

This article reports a series of 114,200 consecutive clinical cases with comprehensive genomic profiles available to identify ALK fusions that could facilitate the use of precision medicine in both non‐small cell lung cancer (NSCLC) and non‐NSCLC malignancies.
Keywords: ALK, Fusion, Crizotinib, Alectinib, Comprehensive genomic profiling, Rearrangement
Abstract
Background.
Genomic fusions of the anaplastic lymphoma kinase gene (ALK) are a well‐established therapy target in non‐small cell lung cancer (NSCLC). From a survey of 114,200 clinical cases, we determined the prevalence of ALK rearrangements (rALK) in non‐NSCLC tumors and report their responsiveness to therapies targeting ALK.
Materials and Methods.
Comprehensive genomic profiling of 114,200 relapsed and metastatic malignancies, including both solid tumors and hematolymphoid cancers, was performed using a hybrid‐capture, adaptor ligation‐based next‐generation sequencing assay.
Results.
Of 114,200 clinical samples, 21,522 (18.8%) were NSCLC and 92,678 (81.2%) were other tumor types. Of the 876 (0.8%) cases with ALK fusions (fALK) or rALK, 675 (77.1%) were NSCLC and 201 (22.9%) were other tumor types. ALK fusions were significantly more frequent in NSCLC (3.1%) than non‐NSCLC (0.2%; p < .0001). Patients with non‐NSCLC tumors harboring fALK were significantly younger (p < .0001) and more often female (p < .0001) than patients with fALK‐positive NSCLC. EML4 was more often the fusion partner in NSCLC (83.5%) versus non‐NSCLC tumors (30.9%; p < .0001).
Conclusion.
ALK rearrangements can be identified in a wide variety of epithelial and mesenchymal malignancies beyond NSCLC. Anti‐ALK therapies can be effective in non‐NSCLC tumors driven by fALK, and further study of therapies targeting ALK in clinical trials involving a wider variety of cancer types appears warranted.
Implications for Practice.
Rearrangements involving the ALK gene have been detected in dozens of cancer types using next‐generation sequencing. Patients whose tumors harbor ALK rearrangements or fusions respond to treatment with crizotinib and alectinib, including tumors not normally associated with ALK mutations, such as non‐Langerhans cell histiocytosis or renal cell carcinoma. Comprehensive genomic profiling using next‐generation sequencing can detect targetable ALK fusions irrespective of tumor type or fusions partner.
摘要
背景.间变性淋巴瘤激酶基因(ALK)融合是非小细胞肺癌(NSCLC)治疗的确定靶点。根据对114 200例临床病例展开的调查, 我们确定了非NSCLC肿瘤患者中ALK重排(rALK)的发生率, 并报告了他们对靶向ALK的疗法的反应。
材料与方法.通过基于杂交捕获和适配器连接的下一代测序法对114 200例复发及转移性恶性肿瘤患者(包括实体瘤和淋巴造血系统癌)进行了全面的基因组测序。
结果.在这114 200例临床病例中, 21 522例(18.8%)罹患NSCLC, 92 678例(81.2%)罹患其他类型肿瘤。876例(0.8%)患者出现ALK融合(fALK)或rALK, 其中675例(77.1%)罹患NSCLC, 201例(22.9%)罹患其他类型肿瘤。NSCLC患者(3.1%)中ALK融合的发生率明显高于非NSCLC患者(0.2%)(p < 0.0001)。与fALK‐阳性 NSCLC患者相比, 负荷fALK的非NSCLC患者的年龄明显更小(p < 0.0001), 并且女性患者的比例通常更高(p < 0.0001)。NSCLC患者(83.5%)中出现融合伴侣EML4的比例通常高于非NSCLC患者(30.9%; p < 0.0001)。
结论.除NSCLC外, 多种上皮和间质恶性肿瘤中均存在ALK重排。抗ALK疗法可有效治疗fALK驱动的非NSCLC肿瘤, 因此, 有必要在涉及各类癌症的临床试验中对靶向ALK的疗法进行进一步研究。
对临床实践的启示:下一代测序技术已被使用对多种癌症类型进行了ALK基因重排检测。负荷ALK重排或融合的肿瘤患者对克唑替尼和alectinib治疗产生反应, 其中包括通常无ALK突变的肿瘤, 例如, 非朗格罕组织细胞增多症或肾细胞癌。使用下一代测序技术进行的全面基因组测序可检测靶向ALK融合, 而这与肿瘤类型或融合伴侣无关。
Introduction
The anaplastic lymphoma kinase gene (ALK) encodes a tyrosine kinase receptor with a major role in neuronal development [1], [2], [3], [4], [5]. The ALK protein, also known as CD246, was originally identified in anaplastic large cell malignant lymphoma (ALCL) and shown to be overexpressed as a result of a t(2;5)(p23;q35) chromosomal translocation [1]. The original ALK fusion partner found in ALCL was NPM1, a nucleophosmin, and this fusion is present in 70%–80% of ALK‐rearranged ALCL [1], [2], [3], [4], [5]. A wide variety of additional fusion partners for ALK have subsequently been described in ALCL, including EML4 in <10% of cases [1], [2], [3], [4], [5]. The vast majority of ALK fusions (fALK) retain the kinase domain and are associated with ALK‐driven tumorigenesis, progression, and metastasis [1], [2], [3], [4], [5]. The first studies investigating fALK as therapy targets in non‐small cell lung cancer (NSCLC) found EML4 as the sole fusion partner, although a variety of less‐common fusion partners have subsequently been identified [6], [7], [8], [9], [10]. Over the past 10 years, therapeutic strategies for ALK‐driven NSCLC have evolved. There are now accepted first‐line treatments targeting ALK, as well as second‐ and third‐generation inhibitors employed to overcome resistance to prior lines of anti‐ALK therapy [6], [7], [8], [9], [10]. Standard of care treatment for NSCLC now includes ALK testing when the disease presents in an advanced stage or has progressed after surgical treatment [11].
