

This phase I study was designed to investigate the safety and pharmacokinetics of pictilisib in combination with erlotinib in patients with advanced solid tumors.
Keywords: Phosphatidylinositol 3‐kinase, Phosphatidylinositol 3‐kinase inhibitor, Pictilisib, Erlotinib, Erlotinib hydrochloride, Epidermal growth factor receptor, Epidermal growth factor receptor inhibitor, Receptor protein‐tyrosine kinases, Phase I clinical trial
Abstract
Background.
Epidermal growth factor receptor (EGFR) and phosphatidylinositol 3‐kinase (PI3K) are involved in the proliferation and survival of many cancer types. Enhanced antitumor activity may be achieved through combined inhibition of these pathways. We report results for pictilisib (GDC‐0941, a class I pan‐PI3K inhibitor) plus erlotinib (an EGFR tyrosine kinase inhibitor) in patients with advanced solid tumors.
Materials and Methods.
A 3 + 3 dose‐escalation study was carried out at a starting daily dose of 60 mg pictilisib on days 1–21 of a 28‐day cycle and 150 mg erlotinib from day 2 of cycle 1. The primary objectives of the study were to assess safety and tolerability, identify dose‐limiting toxicities (DLTs), estimate the maximum tolerated dose, and identify the recommended phase II dose (RP2D). Evaluation of a dose‐expansion cohort at the RP2D was performed.
Results.
Fifty‐seven patients were treated in the study. All patients experienced at least one adverse event (AE). Grade ≥3 AEs, serious AEs, and deaths were reported in 38 (66.7%), 19 (33.3%), and 4 (7.0%) patients, respectively. DLTs occurred in nine patients across eight cohorts and the RP2D was determined to be 340 mg pictilisib on a “5 days on, 2 days off” schedule plus 100 mg erlotinib. Two patients (3.5%) experienced partial response and 19 (33.3%) had stable disease.
Conclusion.
Combining pictilisib with erlotinib in patients with advanced solid tumors is feasible; however, antitumor activity is limited. Additional studies may identify patients likely to benefit from combined inhibition of EGFR and PI3K pathways.
Implications for Practice.
Combining drugs targeting different signaling pathways in cancer growth and survival could overcome drug resistance and improve antitumor activity. In this first‐in‐human study for the combination, addition of the PI3K inhibitor pictilisib to the EGFR tyrosine kinase inhibitor erlotinib resulted in toxicity that led to dose and schedule modifications to identify a tolerable recommended phase II dose of 340 mg pictilisib on a “5 days on, 2 days off” schedule plus 100 mg erlotinib daily. The limited antitumor activity observed, however, suggests that additional studies are needed to identify patients most likely to benefit from combined EGFR and PI3K inhibition.
Introduction
Aberrant regulation of the epidermal growth factor receptor (EGFR) pathway has been implicated in the pathophysiology of many different cancer types [1]. The phosphatidylinositol 3‐kinase (PI3K) pathway plays a major role downstream of receptor tyrosine kinases, such as EGFR [2], and is also frequently dysregulated in cancer through a variety of mechanisms [3], [4], [5], [6], [7]. The EGFR/PI3K/protein kinase B (AKT) pathway is responsible for cell proliferation, survival, migration, and drug resistance [2]. Dysregulated signaling through the PI3K pathway may contribute to resistance to EGFR inhibition in some tumors, and this resistance may be overcome through inhibition of components of the PI3K pathway [8], [9], [10]. Therefore, combined inhibition of both the EGFR and PI3K pathways may provide greater antitumor activity.
Pictilisib (GDC‐0941) is an orally bioavailable class I pan‐PI3K inhibitor [11] that has been shown to inhibit the growth of a variety of human cancers in xenograft models [12]. In humans, pictilisib has linear pharmacokinetics (PK) when administered at doses between 15 and 450 mg daily [13] and has demonstrated single‐agent activity in a phase I study in patients with advanced solid tumors [13]. Erlotinib is an orally active selective inhibitor of the EGFR receptor tyrosine kinase [14], [15] and is approved for locally advanced or metastatic non‐small cell lung cancer (NSCLC) with activating EGFR mutations, and metastatic pancreatic cancer [16].
Activated EGFR signaling can stimulate the PI3K pathway. In addition, synergy between erlotinib and pictilisib has been observed in NSCLC cell lines [17]. Thus, erlotinib in combination with pictilisib may result in greater antitumor activity compared with erlotinib alone by impacting signaling at distinct pathway nodes and thereby preventing signaling reactivation. This phase I study was designed to investigate the safety and PK of erlotinib in combination with pictilisib in patients with advanced solid tumors.
Materials and Methods
Eligibility Criteria
Eligible patients aged ≥18 years had locally advanced or metastatic solid tumors for which there is no standard therapy, or standard therapy had proven ineffective or intolerable for the patient. An Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, adequate hematologic and end organ function, evaluable disease or measurable disease per Response Evaluation Criteria In Solid Tumors (RECIST) Version 1.0, and a baseline diffusing capacity of the lungs for carbon monoxide (DLCO) ≥50% of the predicted value corrected for hemoglobin and alveolar volume were required. Major exclusion criteria were prior PI3K‐inhibitor treatment and diabetes requiring daily medication. Patients with a history of smoking must have stopped ≥2 weeks before the start of treatment.
Study Design and Treatment
This was a phase I, open‐label, multicenter, 3 + 3 dose‐escalation study of pictilisib combined with erlotinib in patients with advanced solid tumors, followed by evaluation of a dose‐expansion cohort, which was planned to include up to 20 patients, at the recommended phase II dose (RP2D).
