---|---|---|
| Overall | N = 176 | - |
| Treatment group | ||
| High intensity | 10/31 (32) | .46 |
| Intermediate intensity | 3/11 (27) | |
| INV/low intensity | 30/134 (22) | |
| Cytogenetics group | ||
| Adverse | 12/70 (17) | .08 |
| Nonadverse | 31/106 (29) | |
| Age, y | ||
| 60-70 | 24/84 (29) | .21 |
| >70 | 19/92 (21) | |
| AHD type | ||
| MDS | 39/163 (23) | .92 |
| Non-MDS | 4/16 (37.5) | |
| t-AML | ||
| Yes | 9/36 (25) | .9 |
| No | 34/140 (26) | |
| WBCs* | ||
| <10 | 29/119 (24) | .17 |
| ≥10 | 6/38 (16) | |
| Platelets* | ||
| ≥50 | 21/103 (20) | .43 |
| <50 | 14/54 (26) | |
| BM blasts | ||
| <30% | 20/72 (27) | .48 |
| ≥30% | 23/104 (22) | |
| AHD therapy | ||
| HMA/lenalidomide/thalidomide | 31/158 (20) | .34 |
| Other | 6/21 (29) | |
Data available on baseline WBCs and platelets in 157 of 176 evaluable patients.
Table 7.
KM analysis of OS by patient and disease characteristics in older AML population
| Univariate K-M survival analysis of OS (N = 200) | Multivariate Cox regression for predictors of OS (N = 157) | |||||
|---|---|---|---|---|---|---|
| Variable | No. of deaths | Median OS (mo) | P | HR | 95% CI | P |
| Overall | 169/200 | 4.7 | — | |||
| Treatment group | ||||||
| High intensity | 32/33 | 3.48 | .22 | |||
| Intermediate intensity | 10/14 | 9.19 | ||||
| INV/low intensity | 127/143 | 4.99 | ||||
| Cyto group | ||||||
| Adverse | 69/79 | 3.08 | .005 | 1.65 | 1.09-2.61 | .018 |
| Nonadverse | 51/60 | 6.89 | ||||
| Age, y | ||||||
| 60-70 | 70/84 | 4.89 | .05 | |||
| >70 | 99/116 | 4.69 | ||||
| AHD type | ||||||
| MDS | 157/182 | 4.76 | .42 | |||
| Non-MDS | 12/18 | 4.4 | ||||
| t-AML status | ||||||
| Yes | 34/39 | 5.09 | .72 | |||
| No | 135/161 | 4.59 | ||||
| WBC* | ||||||
| <10 | 100/119 | 5.4 | .03 | 0.57 | 0.36-0.93 | .02 |
| ≥10 | 33/38 | 4.3 | ||||
| Platelet* | ||||||
| ≥50 | 87/103 | 4.3 | .03 | |||
| <50 | 46/54 | 5.7 | ||||
| BM-blast | ||||||
| <30% | 68/82 | 5.6 | .13 | |||
| ≥30% | 98/114 | 3.9 | ||||
| AHD therapy | ||||||
| HMA/lenalidomide/ thalidomide | 147/177 | 4.69 | .60 | |||
| Other | 22/26 | 5.02 | ||||
Data available on baseline WBC and platelets in 157 of 200 evaluable patients.
Factors predicting response and survival in the overall ts-AML study cohort
Response rates and OS were not influenced by age, baseline hematological indices, t-AML status or the presence of FLT3ITD, NPM1, or RAS mutations. On uni- and multivariate analysis, adverse cytogenetics predicted for decreased response rates and OS. There was trend toward improved OS in the non–FLT3-ITD mutated ts-AML patients (P = .09). Notably, patients with FLT3-ITD mutations that were treated with FLT3-inhibitors experienced an improved survival FLT3 inhibitor regimen vs non-FLT3 inhibitor regimen (median OS: NR vs 4.9 months, P < .001) (supplemental Figure 4).