The impressive efficacy of targeting fALK in NSCLC and ALCL raised interest in ALK alterations in other cancer types. Several studies have shown the efficacy of anti‐ALK therapies in a wide variety of malignancies [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], but these studies are small‐scale and generally limited to individual case reports or case studies. In addition, many of these publications targeted ALK alterations that were not classic gene fusions, and included both ALK gene amplifications and ALK‐activating base substitutions [24], [25], [29]. The following study evaluated a series of 114,200 consecutive clinical cases with comprehensive genomic profiles available to identify fALK that could facilitate the use of precision medicine in both NSCLC and non‐NSCLC malignancies.
Materials and Methods
A series of 114,200 consecutive clinical cases, across a wide variety of disease types, was analyzed using comprehensive genomic profiling (CGP) in a Clinical Laboratory Improvement Amendments‐certified, College of American Pathologists‐accredited laboratory (Foundation Medicine, Cambridge, MA), as previously described [30], [31]. Approval for this study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act of 1996 waiver of authorization, was obtained from the Western Institutional Review Board (Protocol 20152817).
The pathologic diagnosis of each case was confirmed on routine hematoxylin and eosin stained slides and all samples forwarded for DNA extraction contained a minimum of 20% tumor nuclear area. In brief, ≥50 ng DNA was extracted from 40 microns of tumor sample in formalin‐fixed, paraffin‐embedded tissue blocks. The samples were assayed by CGP using adaptor‐ligation and hybrid capture targeting all coding exons in one of three assay versions: 287 (version 1, FoundationOne [Foundation Medicine, Cambridge, MA, https://www.foundationmedicine.com]), 315 (version 2, FoundationOne) or 405 (version 3, FoundationOne Heme) cancer‐related genes plus select introns from 19 (version 1), 28 (version 2), or 31 (version 3) genes frequently rearranged in cancer. A subset of cases was also evaluated by RNA sequencing using ∼3M on‐target unique pairs for 265 genes. Sequencing of captured libraries was performed using Illumina HiSeq (Illumina, San Diego, CA, https://www.illumina.com) technology to a mean exon coverage depth of >600×, and the resultant sequences were analyzed for base substitutions, insertions, deletions, copy number alterations (focal amplifications and homozygous deletions), rearrangements, and select gene fusions.
All three versions of the CGP assay used in this study detect ALK rearrangements (rALK) through DNA sequencing [30], [31]. In addition, version 3 of the assay can detect fALK through RNA sequencing. For the purposes of this study, fALK are defined as genomic rearrangements detected by RNA or DNA sequencing that result in a portion of the ALK gene containing the kinase domain (exons 20–29, NM_004304) being fused in‐strand to the promoter‐containing region of a second gene (at a minimum the 5′ UTR). Fusions detected by RNA were included if they were in‐frame. Fusions detected by DNA were included if an in‐frame product could be generated during mRNA processing, including by exon skipping. ALK rearrangements were defined as any other genomic rearrangement event predicted to separate the exons encoding the ALK kinase domain (exons 20–29, NM_004304) from the upstream exons encoding the regulatory regions.
Tumor mutational burden (TMB) was determined on 0.83 megabase (Mb; version 1), 1.14 Mb (version 2), or 1.23 Mb (version 3) of sequenced DNA using a mutation burden estimation algorithm that, based on the genomic alterations detected, extrapolates to the exome or the genome as a whole (supplemental online Tables 1–3). For purposes of mutation burden estimation, all coding short variant alterations (base substitutions and indels), including synonymous alterations, are counted. Subtracted from this number are functionally oncogenic or germline alterations, as defined below. Germline alterations are those listed in the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP), those with two or more counts in the ExAC database (http://exac.broadinstitute.org), or those predicted by a somatic‐germline zygosity algorithm to be germline in the specimen being assessed [32]. Functionally oncogenic mutations are those occurring as known somatic alterations in the COSMIC database (http://cancer.sanger.ac.uk/cosmic) or with likely functional status (disruptive alterations in tumor suppressor genes). Finally, to calculate the mutation burden per Mb, the total number of relevant mutations is divided by the coding region target territory of the test (0.83 Mb, 1.14 Mb, or 1.23 Mb). Tumor mutational burden is categorized as low (<6 mutation [mut]/Mb), intermediate (6–20 mut/Mb), or high (≥20 mut/Mb) [32], [33].