Pictilisib was administered at a starting dose of 60 mg daily on days 1–21 of a 28‐day cycle (21/28) and erlotinib at a dose of 150 mg daily from day 2 of the first cycle in cohort 1. Due to the risk of overlapping toxicity, especially rash, the starting dose of pictilisib was less than half the highest tolerable dose of single‐agent pictilisib at the time that this study was started [13]. The total number of dose‐escalation cohorts evaluated in the study was eight.
The primary objectives of the study were to assess the safety and tolerability of pictilisib plus erlotinib, to identify dose‐limiting toxicities (DLTs) and estimate the maximum tolerated dose (MTD), and to identify the RP2D in patients with advanced solid tumors. The secondary objectives included characterization of the PKs and antitumor activity of pictilisib and erlotinib.
The study was conducted in full accordance with the guidelines for Good Clinical Practice. Institutional review board/ethics committee approval was obtained for the protocol, patient recruitment material, the Informed Consent Forms, any information given to the patient, and relevant supporting information. The study was registered on ClinicalTrials.gov (NCT00975182).
Safety Evaluations
Patients were assessed by physical examination, vital signs, and hematology and serum chemistry at baseline and throughout the study. An electrocardiogram was performed at baseline, at the end of cycle 1, at study completion, and at any other time as clinically indicated. DLCO was assessed at baseline to determine eligibility, and during the study for any patient with new or worsening dyspnea or cough, decrease of ≥5% in resting oxygen saturation, or change in pulmonary clinical examination.
Treatment‐emergent adverse events (AEs) were recorded and graded according to National Cancer Institute‐Common Terminology Criteria for Adverse Events (NCI‐CTCAE) Version 3.0. A DLT was defined as a study drug‐related toxicity occurring during cycle 1, including the following: (a) grade ≥3 nonhematologic, nonhepatic organ toxicity, excluding grade 3 nausea, vomiting, or diarrhea that resolved to grade ≤1 within 7 days and grade 3 rash that resolved to grade ≤2 within 7 days; (b) grade ≥3 febrile neutropenia; (c) grade ≥4 neutropenia lasting >5 days; (d) grade ≥4 thrombocytopenia lasting >48 hours; (e) grade ≥4 anemia; (f) grade ≥3 elevations in total bilirubin, hepatic transaminase (alanine aminotransferase [ALT] or aspartate aminotransferase [AST]), amylase, or lipase lasting >72 hours (for patients with grade 1 hepatic transaminase at baseline as a result of metastases, hepatic transaminase ≥7.5 × the upper limit of normal was considered a DLT); (g) grade ≥2 DLCO concomitant with a decrease of ≥20% from baseline.
During the DLT assessment window, patients who withdrew from treatment or who missed 5 or more days of treatment without experiencing a DLT were replaced. In addition to this, patients requiring dose modifications of either drug for reasons other than a DLT were considered DLT‐unevaluable and were replaced.
The MTD was defined as the highest dose at which fewer than one‐third of at least six DLT‐evaluable patients had a dose‐limiting toxic event during cycle 1.
Antitumor Assessments
Tumor assessments were carried out at screening and at the end of every other cycle of treatment, beginning in cycle 2. Tumor assessments included high‐resolution computed tomography (CT) scan of the chest and CT scans of the abdomen and pelvis with bone scans and/or brain imaging by CT or magnetic resonance imaging as clinically indicated. The best overall response, based on investigator assessment using RECIST Version 1.0 and confirmed ≥4 weeks after initial documentation, was recorded.
Pharmacokinetic Assessments
In cohort 1, blood samples were taken for PK assessment before and after dosing on days 1, 2, 15, and 16 of cycle 1, and at study completion (samples for both pictilisib and erlotinib analysis), and from cohort 2 onwards, before and after dosing on day 1 (sample for pictilisib analysis only) and then on days 2, 5, 8, and 15 of cycle 1, and at study completion (samples for both pictilisib and erlotinib analyses). Serial blood samples were drawn for up to 3 hours after dosing for cohort 1 and for up to 6 hours after dosing from cohort 2 onwards. The PK of pictilisib was characterized using the following parameters: total exposure, maximum and minimum observed plasma concentrations, and accumulation ratio. For erlotinib, observed predose plasma concentrations were measured and were compared with historical data.
Biomarker Assessments
Deoxyribonucleic acid was extracted from archival and fresh formalin‐fixed paraffin‐embedded tumor tissue and assessed using a quantitative reverse transcription polymerase chain reaction six‐gene mutation panel designed to detect the most common mutations in the EGFR, Kirsten rat sarcoma viral oncogene homolog (KRAS), phosphatidylinositol‐4,5‐bisphosphate 3‐kinase catalytic subunit alpha (PIK3CA), neuroblastoma RAS viral oncogene homolog (NRAS), BRAF, and protein kinase B (AKT1) oncogenes, as previously described [18]. The assays allowed detection of 17 mutations in PIK3CA, 18 in KRAS, and 43 in EGFR (supplemental online Table 1).
Statistical Analysis
Design considerations were not made with regard to explicit power and type I error considerations, but to obtain preliminary safety and PK information. No formal hypotheses were tested in this study, and all analyses were descriptive and exploratory. Data were summarized by dose and schedule unless otherwise stated. The data cutoff was August 29, 2014.
Results
Patient Characteristics
A total of 58 patients were enrolled in the study; 53 of these were enrolled in the dose‐escalation stage, and 5 were enrolled in the cohort‐expansion stage. All but one patient, who was withdrawn due to elevated AST prior to treatment, were treated with pictilisib plus erlotinib. The median age of all treated patients was 58 years (range 32–78) and 56.1% were male (Table 1). Colorectal (13 patients), NSCLC (11 patients), and mesothelioma (5 patients) were the most common primary cancer diagnoses (Table 1). The majority of patients (66.7%) had an ECOG performance status of 1 (Table 1). The most common site of disease involvement was lung (50.9%), followed by liver (36.8%), then mediastinum (24.6%). Patient demographics were similar across all treatment groups in both stages of the study.