There was a significant difference in OS within s-AML by prior treatment of AHD (ts-AML vs s-AML with untreated AHD, 4.2 vs 9.2 months; P = .001) (supplemental Figure 5A). Survival difference was maintained in a subset analysis confined to AML with non-adverse cytogenetics (ts-AML vs s-AML with AHD untreated, 5.9 vs 11.7 months; P = .001) (supplemental Figure 5B). In a multivariate analysis in the overall AML population, ts-AML status associated with significantly inferior survival (supplemental Table 1). In a multivariate sensitivity analysis, after excluding ts-AML with adverse risk cytogenetics, ts-AML still predicted for inferior survival (HR 2.7 [95% CI, 1.8-3.9]; P < .001). Analyzing impact by prior AHD type status, survival was significantly inferior in the ts-AML from prior MDS cohort compared with s-AML, prior MDS untreated (4.8 vs 9.5 months, P = .0002; supplemental Figure 6A). There was trend toward inferior survival in ts-AML from prior MPN cohort compared with s-AML, prior MPN untreated (3.1 vs 8.6 months, P = .24; supplemental Figure 6B).
Discussion
Ts-AML (AML subsequent to AHD which was treated, irrespective of history of previously treated malignancies) selects for a very high risk subset, particularly in the younger aged AML population. Our data finds special relevance in the era of increased recognition and growing inclination toward treating AHDs. Ts-AML represents a growing patient cohort, who while being traditionally classified as ‘untreated’ AML, have had encountered significant prior therapy.
Inclusion of s-AML along with de novo AML in clinical trial study designs finds support in prior conducted studies, such by Ostgard et al, which demonstrated that s-AML, per se, did not impact response rates and survival after accounting for cytogenetics and other relevant deleterious risk factors.24 However, the aforementioned study included a mixed population of s-AML defined by AML arising from prior AHD (including untreated) and t-AML. In our study, survival analysis on s-AML patients by prior treatment of AHD showed a significant survival difference of 5 months, favoring s-AML arising from untreated AHD. This survival disparity was maintained in a subset analysis of s-AML with non-adverse cytogenetics. We argue that s-AML evolving from prior treated AHD should be classified as ‘treated s-AML’, distinct from t-AML, s-AML from untreated AHD, and de novo AML.
s-AML occurs in 5% to 30% of AHD, occurring more frequently in AHD with high risk aberrant karyotype.25-28 Leukemic progression is typically characterized by acquisition by additional genetic changes, involving either chromosomal or molecular mutation abnormalities.29 Acknowledging limited sample size estimates available on molecular characterization data, we noted a high frequency of ASXL1 (38%), TET2 (33%) mutations in the ts-AML cohort, a prominent feature of s-AML.11,30,31 Mutations in these genes occur at a high frequency in MDS as well, suggesting them to be early events in AHD.31 We also observed a high frequency of RUNX1 (31%), FLT3 ITD/TKD (15%), and RAS (20%) mutations the ts-AML cohort. Previously published data indicate that RUNX1, FLT3, RAS mutations occur infrequently in AHD/MDS suggesting that acquisition of this class of mutations plays a transformative role in disease progression.30-32
Outcomes after conventional chemotherapy in patients with AML arising from treated MDS are expectantly poor.33,34 It has been shown that treatment with therapies such as HMAs has a significant effect on disease natural history, effectively protracting disease latency from MDS/MPN to leukemic progression.35 However, it is conceivable that prolonged exposure to these agents may select for the emergence of chemotherapy resistant AML clones. Ts-AML after hypomethylating agent (HMA)/lenalidomide/thalidomide failure is associated with a particularly poor outcome, with a projected median survival of 3-4 months.33,34 Conversely, s-AML arising from MDS managed with supportive care associates with improved median survival of 10.5 months.34 Putting this into perspective, our study cohort included 221 patients with prior MDS, 89% of whom developed AML post HMA/lenalidomide/thalidomide failure. Response rates and survival did not differ statistically by therapy before progression (HMA/lenalidomide/thalidomide vs other) on regression analyses, in either age cohort of our study, suggesting that the prognostic impact may not be selective to prior exposure to HMA or lenalidomide/thalidomide therapy. We noted a nonsignificant trend toward inferior survival in patients with ts-AML from prior MPN compared with MPN-untreated s-AML patients. The impact of other AHD therapies on subsequent AML behavior must be explored in a larger study cohort.