Results
Of the 114,200 clinical samples evaluated with CGP in this study, 21,522 (18.8%) were NSCLC and 92,678 (81.2%) were other tumor types (Table 1). The gender and age characteristics of these cohorts are shown in Table 1. Patients with non‐NSCLC tumors were significantly younger (p < .0001) and more often female (p < .0001) than the NSCLC ALK‐fusion positive patients. ALK genomic alterations were found in 1,313 (1.1%) cases, of which 896 (0.8%) harbored rALK (10.9%) or fALK (89.1%) with EML4 or another partner (Table 2). The ALK‐altered cases that lacked gene fusions included ALK amplifications and a wide variety of short variants, including base substitutions and short insertions and deletions. Of the 876 (0.8%) cases with fALK or rALK, 675 (77.1%) were NSCLC and 201 (22.9%) were other tumor types, and fALK were significantly more frequent in NSCLC (3.1%) than non‐NSCLC (0.2%; p < .0001). Patients with non‐NSCLC tumors harboring fALK were significantly older (p < .0001) and more often female (p < .0001) than patients with fALK‐positive NSCLC. The distribution of tumor histologies is shown in supplemental online Tables 4 and 5. Outside of NSCLC, rALK were most often found in carcinomas (67), sarcomas (39), and hematolymphoid malignancies (24).
Table 1. Clinical and genomic features of ALK fusion‐positive and ALK fusion‐negative NSCLC and non‐NSCLC cases.
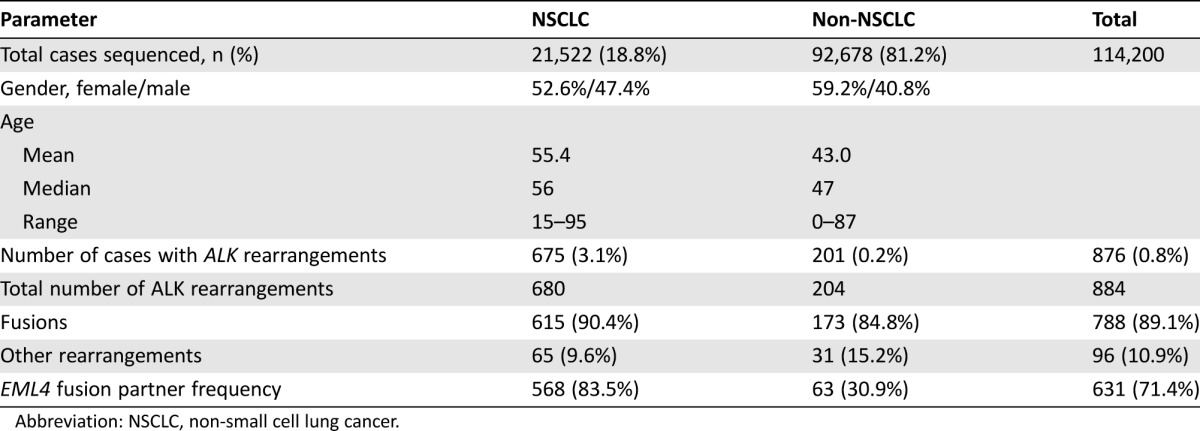
Abbreviation: NSCLC, non‐small cell lung cancer.
Table 2. Recurrent fusions partners in NSCLC and non‐NSCLC samples.
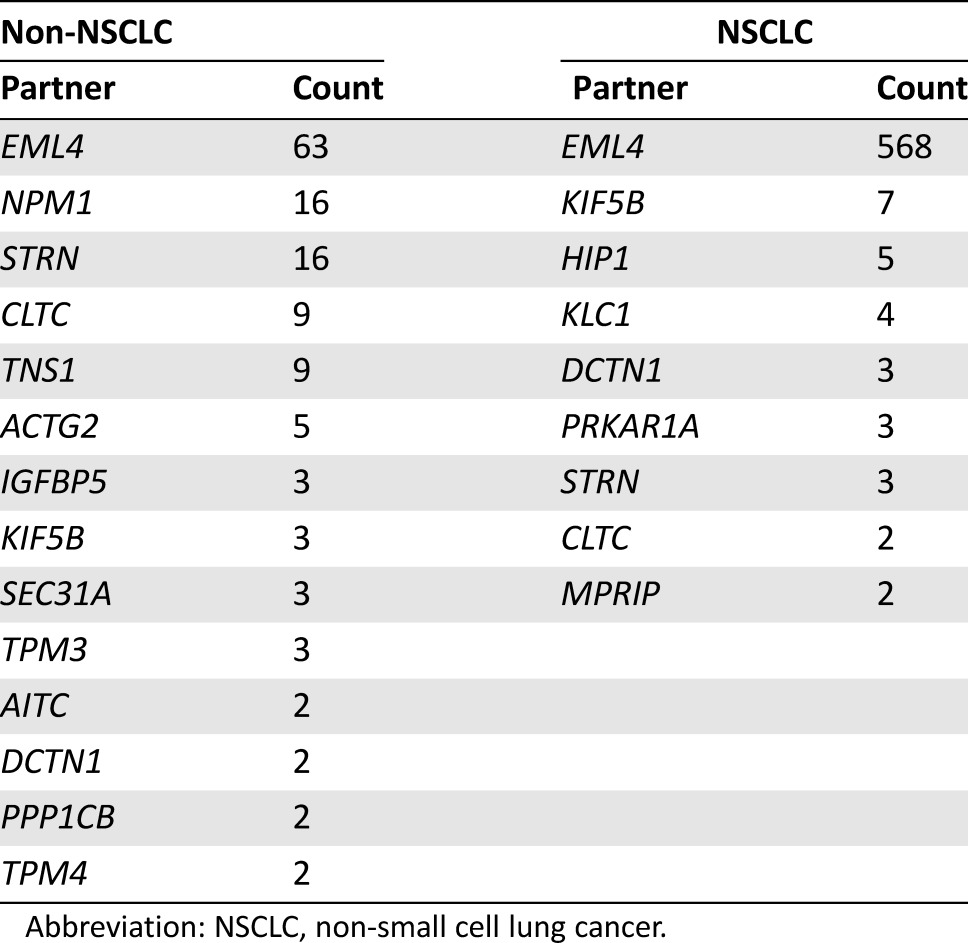
Abbreviation: NSCLC, non‐small cell lung cancer.