Table 1. Baseline demographics.
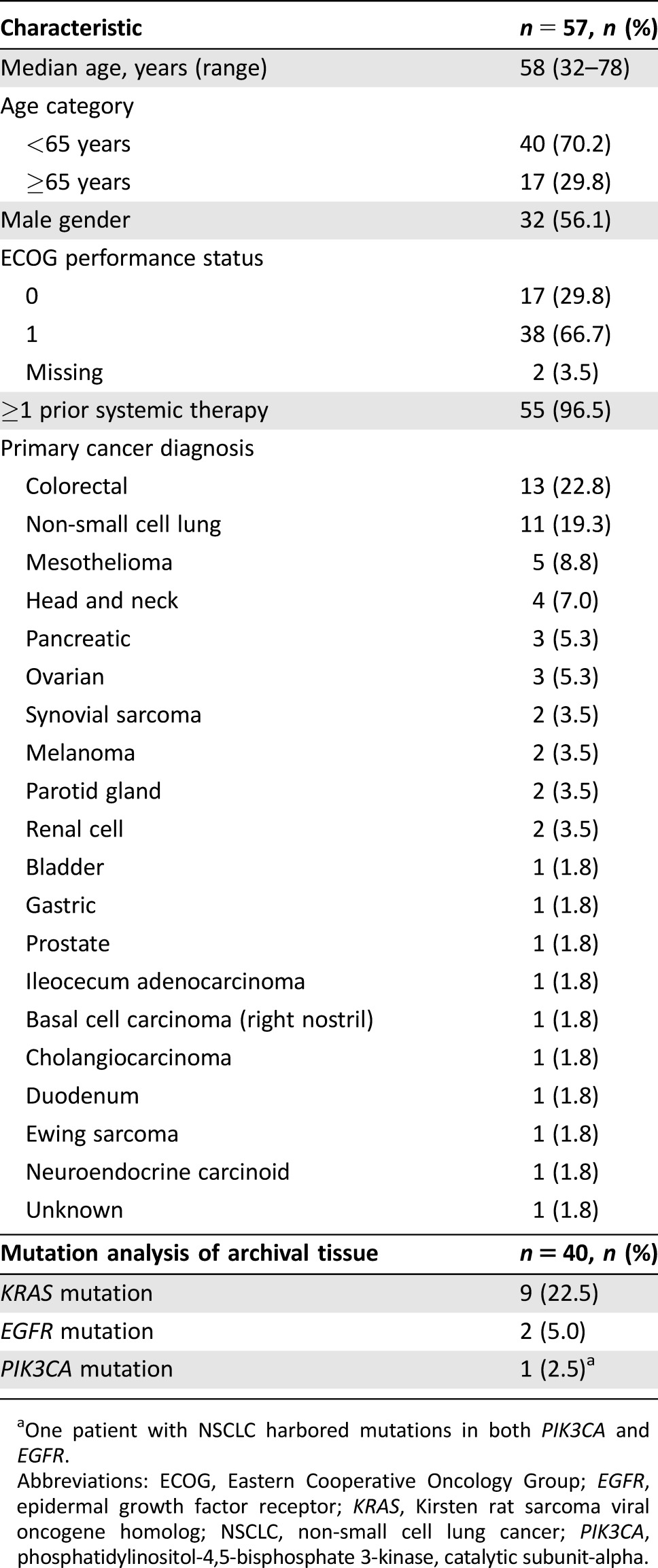
One patient with NSCLC harbored mutations in both PIK3CA and EGFR.
Abbreviations: ECOG, Eastern Cooperative Oncology Group; EGFR, epidermal growth factor receptor; KRAS, Kirsten rat sarcoma viral oncogene homolog; NSCLC, non‐small cell lung cancer; PIK3CA, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit‐alpha.
Safety and Dose‐Limiting Toxicities
Eight cohorts were evaluated in the dose‐escalation stage of the study. An overview of the doses and schedules of pictilisib and erlotinib evaluated in each of these cohorts and in the cohort‐expansion stage is provided in Figure 1. Patients in cohorts 1–6 were treated with pictilisib capsules, whereas those in cohorts 7 and 8, and in the cohort‐expansion stage of the study, received pictilisib tablets.
Figure 1.
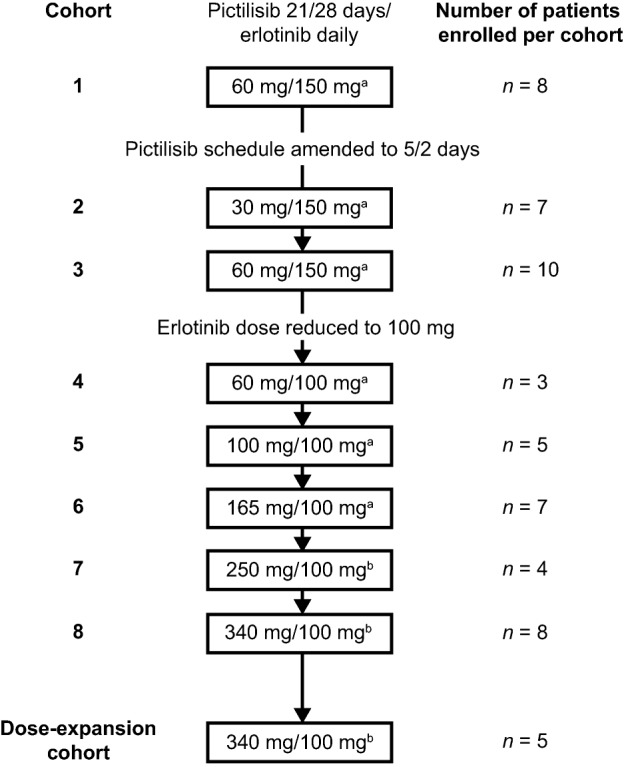
Dosing cohorts and dose levels. aPatients were treated with pictilisib capsules. bPatients were treated with pictilisib tablets.