Treatment of AHD may result in various genetic and epigenetic alterations paving the way for biologically aggressive, chemo-insensitive leukemic disease less amenable to response with the currently applied therapeutic approaches.36 Poor responses to conventional induction therapy in this form of AML are related partly to the overexpression of several multidrug resistant gene products. Prior treatments with agents such as decitabine have been shown to result in multidrug resistance protein-1 (MDR1) gene activation through MDR1 demethylation.37 Overexpression of MDR-1 confers resistance to multiple antileukemic agents including cytarabine, anthracyclines, and epipodophyllotoxins.38,39 Also, leukemic cells in s-AML are characterized by a far lower expression levels of Ara-C transporting proteins, such as hENT1. Expression levels of hENT1correlate with responses to cytarabine-based intensive chemotherapy.40
Acknowledging genetic/biological differences, and dissimilarities in the nature of chemotherapy regimens and their intensities, outcome analyses were performed separately for younger and older AML cohorts. Irrespective of the cohort, we noted no statistically significant difference in response rates or OS across treatment regimens. CR/CRp rates in the younger age cohort were low at 32%, and 8 week mortality rates high at 27%. The comparatively inferior survival rates in ts-AML are related in part to significantly lower response rates and higher 8-week mortality rates as compared with t-AML and de novo AML groups. A high frequency of early deaths (11 of 17 overall) occurred in patients induced with high intensity regimens (ie, HiDAC/intensive chemotherapy). In this regard, benefits of cytarabine dose escalation in affecting responses in refractory AML are isolated to a few small studies.41,42 Combinatorial regimens built around a high-dose cytarabine backbone such as fludarabine, Ara-C, idarubicin, and granulocyte colony-stimulating factor and cladribine-Ara-C, purportedly improve upon the cytotoxic effects of HiDAC, and are fairly equivalent in efficacy.43-45 However, there is mounting evidence to suggest that responses, even to escalated doses of Ara-C, are heavily dependent upon the underlying cytogenetic and molecular signatures.46 There is an urgent need for genetic annotation to define patients who best benefit from cytarabine-based therapy. The much favored high-intensity Ara-C–based induction regimens in younger AML patients should be reconsidered in the difficult-to-treat setting of ts-AML. The role of novel approaches, including CPX-351, merits reevaluation in this setting.47 Treatment with targeted therapies such as small molecule inhibitors (for example, FLT3) may provide another efficacious approach even in this high-risk ts-AML subset (supplemental Figure 4). Other emerging therapies, including antibody-drug conjugates, immune check point inhibitors, and molecularly targeted agents inhibiting deranged signaling and metabolic pathways, should be explored either alone or in rational combinations with other agents of nonoverlapping mechanisms of action.48 Similar to previous studies,49 our data showed a significant improvement in survival with proceeding to transplant (range, 9.5-11.6 months), suggesting that transplant can significantly alter the natural history of the disease.
Prior confirmatory studies have upheld the prognostic utility of European Leukemia Net (ELN) cytogenetic classification in younger and older AML alike.21,50 To put ts-AML outcomes in context, we compared ts-AML against these other high-risk subgroups. Although we did not discern a survival difference between ts-AML and any of the aforementioned ELN adverse subgroups in older patients, survival analyses clearly showed ts-AML’s prognostic separation from other adverse risk subgroups in younger AML. The 2.7- to 2.9-month median survival difference between ts-AML and other ELN adverse risk groups’ merits significance given that 41% of younger age AML did not have an adverse karyotype. The associated presence of an adverse risk karyotype such as 3q, or 5/7 chromosomal abnormality in the setting of ts-AML. identified an even higher risk entity (median survival rates not exceeding 3 months, irrespective of age). This not only reaffirms our perspectives of the clinically aggressive nature of ts-AML, but also helps identify it as poor risk entity extending beyond the prognostic informativeness of adverse cytogenetics.
An important strength of our study is the assessment of ts-AML impact after its categorization by age. This helped demonstrate that outcomes are uniformly dismal regardless of age. An important limitation to our study includes its retrospective study design. Our ts-AML cohort was overrepresented by patients with prior MDS, which constituted 85% of the study population. Small sample size estimates in certain population subsets were unconducive to statistical analyses, potentially limiting a reliable estimation for differences that may have existed. We did not have molecular mutation information on most patients and hence were not able to incorporate molecular mutation profile data in our prediction analysis for individual age cohorts. Our data lacked the level of granularity sufficient to analyze the effects of varying temporal dynamics of AHD-AML transition on ts-AML outcomes. Our sample cohort included a heterogeneous population characterized by varying temporal evolution profiles from time of AHD treatment to AML progression. Assuming ts-AML arising early into AHD treatment behaves along the lines of s-AML from untreated AHD and factoring time from AHD treatment to leukemia onset would identify for even higher risk subgroups within ts-AML.