EML4 was by far the most common fusion partner in NSCLC (568; 83.5%), with a significant difference versus non‐NSCLC tumors (63; 30.9%; p < .0001; Table 2). A wide variety of other fusion partners were identified in non‐NSCLC tumors, the most frequent being STRN and NPM1 (16 cases each), TNS1 and CLTC (9 cases each), and ACTG2 (5 cases; Table 2).
The genes most frequently coaltered with fALK in these cohorts are shown in Figure 1. By far the most common were loss of CDKN2A and/or CDKN2B and inactivation of TP53. Less than 1% of ALK‐rearranged NSCLC featured high TMB (≥20 mut/Mb), compared with 3.5% of non‐NSCLC samples with rALK (Table 1). The lower frequency of high TMB in fALK‐positive NSCLC versus fALK‐positive non‐NSCLC was significant (p = .004).
Figure 1.

Mutation frequencies of genes coaltered with ALK fusions. Note that cases may have both ALK fusions and ALK nonfusion alterations in the same tumor. All cases feature at least a single ALK fusion genomic alteration.
Abbreviation: NSCLC, non‐small cell lung cancer.
Multiple examples have been published of clinical responses to selected anti‐ALK targeted therapy for patients whose non‐NSCLC tumors harbor fALK [12], [14], [16], [17], [19], [20], [22], [23], [26]. In this cohort of patients, novel examples of responses to ALK‐targeted therapy include case Non‐NSCLC 092, a non‐Langerhans cell histiocytosis with a robust response to crizotinib (Figure 2), and Non‐NSCLC 037, a renal cell carcinoma with a significant response to alectinib (Figure 3).
Figure 2.

A series of positron emission tomography scans showing a robust and sustained response to a crizotinib‐based treatment regimen. The patient received cervical radiotherapy, one cycle of chemotherapy (cytoxan 750 mg/m2, etoposide 100 mg/m2 days 1–3, and prednisone 100 mg days 1–5), and crizotinib 250 mg daily from July 2015 to present. Shown here are scans taken prior to treatment initiation (far left) and at 20 weeks (center) and 35 weeks (right) after treatment initiation.
Figure 3.

A representative response in a mesenteric lymph node to alectinib for a patient with renal cell carcinoma harboring an EML4‐ALK fusion.
Figure 2 illustrates a prolonged, robust response to treatment with crizotinib for Non‐NSCLC 092, a non‐Langerhans cell histiocytosis. A 40‐year‐old male presented with severe interscapular pain. Magnetic resonance imaging (MRI) of the C‐spine revealed a homogenous lesion in the cervical cord at C3‐C4 measuring 2.5 × 1 × 1 cm with avid enhancement. This was thought to be neoplastic; hence, a CT scan of the thorax/abdomen/pelvis was obtained, which revealed heterogeneous enhancement of the hepatic parenchyma, numerous peritoneal and omental lesions, numerous osseous lesions (some of which were sclerotic whereas others were lytic), and scattered subcutaneous nodules. Comprehensive genomic profiling of a histiocytic biopsy sample from the liver revealed a KIF5B‐ALK fusion. Based on the CGP results and the aggressive malignant clinical picture, treatment was started, which included cervical radiotherapy, one cycle of chemotherapy (cytoxan 750 mg/m2, etoposide 100 mg/m2 days 1–3, and prednisone 100 mg days 1–5), and finally crizotinib 250 mg daily from July 2015 to present. Comparison of a pretreatment positron emission tomography scan to scans obtained at 20 and 35 weeks shows a robust response to this crizotinib‐based treatment regimen (Figure 2).
Successful targeting of an ALK fusion has also been achieved in the context of renal cell carcinoma (non‐NSCLC 037), as illustrated by Figure 3. A 65‐year‐old male presented with a renal mass and pulmonary metastases. Nephrectomy was performed and pathology revealed a mixed histology in the renal specimen, comprising 70% papillary and 30% clear cell disease. The patient was initially treated with pazopanib but developed elevated transaminases. He then received an investigational MET inhibitor that was discontinued due to dermatologic toxicities. Following this course, he received everolimus with progression and development of interstitial changes consistent with pneumonitis. The patient was then placed on nivolumab with progression of new thyroid metastases, followed by cabozantinib. Within a short time frame, he was noted to have substantial headaches and hemorrhaging brain metastases were identified on MRI of the brain. Comprehensive genomic profiling evaluation of both circulating tumor DNA and a tissue biopsy of the kidney (October 2013) revealed an EML4‐ALK translocation. He completed stereotactic radiotherapy to brain metastases, and then began therapy with alectinib. Both brain metastases and lung metastases displayed a substantial response to treatment. Shown here is a representative response in a mesenteric lymph node.