Overall, the median duration of exposure for both erlotinib and pictilisib was 56 days (range 7–910 days). Table 2 shows the all‐grade AEs regardless of attribution that occurred in ≥20% of patients and the corresponding grade ≥3 AEs by preferred term for each cohort in the study. All patients in the study experienced at least one AE. The most common AEs were diarrhea (70.2%), nausea (54.4%), fatigue (54.4%), and rash (49.1%). Grade ≥3 AEs occurred in 38 patients (66.7%); the most common of which were rash and increased ALT, which occurred in six patients (10.5%) each, and lymphopenia, which occurred in five patients (8.8%). Serious AEs (SAEs) were reported in a total of 19 patients (33.3%; Table 3). Three patients each experienced SAEs of abdominal pain and dyspnea. Diarrhea, pulmonary embolism, and pneumonia occurred in two patients each. No other SAE occurred in more than one patient.
Table 2. All‐grade AEs occurring in ≥20% of all patients and corresponding grade ≥3 AEs.

Patients were treated with pictilisib capsules on a 21/28‐day dosing schedule.
Patients were treated with pictilisib capsules on a “5 days on, 2 days off” dosing schedule.
Patients were treated with pictilisib tablets on a “5 days on, 2 days off” dosing schedule.
Abbreviations: AE, adverse event; AST, aspartate aminotransferase.
Table 3. Summary of AEs.
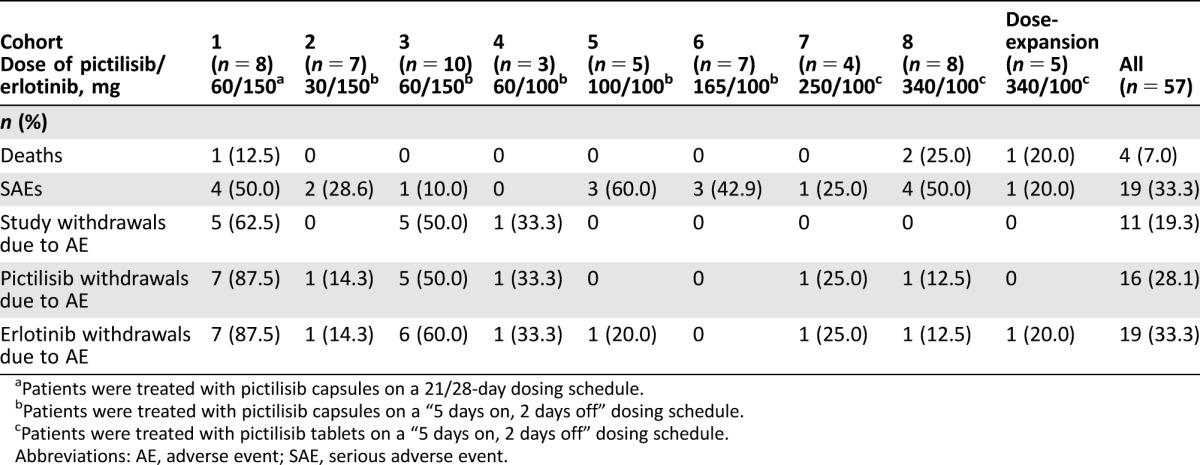
Patients were treated with pictilisib capsules on a 21/28‐day dosing schedule.
Patients were treated with pictilisib capsules on a “5 days on, 2 days off” dosing schedule.
Patients were treated with pictilisib tablets on a “5 days on, 2 days off” dosing schedule.
Abbreviations: AE, adverse event; SAE, serious adverse event.
At the time of data cutoff, all patients had discontinued from the study. The reasons for study discontinuation were disease progression (n = 35 patients; 61.4%), AEs (n = 11; 19.3%), patient or physician decision (n = 8; 14.1%), or death (n = 3; 5.3%). The 11 patients (19.3%) who discontinued the study due to AEs included 10 of the 25 patients treated with 150 mg erlotinib (cohorts 1–3) and one of the 32 patients treated with 100 mg erlotinib (cohorts 4–8 and the dose‐expansion cohort). Sixteen (28.1%) and 19 (33.3%) patients discontinued pictilisib and erlotinib, respectively, due to an AE. A total of 20 AEs led to discontinuation of both study drugs in 15 patients. These were increased ALT levels (four events), rash (two events), and acute renal failure, ascites, increased AST, cerebral ischemia, dehydration, diarrhea, dyspnea, fatigue, malaise, disease progression, pulmonary embolism, pyrexia, syncope, and ulcerative colitis (one event each).
In total, four patients (7.0%) died during the study (Table 3); the causes of death were disease progression, arterial hemorrhage, dyspnea, and cardiac failure. The event of arterial hemorrhage was assessed by the investigator as related to study treatment and occurring as a result of tumor regression at the right pulmonary artery. All other fatal events were considered unrelated to study treatment by the investigators.
Cohort 1 (pictilisib 60 mg 21/28 and erlotinib 150 mg) exceeded the MTD, with three of the eight patients experiencing DLTs (Table 4). A 64‐year‐old female with melanoma experienced grade 3 dyspnea on day 12 and discontinued all study treatment. A 54‐year‐old female with ovarian cancer had grade 2 increased ALT on day 22, which worsened to grade 3 on day 29. Study treatment was interrupted due to increased ALT levels that subsequently improved to grade 1 on day 36. Upon retreatment, this patient experienced recurrence of grade 3 increased ALT levels, and treatment was discontinued on day 43. Finally, a 48‐year‐old female with colorectal cancer experienced grade 4 ulcerative colitis on day 9 that resolved on day 23; treatment was permanently discontinued on day 38 due to the event.