In conclusion, poor outcomes in ts-AML are a combined translation of low response rates, high early mortality, and higher risk of early disease relapse. Future investigational treatment approaches including multidrug modulators, antibody-drug conjugates, and molecularly targeted inhibitors should be considered in treating these patients. Although data on an increasing number of effective novel combinations in high-risk AML is emerging in trials, there is an urgent need to evaluate their efficacy in this subset. We propose that the definition of s-AML should be narrowed to define ts-AML, a category of AML typically less responsive to currently applied treatment approaches.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This work was supported in part by the National Institutes of Health, National Cancer Institute (grant CA016672 to the MD Anderson Cancer Center and grant P01 CA049639).
Authorship
Contribution: P.B. and T.M.K. were involved in designing study, collecting data, and writing and reviewing the manuscript; S.R.P. provided data for the study and reviewed the manuscript; and H.M.K., J.E.C., G.G.-M., F.R., S.V., E.J., G.B., M.K., K.N.B., N.D., C.D.D., C.B.B., K.T., Z.E., and M.A. provided intellectual content and patients for the study, and wrote and reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Tapan M. Kadia, Department of Leukemia, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Box 428, Houston, TX 77030; e-mail: tkadia@mdanderson.org.
References
- 1.Bennett JM. Secondary acute myeloid leukemia. Leuk Res. 1995;19(4):231-232. [DOI] [PubMed] [Google Scholar]
- 2.Larson RA. Is secondary leukemia an independent poor prognostic factor in acute myeloid leukemia? Best Pract Res Clin Haematol. 2007;20(1):29-37. [DOI] [PubMed] [Google Scholar]
- 3.Granfeldt Østgård LS, Medeiros BC, Sengeløv H, et al. . Epidemiology and clinical significance of secondary and therapy-related acute myeloid leukemia: a national population-based cohort study. J Clin Oncol. 2015;33(31):3641-3649. [DOI] [PubMed] [Google Scholar]
- 4.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019-5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinberg OK, Seetharam M, Ren L, et al. . Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood. 2009;113(9):1906-1908. [DOI] [PubMed] [Google Scholar]
- 6.Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. [DOI] [PubMed] [Google Scholar]
- 7.Leone G, Pagano L, Ben-Yehuda D, Voso MT. Therapy-related leukemia and myelodysplasia: susceptibility and incidence. Haematologica. 2007;92(10):1389-1398. [DOI] [PubMed] [Google Scholar]
- 8.Larson RA. Etiology and management of therapy-related myeloid leukemia. Hematology Am Soc Hematol Educ Program. 2007;2007:453-459. [DOI] [PubMed] [Google Scholar]
- 9.Czader M, Orazi A. Therapy-related myeloid neoplasms. Am J Clin Pathol. 2009;132(3):410-425. [DOI] [PubMed] [Google Scholar]
- 10.Kayser S, Döhner K, Krauter J, et al. ; German-Austrian AMLSG. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood. 2011;117(7):2137-2145. [DOI] [PubMed] [Google Scholar]
- 11.Lindsley RC, Mar BG, Mazzola E, et al. . Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367-1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Appelbaum FR, Gundacker H, Head DR, et al. . Age and acute myeloid leukemia. Blood. 2006;107(9):3481-3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kantarjian H, O’brien S, Cortes J, et al. . Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer. 2006;106(5):1090-1098. [DOI] [PubMed] [Google Scholar]
- 14.Foucar K. Myelodysplastic/myeloproliferative neoplasms. Am J Clin Pathol. 2009;132(2):281-289. [DOI] [PubMed] [Google Scholar]
- 15.Kojima S, Ohara A, Tsuchida M, et al. ; Japan Childhood Aplastic Anemia Study Group. Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children. Blood. 2002;100(3):786-790. [DOI] [PubMed] [Google Scholar]