Discussion
The dramatic therapeutic benefit of therapies targeting ALK for patients with NSCLC driven by fALK is now widely accepted. The ALK inhibitors crizotinib, ceritinib, and alectinib are all approved for the treatment of patients with NSCLC whose tumors test positive for ALK rearrangement [34], [35]. In addition, ALK inhibitors, including ceritinib, alectinib, brigatinib, and lorlatinib, are under evaluation for patients with ALK‐rearranged NSCLC that may have developed resistance to crizotinib [34], [35], [36]. The EML4‐ALK gene fusion has been observed in 3%–7% of NSCLC cases, more frequently in younger patients, nonsmokers, and men [37], [38]. As previously reported and seen in the present study, different EML4‐ALK variants have been identified in a variety of both NSCLC and non‐NSCLC cases, all of which contain the intracellular tyrosine kinase domain of ALK [39].
In this study of more than 114,000 clinical cases, fALK are extremely uncommon in tumors other than NSCLC, with a frequency of only 0.2% in a series of more than 90,000 cases. These non‐NSCLC, fALK‐driven tumors are significantly more often from female patients or younger patients, and less often have EML4 as the ALK fusion partner, although the impact of fusion partner on the sensitivity to therapies remains under investigation [40]. In addition, this data shows that fALK can be found in a wide variety of tumor types, including carcinomas, sarcomas, and hematolymphoid malignancies. Several of these tumor types warrant more detailed discussion. The presence of fALK in tubular gastrointestinal cancers, especially colorectal cancer (CRC), is noteworthy, as are the diverse group of unknown primary cases featuring fusions. In sarcomas, fALK appear to segregate with smooth muscle differentiation. Finally, as expected, fALK are more commonly identified in B‐cell lymphomas compared with other hematological cancers (supplemental online Table 5).
ALK fusions were first identified in CRC in 2009 [41] using exon array profiling, and have been subsequently investigated by hybrid capture‐based CGP [42] and indirectly detected by immunohistochemistry [43]. The 11 fALK‐positive CRC cases listed in supplementary online Table 2 represent only 0.1% of the more than 9,000 CRC cases profiled in this study. However, the clinical response to ceritinib treatment experienced by a patient with a STRN‐ALK fusion [14] highlights that, however uncommon, the identification of an ALK fusion in a non‐NSCLC tumor can lead to benefits from targeted therapy treatment [14]. Responses to the TRK/ALK/ROS1 inhibitor entrectinib have been reported for 2/2 CRC patients with ALK alterations in an ongoing phase 1/2 study [44] and for a patient whose CRC tumor harbored a novel CAD‐ALK fusion [16]. Preclinical models involving established cell lines, patient‐derived cell lines, and xenografts of fALK‐driven CRC have shown responsiveness to agents targeting ALK [45].
In the current study, 44 cases of cancer of unknown primary source (CUP) harbored fALK. These tumors were classified as unknown primary site by the submitting clinician due to a lack of pulmonary involvement and/or insufficient pathology confirmation, such as being negative for TTF1 immunostaining. In this clinical context, the possibility of lung origin cannot always be discounted, and the finding of an ALK fusion to justify reclassification of CUP to an occult NSCLC is now well recognized [46]. The case Non‐NSCLC 166 was submitted as a CUP with massive secondary pulmonary involvement and showed no expression of epithelial markers (cytokeratin, TTF1, etc.). Based on subsequent analysis, this tumor is likely a sarcoma and possibly a high‐grade variant of inflammatory myofibroblastic tumor metastatic to the lung from a soft tissue origin [19]. Thus, for this CUP case, reclassification as a form of sarcoma appears warranted.
ALK fusions are identified in a variety of sarcomas, but are predominantly found in tumors with smooth muscle differentiation including leiomyosarcomas, epithelioid leiomyosarcomas, myxoid leiomyosarcomas, inflammatory myofibroblastic tumors, and smooth muscle tumors of uncertain malignant potential [20], [21], [22], [23]. As seen in supplementary online Table 4, these smooth muscle sarcomas may be derived from soft tissues, including both superficial sites and deep sites such as the retroperitoneum, walls of the intestine, and the uterus. Evidence has also emerged that these ALK fusion driven sarcomas are responsive to both first‐generation therapies, such as crizotinib, as well as second‐ and third‐generation inhibitors [19], [20], [21].
ALK fusions were initially described in anaplastic large‐cell lymphoma [1], [2], [3], [4], [5] and are generally restricted to large B‐cell‐type non‐Hodgkin lymphoma (NHL), but are seen in the current study to occur in differentiated B‐cell malignancies, such as myeloma, as well as histiocytic disorders [47]. ALK‐driven B‐cell malignancies are typically treated with standard chemotherapy plus, when indicated, anti‐CD20 antibody infusion and bone marrow transplantation [48]. It is relatively uncommon for patients with NHL to receive anti‐ALK targeted therapies, but recent evidence has emerged that chemorefractory fALK‐positive NHL is sensitive to inhibitors such as crizotinib [49].