Table 4. Summary of DLTs observed during the study.
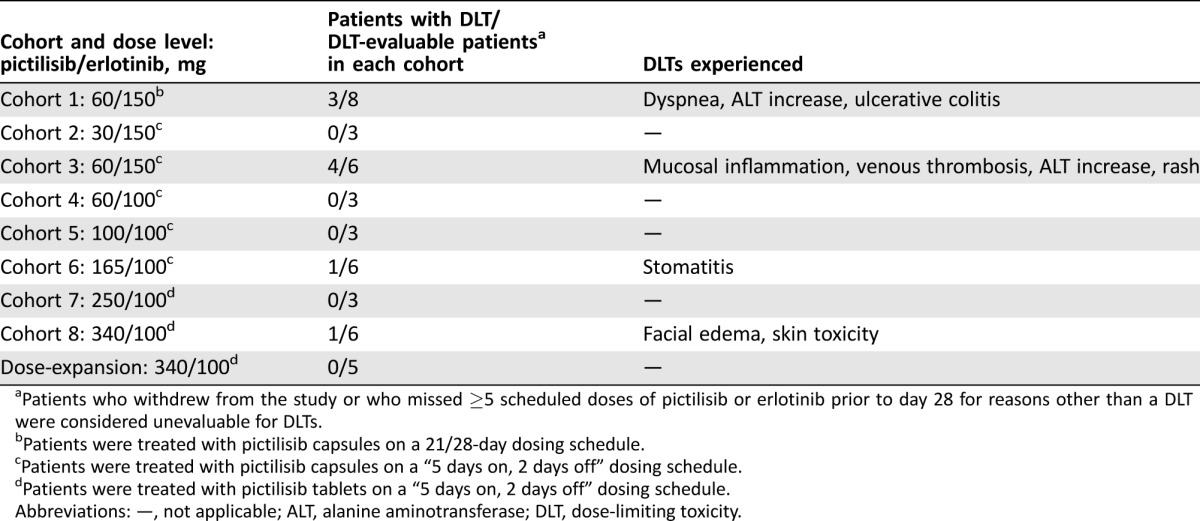
Patients who withdrew from the study or who missed ≥5 scheduled doses of pictilisib or erlotinib prior to day 28 for reasons other than a DLT were considered unevaluable for DLTs.
Patients were treated with pictilisib capsules on a 21/28‐day dosing schedule.
Patients were treated with pictilisib capsules on a “5 days on, 2 days off” dosing schedule.
Patients were treated with pictilisib tablets on a “5 days on, 2 days off” dosing schedule.
Abbreviations: —, not applicable; ALT, alanine aminotransferase; DLT, dose‐limiting toxicity.
As the MTD was exceeded in cohort 1, the pictilisib dosing schedule was changed to 5 consecutive days of treatment, followed by 2 consecutive dose holidays (“5 days on, 2 days off” [5/2]) in subsequent cohorts, starting with a dose of 30 mg in cohort 2. The dose of erlotinib in cohorts 2 and 3 was 150 mg. No DLTs were observed in cohort 2 (pictilisib 30 mg 5/2 and erlotinib 150 mg).
Four of six DLT‐evaluable patients experienced a DLT in cohort 3 (pictilisib 60 mg 5/2 and erlotinib 150 mg) and thus this combined dose exceeded the MTD (Table 4). Grade 3 mucosal inflammation occurred on day 19 in a 76‐year‐old male patient with NSCLC. With treatment interruption, the mucosal inflammation improved to grade 2 on day 24. Grade 4 venous thrombosis of the left leg was experienced on day 29 by a 58‐year‐old male patient with gastric cancer. Treatment was initiated with nadroparin and antiembolism stockings and study treatment was withheld until day 43. Grade 3 elevations in ALT occurred in a 66‐year‐old female patient with colorectal cancer on day 29, which led to permanent discontinuation of study treatment on day 43. Grade 2 rash was reported on day 3 in a 38‐year‐old male patient with synovial sarcoma, which worsened to grade 3 rash on day 8, resulting in study treatment being permanently discontinued.
Given that pictilisib 60 mg 5/2 and erlotinib 150 mg (cohort 3) exceeded the MTD, the erlotinib dose was reduced to 100 mg starting in cohort 4. There were no DLTs in cohort 4 (pictilisib 60 mg 5/2 and erlotinib 100 mg) or cohort 5 (pictilisib 100 mg 5/2 and erlotinib 100 mg). One patient (a 47‐year‐old male with colorectal cancer) in cohort 6 (pictilisib 165 mg 5/2 and erlotinib 100 mg) had a DLT of grade 3 abdominal pain (day 16). An x‐ray on day 20 revealed constipation. The patient received sodium phosphate dibasic, laxatives, morphine, and fentanyl, and remained on study treatment. No DLTs were observed in cohort 7 (pictilisib 250 mg and erlotinib 100 mg). One patient (a 61‐year‐old female with NSCLC) in cohort 8 (pictilisib 340 mg 5/2 and erlotinib 100 mg) experienced DLTs of grade 3 facial edema and skin toxicity on day 12 that resulted in treatment interruption. The patient received prednisolone for facial edema, which resolved on day 13; the remainder of the skin toxicity was considered resolved on day 38 (Table 4).
On the 5/2 pictilisib dosing schedule with 100 mg erlotinib, the MTD of pictilisib was not reached at doses evaluated up to the single‐agent MTD of 340 mg. Therefore, 340 mg pictilisib 5/2 was the maximum‐administered dose used in this study. Additional patients were enrolled to a cohort‐expansion stage to assess the tolerability and antitumor activity of 340 mg pictilisib 5/2 schedule plus 100 mg erlotinib daily in 28‐day cycles. Safety in the cohort‐expansion stage of the study was comparable to that observed in cohort 8 (pictilisib 340 mg 5/2 and erlotinib 100 mg) of the dose‐escalation stage.