- 16.Shukron O, Vainstein V, Kündgen A, Germing U, Agur Z. Analyzing transformation of myelodysplastic syndrome to secondary acute myeloid leukemia using a large patient database. Am J Hematol. 2012;87(9):853-860. [DOI] [PubMed] [Google Scholar]
- 17.Shi J, Shao ZH, Liu H, et al. . Transformation of myelodysplastic syndromes into acute myeloid leukemias. Chin Med J (Engl). 2004;117(7):963-967. [PubMed] [Google Scholar]
- 18.Björkholm M, Derolf AR, Hultcrantz M, et al. . Treatment-related risk factors for transformation to acute myeloid leukemia and myelodysplastic syndromes in myeloproliferative neoplasms. J Clin Oncol. 2011;29(17):2410-2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casás-Selves M, Degregori J. How cancer shapes evolution, and how evolution shapes cancer. Evolution (N Y). 2011;4(4):624-634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Döhner H, Estey EH, Amadori S, et al. ; European LeukemiaNet. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453-474. [DOI] [PubMed] [Google Scholar]
- 21.Mrózek K, Marcucci G, Nicolet D, et al. . Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012;30(36):4515-4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu Y, Chen ZM, Xu WL, Mu QT, Jin J. Clinical analysis of 24 cases of acute myeloid leukemia with 3q abnormalities [in Chinese]. Zhejiang Da Xue Xue Bao Yi Xue Ban. 2010;39(3):241-245. [DOI] [PubMed] [Google Scholar]
- 23.Jabbour E, Garcia-Manero G, Cortes J, et al. . Twice-daily fludarabine and cytarabine combination with or without gentuzumab ozogamicin is effective in patients with relapsed/refractory acute myeloid leukemia, high-risk myelodysplastic syndrome, and blast- phase chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2012;12(4):244-251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ostgård LS, Kjeldsen E, Holm MS, et al. . Reasons for treating secondary AML as de novo AML. Eur J Haematol. 2010;85(3):217-226. [DOI] [PubMed] [Google Scholar]
- 25.Preiss BS, Bergmann OJ, Friis LS, et al. ; AML Study Group of Southern Denmark. Cytogenetic findings in adult secondary acute myeloid leukemia (AML): frequency of favorable and adverse chromosomal aberrations do not differ from adult de novo AML [published correction appears in Cancer Genet Cytogenet. 2011;204(2):111]. Cancer Genet Cytogenet. 2010;202(2):108-122. [DOI] [PubMed] [Google Scholar]
- 26.Padua RA, McGlynn A, McGlynn H. Molecular, cytogenetic and genetic abnormalities in MDS and secondary AML. Cancer Treat Res. 2001;108:111-157. [DOI] [PubMed] [Google Scholar]
- 27.Heaney ML, Soriano G. Acute myeloid leukemia following a myeloproliferative neoplasm: clinical characteristics, genetic features and effects of therapy. Curr Hematol Malig Rep. 2013;8(2):116-122. [DOI] [PubMed] [Google Scholar]
- 28.Barrett J, Saunthararajah Y, Molldrem J. Myelodysplastic syndrome and aplastic anemia: distinct entities or diseases linked by a common pathophysiology? Semin Hematol. 2000;37(1):15-29. [DOI] [PubMed] [Google Scholar]
- 29.Flach J, Dicker F, Schnittger S, et al. . An accumulation of cytogenetic and molecular genetic events characterizes the progression from MDS to secondary AML: an analysis of 38 paired samples analyzed by cytogenetics, molecular mutation analysis and SNP microarray profiling. Leukemia. 2011;25(4):713-718. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Mercado M, Yip BH, Pellagatti A, et al. . Mutation patterns of 16 genes in primary and secondary acute myeloid leukemia (AML) with normal cytogenetics. PLoS One. 2012;7(8):e42334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dicker F, Haferlach C, Sundermann J, et al. . Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia. 2010;24(8):1528-1532. [DOI] [PubMed] [Google Scholar]
- 32.Takahashi K, Jabbour E, Wang X, et al. . Dynamic acquisition of FLT3 or RAS alterations drive a subset of patients with lower risk MDS to secondary AML. Leukemia. 2013;27(10):2081-2083. [DOI] [PubMed] [Google Scholar]