The results of this study suggest that CGP may provide increased sensitivity for ALK fusion detection across a variety of tumor types. Recent evidence challenges the sensitivity of fluorescence in situ hybridization (FISH) as the standard of care for detecting fALK in NSCLC [50], and although some investigators have suggested that the addition of immunohistochemistry screening (IHC) for ALK protein overexpression could prevent false‐negative FISH results [51], IHC is prone to technical issues and the ability of IHC to detect all tumors driven by fALK remains controversial. It should be noted that the non‐NSCLC fALK‐driven cancers uncovered in the current study are significantly less likely to feature EML4 as the ALK fusion partner and thus could present a greater challenge for detection if FISH were the only technique employed [50]. The CGP assay used in the current study detects all subtypes of EML4‐ALK fusions, as well as fALK with a wide range of non‐EML4 partners. Indeed, the first use of this assay in clinical practice was for a patient with widespread NSCLC, including brain metastases, whose tumor was negative for ALK fusion by FISH. Comprehensive genomic profiling detected an alternative EML4‐ALK fusion and the treatment strategy was shifted from standard of care chemotherapy to anti‐ALK targeted therapy. The patient achieved long‐term durable disease control extending beyond 4 years [52], [53].
The inverse association between high TMB and the presence of an ALK fusion in both NSCLC and non‐NSCLC is of potential interest when designing a precision therapy strategy for these patients. Tumor mutational burden calculated from CGP results has been directly linked to the efficacy of immune checkpoint‐inhibitor drugs in NSCLC [54], urinary bladder urothelial carcinoma [33], and metastatic melanoma [55]. Although IHC‐based analysis of programmed death‐ligand 1 expression in the ALK‐positive tumors presented in this study, both NSCLC and non‐NSCLC, is not available, the observation that high TMB is rare in these patients suggests that immunotherapies may not be as broadly relevant compared with ALK fusion wild‐type tumors.
Conclusion
ALK fusions can be identified in small sets of non‐NSCLC patients and are found in a wide variety of epithelial and mesenchymal malignancies. The non‐NSCLC cases harboring fALK are more often identified in slightly older female patients, are more likely to have ALK fusion partners other than EML4, and are less likely to have high TMB than fALK‐negative malignancies. Initial evidence now strongly favors that anti‐ALK therapies can be effective in a variety of tumor types driven by fALK and further study in basket trials including various cancer types appears warranted.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Footnotes
For Further Reading: Yoko Shimada, Takashi Kohno, Hideki Ueno et al. An Oncogenic ALK Fusion and an RRAS Mutation in KRAS Mutation‐Negative Pancreatic Ductal Adenocarcinoma. The Oncologist 2017;22:158–164; first published on February 6, 2017.
Implications for Practice: The oncogenic DCTN1‐ALK fusion and the RRAS mutation were associated with the development of pancreatic ductal adenocarcinoma (PDAC) in the absence of the KRAS mutation. Constitutional activation of DCTN1‐ALK fusion protein was suppressed by the anaplastic lymphoma kinase tyrosine kinase inhibitors crizotinib and alectinib. Thus, a small subset of PDAC patients might benefit from therapy using these inhibitors.
Author Contributions
Conception/design: Jeffrey S. Ross, Siraj M. Ali, Philip J. Stephens, Laurie M. Gay
Provision of study material or patients: Omotayo Fasan, Jared Block, Sumanta Pal
Collection and/or assembly of data: Omotayo Fasan, Jared Block, Sumanta Pal
Data analysis and interpretation: Jeffrey S. Ross, Laurie M. Gay
Manuscript writing: Jeffrey S. Ross, Alexa B. Schrock, Laurie M. Gay
Final approval of manuscript: Jeffrey S. Ross, Siraj M. Ali, Omotayo Fasan, Jared Block, Sumanta Pal, Julia A. Elvin, Alexa B. Schrock, James Suh, Sahar Nozad, Sungeun Kim, Hwa Jeong Lee, Christine E. Sheehan, David M. Jones, Jo‐Anne Vergilio, Shakti Ramkissoon, Eric Severson, Sugganth Daniel, David Fabrizio, Garrett Frampton, Vince A. Miller, Philip J. Stephens, Laurie M. Gay
Disclosures
Jeffrey S. Ross: Foundation Medicine (E, RF, OI); Siraj M. Ali: Foundation Medicine (E, IP, OI); Sumanta Pal: Aveo, Bristol Myers Squibb, Exelixis, Genentech, Myriad Pharmaceuticals, Novartis, Pfizer (C/A) Astellas Pharma, Medivation, Novartis (H); Julia A. Elvin: Foundation Medicine (E, OI); Alexa B. Schrock: Foundation Medicine (E, OI); James Suh: Foundation Medicine (E, OI); Christine E. Sheehan: Foundation Medicine (C/A); Jo‐Anne Vergilio: Foundation Medicine (E, OI); Shakti Ramkissoon: Foundation Medicine (E), Bristol‐Myers Squibb (C/A); Eric Severson: Foundation Medicine (E, OI); Sugganth Daniel: Foundation Medicine (E, OI); David Fabrizio: Foundation Medicine, Juno Therapeutics, Seattle Genetics (OI), Foundation Medicine (E); Garrett Frampton: Foundation Medicine (E, OI); Vince A. Miller: Foundation Medicine (E, OI); Philip J. Stephens: Foundation Medicine (E, OI); Laurie M. Gay: Foundation Medicine (E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Chiarle R, Voena C, Ambrogio C et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer 2008;8:11–23. [DOI] [PubMed] [Google Scholar]