Antitumor Activity
The best confirmed response was partial response in 2 patients (3.5%) and stable disease in 19 patients (33.3%; Table 5). Of the two patients who experienced an objective response, one patient had mesothelioma and the other had cancer of the parotid gland. The duration of time on study for each patient included in the study is shown in Figure 2. The two patients with the longest time on study (>900 days) were from cohort 2 (pictilisib 30 mg 5/2 and erlotinib 150 mg) and cohort 7 (pictilisib 250 mg 5/2 and erlotinib 100 mg). Mutation analysis was carried out on archival tissue from a total of 40 patients. From the available archival tissue, nine patients (22.5%) had a KRAS mutation, one (2.5%) had an EGFR mutation, and one (2.5%) had both an EGFR and a PIK3CA mutation (Table 1). No KRAS, EGFR, or PIK3CA mutations were present in the archival tumor tissue from either of the patients who experienced a partial response (Fig. 2). For the two patients who were on the study for the longest duration, no mutations were identified in the archival tissue from one patient, and no archival tissue was available for testing for the second patient (Fig. 2).
Table 5. Best confirmed response.
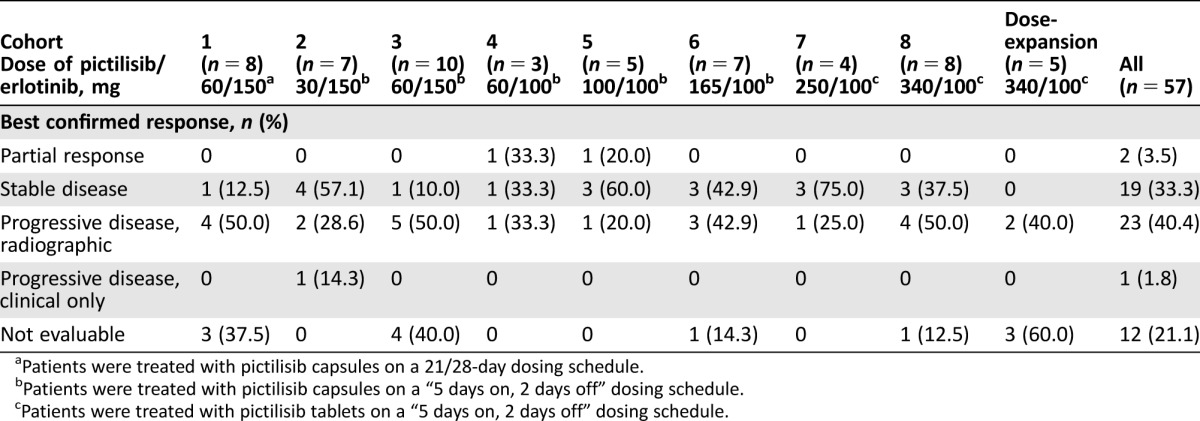
Patients were treated with pictilisib capsules on a 21/28‐day dosing schedule.
Patients were treated with pictilisib capsules on a “5 days on, 2 days off” dosing schedule.
Patients were treated with pictilisib tablets on a “5 days on, 2 days off” dosing schedule.
Figure 2.

Time on study by dose regimen.
aPatients were treated with pictilisib capsules.
bPatients were treated with pictilisib tablets.
cPatients who had a best confirmed response of partial response.
Abbreviations: –, no mutation detected; B, bladder; Ba, basal cell carcinoma; C, colorectal; Ch, cholangiocarcinoma; D, duodenum; E, Ewing sarcoma; EGFR, epidermal growth factor receptor; G, gastric; H, head and neck; I, ileocecum carcinoma; KRAS, Kirsten rat sarcoma viral oncogene homolog; L, non‐small cell lung; M, melanoma; Me, mesothelioma; N, neuroendocrine carcinoid; NT, not tested (mutation status); O, ovarian; Pa, pancreatic; Pg, parotid gland; PIK3CA, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase, catalytic subunit‐alpha; Pr, prostate; R, renal cell; S, synovial sarcoma; U, unknown.
Pharmacokinetics
Pharmacokinetics data were collected to evaluate whether there was any interaction between the study drugs. Preliminary PK data showed no evidence of a drug‐drug interaction between erlotinib and pictilisib (data not shown; to be published separately).
Discussion
Pictilisib inhibits all four isoforms of PI3K class I (α, β, γ, and δ) [19], [20]. In the single‐agent phase I study, the RP2D for pictilisib capsules was 330 mg daily (equivalent to 340 mg tablet formulation) with no dosing holiday with on‐target pharmacodynamic activity [13]. Grade 1–2 nausea, rash, and fatigue were the most common toxicities, and there was evidence of antitumor activity in patients with and without PI3K pathway dysregulation [13].
In this phase Ib study of pictilisib in combination with erlotinib in patients with advanced solid tumors, the initial pictilisib dosing schedule (60 mg on a 21/28 schedule) was poorly tolerated when combined with 150 mg erlotinib. The change in the pictilisib dosing schedule to a 5/2 schedule together with reduction of the erlotinib dose to 100 mg improved tolerability such that doses of pictilisib up to 340 mg (the single‐agent MTD) were tolerated. Thus, the RP2D of 340 mg pictilisib on a 5/2 schedule with daily 100 mg erlotinib had an acceptable safety profile in the treatment of patients with advanced solid tumors. The AE profile of this treatment combination was in line with previous reports of these drugs as single agents and there was no evidence of drug‐drug interactions. However, combining pictilisib with erlotinib exhibited only modest antitumor activity, with 2 of the 57 patients included in the study experiencing an objective response.