- 33.Prébet T, Gore SD, Thépot S, et al. . Outcome of acute myeloid leukaemia following myelodysplastic syndrome after azacitidine treatment failure. Br J Haematol. 2012;157(6):764-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bello C, Yu D, Komrokji RS, et al. . Outcomes after induction chemotherapy in patients with acute myeloid leukemia arising from myelodysplastic syndrome. Cancer. 2011;117(7):1463-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nimer SD. Myelodysplastic syndromes. Blood. 2008;111(10):4841-4851. [DOI] [PubMed] [Google Scholar]
- 36.Qin T, Jelinek J, Si J, Shu J, Issa JP. Mechanisms of resistance to 5-aza-2′-deoxycytidine in human cancer cell lines. Blood. 2009;113(3):659-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kantharidis P, El-Osta A, deSilva M, et al. . Altered methylation of the human MDR1 promoter is associated with acquired multidrug resistance. Clin Cancer Res. 1997;3(11):2025-2032. [PubMed] [Google Scholar]
- 38.Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385-427. [DOI] [PubMed] [Google Scholar]
- 39.Marie JP, Zittoun R, Sikic BI. Multidrug resistance (mdr1) gene expression in adult acute leukemias: correlations with treatment outcome and in vitro drug sensitivity. Blood. 1991;78(3):586-592. [PubMed] [Google Scholar]
- 40.Bellos F, Davis BH, Culp NB, et al. . Assessment of human equilibrative nucleoside transporter 1 (hENT1) – expression by flow cytometry in acute myeloid leukemia and myelodysplastic syndromes and correlation to clinical outcome in acute myeloid leukemia patients treated with intensive chemotherapy. Blood. 2013;122(21):2582.23943651 [Google Scholar]
- 41.Fey M. High-dose cytosine arabinoside therapy for refractory leukemia. Blood. 1984;64(3):758-759. [PubMed] [Google Scholar]
- 42.Herzig RH, Wolff SN, Lazarus HM, Phillips GL, Karanes C, Herzig GP. High-dose cytosine arabinoside therapy for refractory leukemia. Blood. 1983;62(2):361-369. [PubMed] [Google Scholar]
- 43.Wierzbowska A, Robak T, Pluta A, et al. ; Polish Adult Leukemia Group. Cladribine combined with high doses of arabinoside cytosine, mitoxantrone, and G-CSF (CLAG-M) is a highly effective salvage regimen in patients with refractory and relapsed acute myeloid leukemia of the poor risk: a final report of the Polish Adult Leukemia Group. Eur J Haematol. 2008;80(2):115-126. [DOI] [PubMed] [Google Scholar]
- 44.Pastore D, Specchia G, Carluccio P, et al. . FLAG-IDA in the treatment of refractory/relapsed acute myeloid leukemia: single-center experience. Ann Hematol. 2003;82(4):231-235. [DOI] [PubMed] [Google Scholar]
- 45.Reese ND, Schiller GJ. High-dose cytarabine (HD araC) in the treatment of leukemias: a review. Curr Hematol Malig Rep. 2013;8(2):141-148. [DOI] [PubMed] [Google Scholar]
- 46.Bloomfield CD, Lawrence D, Byrd JC, et al. . Frequency of prolonged remission duration after high-dose cytarabine intensification in acute myeloid leukemia varies by cytogenetic subtype. Cancer Res. 1998;58(18):4173-4179. [PubMed] [Google Scholar]
- 47.Isidori A, Venditti A, Maurillo L, et al. . Alternative novel therapies for the treatment of elderly acute myeloid leukemia patients. Expert Rev Hematol. 2013;6(6):767-784. [DOI] [PubMed] [Google Scholar]
- 48.DeAngelo DJ, Stein EM, Ravandi F. Evolving therapies in acute myeloid leukemia: progress at last? Am Soc Clin Oncol Educ Book. 2016;35:e302-e312. [DOI] [PubMed] [Google Scholar]
- 49.Shin SH, Yahng SA, Yoon JH, et al. . Survival benefits with transplantation in secondary AML evolving from myelodysplastic syndrome with hypomethylating treatment failure. Bone Marrow Transplant. 2013;48(5):678-683. [DOI] [PubMed] [Google Scholar]
- 50.Fröhling S, Schlenk RF, Kayser S, et al. ; German-Austrian AML Study Group. Cytogenetics and age are major determinants of outcome in intensively treated acute myeloid leukemia patients older than 60 years: results from AMLSG trial AML HD98-B. Blood. 2006;108(10):3280-3288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.