- 2. Barreca A, Lasorsa E, Riera L et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol 2011;47:R11–R23. [DOI] [PubMed] [Google Scholar]
- 3. Mano H. The EML4‐ALK oncogene: Targeting an essential growth driver in human cancer. Proc Jpn Acad Ser B Phys Biol Sci 2015;91:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hallberg B, Palmer RH. The role of the ALK receptor in cancer biology. Ann Oncol 2016;27(suppl 3):iii4–iii15. [DOI] [PubMed] [Google Scholar]
- 5. Zhao Z, Verma V, Zhang M. Anaplastic lymphoma kinase: Role in cancer and therapy perspective. Cancer Biol Ther 2015;16:1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Passaro A, Lazzari C, Karachaliou N et al. Personalized treatment in advanced ALK‐positive non‐small cell lung cancer: From bench to clinical practice. Onco Targets Ther 2016;9:6361–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blackhall F, Cappuzzo F. Crizotinib: From discovery to accelerated development to front‐line treatment. Ann Oncol 2017;27(suppl 3):iii35–iii41. [DOI] [PubMed] [Google Scholar]
- 8. Sullivan I, Planchard D. ALK inhibitors in non‐small cell lung cancer: The latest evidence and developments. Ther Adv Med Oncol 2016;8:32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Minguet J, Smith KH, Bramlage P. Targeted therapies for treatment of non‐small cell lung cancer–Recent advances and future perspectives. Int J Cancer 2016;138:2549–2561. [DOI] [PubMed] [Google Scholar]
- 10. Iams WT, Lovly CM. Anaplastic lymphoma kinase as a therapeutic target in non‐small cell lung cancer. Cancer J 2015;21:378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ettinger DS, Wood DE, Akerley W et al. NCCN guidelines insights: Non‐small cell lung cancer, version 4.2016. J Natl Compr Cancer Netw 2016;14:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu C, Ding L, Sun B et al. Bilateral breast adenocarcinomas with EML4‐ALK fusion in a patient with multiple metastases successfully treated with crizotinib: Is lung the primary site? Onco Targets Ther 2016;9:3589–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fernandez SV, Robertson FM, Pei J et al. Inflammatory breast cancer (IBC): Clues for targeted therapies. Breast Cancer Res Treat 2013;140:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yakirevich E, Resnick MB, Mangray S et al. Oncogenic ALK fusion in rare and aggressive subtype of colorectal adenocarcinoma as a potential therapeutic target. Clin Cancer Res 2016;22:3831–3840. [DOI] [PubMed] [Google Scholar]
- 15. Lee J, Kim HC, Hong JY et al. Detection of novel and potentially actionable anaplastic lymphoma kinase (ALK) rearrangement in colorectal adenocarcinoma by immunohistochemistry screening. Oncotarget 2015;6:24320–24332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Amatu A, Somaschini A, Cerea G et al. Novel CAD‐ALK gene rearrangement is drugable by entrectinib in colorectal cancer. Br J Cancer 2015;113:1730–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Diamond EL, Durham BH, Haroche J et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov 2016;6:154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ross JS, Wang K, Gay L et al. Comprehensive genomic profiling of carcinoma of unknown primary site: New routes to targeted therapies. JAMA Oncol 2015;1:40–49. [DOI] [PubMed] [Google Scholar]
- 19. Chung JH, Ali SM, Davis J et al. A poorly differentiated malignant neoplasm lacking lung markers harbors an EML4‐ALK rearrangement and responds to crizotinib. Case Rep Oncol 2014;7:628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Subbiah V, McMahon C, Patel S et al. STUMP un“stumped”: anti‐tumor response to anaplastic lymphoma kinase (ALK) inhibitor based targeted therapy in uterine inflammatory myofibroblastic tumor with myxoid features harboring DCTN1‐ALK fusion. J Hematol Oncol 2015;8:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lovly CM, Gupta A, Lipson D et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov 2014;4:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee JC, Li CF, Huang HY et al. ALK oncoproteins in atypical inflammatory myofibroblastic tumours: Novel RRBP1‐ALK fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol 2017;241:316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parra‐Herran C, Schoolmeester JK, Yuan L et al. Myxoid leiomyosarcoma of the uterus: A clinicopathologic analysis of 30 cases and review of the literature with reappraisal of its distinction from other uterine myxoid mesenchymal neoplasms. Am J Surg Pathol 2016;40:285–301. [DOI] [PubMed] [Google Scholar]