Pictilisib has also been studied in combination with hormone therapies and chemotherapy in hormone receptor (HR) positive breast cancer [21], [22], [23] and NSCLC (NCT01493843, NCT00974584). When pictilisib was administered at the single‐agent MTD (340 mg) in combination with approved doses of fulvestrant in patients with HR positive breast cancer in the FERGI study, the safety profile was similar to that observed for pictilisib in previous studies [21]. During the course of the study, the dose of pictilisib was lowered to 260 mg in an effort to improve long‐term tolerability [21]. Similarly, pictilisib was administered at a dose of 260 mg in combination with paclitaxel in the PEGGY study [22], and the OPPORTUNE study demonstrated a significantly more favorable skin toxicity profile with 260 mg pictilisib in combination with anastrozole versus the same combination with 360 mg pictilisib in patients with preoperative early breast cancer [23]. In a phase I trial of pictilisib with various standard‐of‐care chemotherapy backbones in patients with NSCLC, pictilisib was tolerated at 340 mg (NCT00974584; publication pending). Pictilisib was subsequently examined at this dose in combination with carboplatin and paclitaxel in patients with squamous NSCLC, and at 340 mg and 260 mg in combination with carboplatin, paclitaxel, and bevacizumab in patients with nonsquamous NSCLC (NCT01493843; publication pending). However, despite evaluating two different doses in combination with multiple standard‐of‐care therapies in breast and lung cancer, the addition of pictilisib has not led to any significant benefit. Overall, combination toxicity appears to limit long‐term pictilisib dosing, and thus may result in lack of additional benefit for the pictilisib combinations when compared with the active standard‐of‐care comparators [21]. In our study, the combination of pictilisib with erlotinib required a reduction in the dose of erlotinib from 150 mg (the recommended dose for NSCLC harboring mutations in EGFR) to 100 mg before acceptable toxicity was achieved. This further limits the potential for the combination moving forward.
One limitation of this study is the lack of pharmacodynamic assessments, which means that the impact of pictilisib dose and schedule in combination with 100 mg erlotinib on pathway inhibition remains unknown. Extensive pharmacodynamic assessment previously performed with single‐agent pictilisib [13] showed consistent pathway modulation only in a subset of patients receiving 330–450 mg pictilisib, but the effects of combining pictilisib and erlotinib on downstream pathway markers still requires investigation. Another limitation is the lack of extensive genomic profiling, which could provide some insight into other mechanisms that contribute to PI3K pathway activation that were not assessed in this study.
In addition to pictilisib, another pan‐PI3K class I inhibitor, pilaralisib, has been investigated in a phase I study in combination with erlotinib for the treatment of solid tumors [24]. In contrast to the current study, 400 mg pilaralisib (less than the single‐agent MTD of 600 mg pilaralisib) was tolerated in combination with 150 mg erlotinib and was determined to be the MTD for the combination [24].
Phosphatidylinositol 3‐kinase inhibitor treatment combinations, including those with erlotinib, are feasible but are often associated with additional toxicity. Despite ongoing efforts to target therapy to actionable mutations in trials such as NCI MATCH (NCT02465060), it is notable that neither of the patients who had a partial response in our study had any EGFR, KRAS, or PIK3CA mutations. To achieve a broader therapeutic window, alternative strategies may include combination with a monoclonal antibody, use of a more selective PI3K inhibitor in a biomarker‐defined patient population, or identification of additional predictive biomarkers.
Conclusion
Combining pictilisib at a dose of 340 mg daily on a 5/2 schedule with erlotinib at 100 mg daily to achieve dual inhibition of the PI3K and EGFR pathways is feasible in patients with advanced solid tumors. However, given the limited antitumor activity observed, additional studies are needed to identify the patients most likely to derive benefit from combined EGFR and PI3K inhibition.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This study was funded by Genentech, Inc. Support for third‐party writing assistance for this manuscript, furnished by Katie Wilson, Ph.D., of Health Interactions, was provided by F. Hoffmann‐La Roche Ltd, Basel, Switzerland.
Rebecca A. Moss completed all work for this study at the Rutgers Cancer Institute of New Jersey. She joined Bristol‐Myers Squibb, Princeton, New Jersey, USA, and became a Volunteer Clinical Faculty member at the Rutgers Cancer Institute of New Jersey in October 2015. Emile E. Voest worked at the University Medical Center Utrecht until December 31, 2013.
These analyses have been presented in part in Lolkema MP, Leong S, Voest EE et al. A phase 1b open‐label, dose‐escalation study of the safety and pharmacology of GDC‐0941 in combination with erlotinib in patients with advanced solid tumors. Presented at: 24th EORTC‐NCI‐AACR symposium; November 6–9, 2012; Dublin, Ireland. Poster presentation 365.
Footnotes
See the related article on page 1421.