- 24. Infarinato NR, Park JH, Krytska K et al. The ALK/ROS1 inhibitor PF‐06463922 overcomes primary resistance to crizotinib in ALK‐driven neuroblastoma. Cancer Discov 2016;6:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bresler SC, Weiser DA, Huwe PJ et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell 2014;26:682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olsen TK, Panagopoulos I, Meling TR et al. Fusion genes with ALK as recurrent partner in ependymoma‐like gliomas: A new brain tumor entity? Neuro Oncol 2015;17:1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wallace GC 4th, Dixon‐Mah YN, Vandergrift WA 3rd et al. Targeting oncogenic ALK and MET: A promising therapeutic strategy for glioblastoma. Metab Brain Dis 2013;28:355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ji JH, Oh YL, Hong M et al. Identification of driving ALK fusion genes and genomic landscape of medullary thyroid cancer. PLoS Genet 2015;11:e1005467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guan J, Tucker ER, Wan H et al. The ALK inhibitor PF‐06463922 is effective as a single agent in neuroblastoma driven by expression of ALK and MYCN. Dis Model Mech 2016;9:941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He J, Abdel‐Wahab O, Nahas MK et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016;127:3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chalmers ZR, Connelly CF, Fabrizio D et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosenberg JE, Hoffman‐Censits J, Powles T et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: A single‐arm, multicentre, phase 2 trial. Lancet 2016;387:1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shaw AT, Kim DW, Mehra R et al. Ceritinib in ALK‐rearranged non‐small‐cell lung cancer. N Engl J Med 2014;370:1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ou SH, Ahn JS, De Petris L et al. Alectinib in crizotinib‐refractory ALK‐rearranged non‐small‐cell lung cancer: A phase II global study. J Clin Oncol 2016;34:661–668. [DOI] [PubMed] [Google Scholar]
- 36. Gadgeel SM, Gandhi L, Riely GJ et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib‐resistant ALK‐rearranged non‐small‐cell lung cancer (AF‐002JG): Results from the dose‐finding portion of a phase 1/2 study. Lancet Oncol 2014;15:1119–1128. [DOI] [PubMed] [Google Scholar]
- 37. Shaw AT, Yeap BY, Mino‐Kenudson M et al. Clinical features and outcome of patients with non‐small‐cell lung cancer who harbor EML4‐ALK. J Clin Oncol 2009;27:4247–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Takahashi T, Sonobe M, Kobayashi M et al. Clinicopathologic features of non‐small‐cell lung cancer with EML4‐ALK fusion gene. Ann Surg Oncol 2010;17:889–897. [DOI] [PubMed] [Google Scholar]
- 39. Peters S, Taron M, Bubendorf L et al. Treatment and detection of ALK‐rearranged NSCLC. Lung Cancer 2013;81:145–154. [DOI] [PubMed] [Google Scholar]
- 40. Lin JJ, Shaw AT. Differential sensitivity to crizotinib: Does EML4‐ALK fusion variant matter? J Clin Oncol 2016;34:3363–3365. [DOI] [PubMed] [Google Scholar]
- 41. Lin E, Li L, Guan Y et al. Exon array profiling detects EML4‐ALK fusion in breast, colorectal, and non‐small cell lung cancers. Mol Cancer Res 2009;7:1466–1476. [DOI] [PubMed] [Google Scholar]
- 42. Lipson D, Capelletti M, Yelensky R et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 2012;18:382–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nozad S, Kim S, Lee H et al. Detection of anaplastic lymphoma kinase (ALK) rearrangement in colorectal cancer: An immunohistochemical study of 128 cases. Mod Pathol 2017;30:157–210. 28134903 [Google Scholar]
- 44. De Braud FG, Niger M, Damian S et al. Alka‐372‐001: First‐in‐human, phase I study of entrectinib – an oral pan‐trk, ROS1, and ALK inhibitor – in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol 2015;33:2517. [Google Scholar]
- 45. Medico E, Russo M, Picco G et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat Commun 2015;6:7002. [DOI] [PubMed] [Google Scholar]
- 46. Hainsworth JD, Anthony Greco F. Lung adenocarcinoma with anaplastic lymphoma kinase (ALK) rearrangement presenting as carcinoma of unknown primary site: Recognition and treatment implications. Drugs Real World Outcomes 2016;3:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Drexler HG, Gignac SM, von Wasielewski R et al. Pathobiology of NPM‐ALK and variant fusion genes in anaplastic large cell lymphoma and other lymphomas. Leukemia 2000;14:1533–1559. [DOI] [PubMed] [Google Scholar]
- 48. Hapgood G, Savage KJ. The biology and management of systemic anaplastic large cell lymphoma. Blood 2015;126:17–25. [DOI] [PubMed] [Google Scholar]
- 49. Gambacorti Passerini C, Farina F, Stasia A et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase‐positive lymphoma patients. J Natl Cancer Inst 2014;106:djt378. [DOI] [PubMed] [Google Scholar]
- 50. Ali SM, Hensing T, Schrock AB et al. Comprehensive genomic profiling identifies a subset of crizotinib‐responsive ALK‐rearranged non‐small cell lung cancer not detected by fluorescence in situ hybridization. The Oncologist 2016;21:762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thunnissen E, Bubendorf L, Dietel M et al. EML4‐ALK testing in non‐small cell carcinomas of the lung: A review with recommendations. Virchows Arch 2012;461:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peled N, Palmer G, Hirsch FR et al. Next‐generation sequencing identifies and immunohistochemistry confirms a novel crizotinib‐sensitive ALK rearrangement in a patient with metastatic non–small‐cell lung cancer. J Thorac Oncol 2012;7:e14–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dudnik E, Siegal T, Zach L et al. Durable brain response with pulse‐dose crizotinib and ceritinib in ALK‐positive non‐small cell lung cancer compared with brain radiotherapy. J Clin Neurosci 2016;26:46–49. [DOI] [PubMed] [Google Scholar]
- 54. Rizvi NA, Hellmann MD, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Johnson DB, Frampton GM, Rioth MJ et al. Targeted next generation sequencing identifies markers of response to PD‐1 blockade. Cancer Immunol Res 2016;4:959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.