Author Contributions
Conception/design: Mark R. Lackner, Emile E. Voest, Jan H.M. Schellens
Provision of study material or patients: Stephen Leong, Rebecca A. Moss, Daniel W. Bowles, Emile E. Voest, Jan H.M. Schellens
Collection and/or assembly of data: Stephen Leong, Rebecca A. Moss, Ruud van der Noll, Emile E. Voest
Data analysis and interpretation: Stephen Leong, Rebecca A. Moss, Daniel W. Bowles, Joseph Ware, Jing Zhou, Jill M. Spoerke, Mark R. Lackner, Geetha Shankar, Jennifer L. Schutzman, Emile E. Voest, Jan H.M. Schellens
Manuscript writing: Stephen Leong, Daniel W. Bowles, Jill M. Spoerke, Mark R. Lackner, Geetha Shankar, Jennifer L. Schutzman, Emile E. Voest, Jan H.M. Schellens
Final approval of manuscript: Stephen Leong, Rebecca A. Moss, Daniel W. Bowles, Joseph Ware, Jing Zhou, Jill M. Spoerke, Mark R. Lackner, Geetha Shankar, Jennifer L. Schutzman, Ruud van der Noll, Emile E. Voest, Jan H.M. Schellens
Disclosures
Rebecca A. Moss: Bristol‐Myers Squibb (E, OI); Joseph Ware: Genentech/Roche (E); Jing Zhou: Genentech/Roche (E); Jill M. Spoerke: Genentech/Roche (E); Mark R. Lackner: Genentech/Roche (E); Geetha Shanker: Genentech/Roche (E); Jennifer L. Schutzman: Genentech/Roche (E). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Rowinsky EK. The erbB family: Targets for therapeutic development against cancer and therapeutic strategies using monoclonal antibodies and tyrosine kinase inhibitors. Annu Rev Med 2004;55:433–457. [DOI] [PubMed] [Google Scholar]
- 2. Engelman JA. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat Rev Cancer 2009;9:550–562. [DOI] [PubMed] [Google Scholar]
- 3. Bellacosa A, de Feo D, Godwin AK et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer 1995;64:280–285. [DOI] [PubMed] [Google Scholar]
- 4. Li J, Yen C, Liaw D et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997;275:1943–1947. [DOI] [PubMed] [Google Scholar]
- 5. Steck PA, Pershouse MA, Jasser SA et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997;15:356–362. [DOI] [PubMed] [Google Scholar]
- 6. Dahia PL. PTEN, a unique tumor suppressor gene. Endocr Relat Cancer 2000;7:115–129. [DOI] [PubMed] [Google Scholar]
- 7. Angulo B, Suarez‐Gauthier A, Lopez‐Rios F et al. Expression signatures in lung cancer reveal a profile for EGFR‐mutant tumours and identify selective PIK3CA overexpression by gene amplification. J Pathol 2008;214:347–356. [DOI] [PubMed] [Google Scholar]
- 8. D'Amato V, Rosa R, D'Amato C et al. The dual PI3K/mTOR inhibitor PKI‐587 enhances sensitivity to cetuximab in EGFR‐resistant human head and neck cancer models. Br J Cancer 2014;110:2887–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Donev IS, Wang W, Yamada T et al. Transient PI3K inhibition induces apoptosis and overcomes HGF‐mediated resistance to EGFR‐TKIs in EGFR mutant lung cancer. Clin Cancer Res 2011;17:2260–2269. [DOI] [PubMed] [Google Scholar]
- 10. Yi YW, Hong W, Kang HJ et al. Inhibition of the PI3K/AKT pathway potentiates cytotoxicity of EGFR kinase inhibitors in triple‐negative breast cancer cells. J Cell Mol Med 2013;17:648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Folkes AJ, Ahmadi K, Alderton WK et al. The identification of 2‐(1H‐indazol‐4‐yl)‐6‐(4‐methanesulfonyl‐piperazin‐1‐ylmethyl)‐4‐morpholin‐4‐yl‐thieno[3,2‐d]pyrimidine (GDC‐0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem 2008;51:5522–5532. [DOI] [PubMed] [Google Scholar]
- 12. O'Brien C, Wallin JJ, Sampath D et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3' kinase inhibitor GDC‐0941 in breast cancer preclinical models. Clin Cancer Res 2010;16:3670–3683. [DOI] [PubMed] [Google Scholar]
- 13. Sarker D, Ang JE, Baird R et al. First‐in‐human phase I study of pictilisib (GDC‐0941), a potent pan‐class I phosphatidylinositol‐3‐kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2015;21:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moyer JD, Barbacci EG, Iwata KK et al. Induction of apoptosis and cell cycle arrest by CP‐358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res 1997;57:4838–4848. [PubMed] [Google Scholar]
- 15. Pollack VA, Savage DM, Baker DA et al. Inhibition of epidermal growth factor receptor‐associated tyrosine phosphorylation in human carcinomas with CP‐358,774: Dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther 1999;291:739–748. [PubMed] [Google Scholar]
- 16.Tarceva® (erlotinib). Summary of Product Characteristics. Roche Registration Ltd. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000618/WC500033994.pdf. Accessed July 18, 2017.
- 17. Spoerke JM, O'Brien C, Huw L et al. Phosphoinositide 3‐kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin Cancer Res 2012;18:6771–6783. [DOI] [PubMed] [Google Scholar]
- 18. Patel R, Tsan A, Tam R et al. Mutation scanning using MUT‐MAP, a high‐throughput, microfluidic chip‐based, multi‐analyte panel. PloS One 2012;7:e51153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fruman DA, Rommel C. PI3K and cancer: Lessons, challenges and opportunities. Nat Rev Drug Discov 2014;13:140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martini M, Ciraolo E, Gulluni F et al. Targeting PI3K in cancer: Any good news? Front Oncol 2013;3:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krop IE, Mayer IA, Ganju V et al. Pictilisib for oestrogen receptor‐positive, aromatase inhibitor‐resistant, advanced or metastatic breast cancer (FERGI): A randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Oncol 2016;17:811–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vuylsteke P, Huizing M, Petrakova K et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3‐kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor‐positive, HER2‐negative, locally recurrent, or metastatic breast cancer: Interim analysis of the multicentre, placebo‐controlled, phase II randomised PEGGY study. Ann Oncol 2016;27:2059–2066. [DOI] [PubMed] [Google Scholar]
- 23. Schmid P, Pinder SE, Wheatley D et al. Phase II randomized preoperative window‐of‐opportunity study of the PI3K inhibitor pictilisib plus anastrozole compared with anastrozole alone in patients with estrogen receptor‐positive breast cancer. J Clin Oncol 2016;34:1987–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soria JC, LoRusso P, Bahleda R et al. Phase I dose‐escalation study of pilaralisib (SAR245408, XL147), a pan‐class I PI3K inhibitor, in combination with erlotinib in patients with solid tumors. The Oncologist 2015;20:245–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.