

Key Points
The combination of elotuzumab and an anti–PD-1 antibody leads to enhanced antitumor efficacy in mouse models.
Enhanced antitumor activity is likely due to the promotion of tumor-infiltrating NK and T-cell activity.
Abstract
Elotuzumab, a humanized monoclonal antibody that binds human signaling lymphocytic activation molecule F7 (hSLAMF7) on myeloma cells, was developed to treat patients with multiple myeloma (MM). Elotuzumab has a dual mechanism of action that includes the direct activation of natural killer (NK) cells and the induction of NK cell–mediated antibody-dependent cellular cytotoxicity. This study aimed to characterize the effects of elotuzumab on NK cells in vitro and in patients with MM and to determine whether elotuzumab antitumor activity was improved by programmed death receptor-1 (PD-1) blockade. Elotuzumab promoted NK cell activation when added to a coculture of human NK cells and SLAMF7-expressing myeloma cells. An increased frequency of activated NK cells was observed in bone marrow aspirates from elotuzumab-treated patients. In mouse tumor models expressing hSLAMF7, maximal antitumor efficacy of a murine immunoglobulin G2a version of elotuzumab (elotuzumab-g2a) required both Fcγ receptor–expressing NK cells and CD8+ T cells and was significantly enhanced by coadministration of anti–PD-1 antibody. In these mouse models, elotuzumab-g2a and anti–PD-1 combination treatment promoted tumor-infiltrating NK and CD8+ T-cell activation, as well as increased intratumoral cytokine and chemokine release. These observations support the rationale for clinical investigation of elotuzumab/anti–PD-1 combination therapy in patients with MM.
Visual Abstract
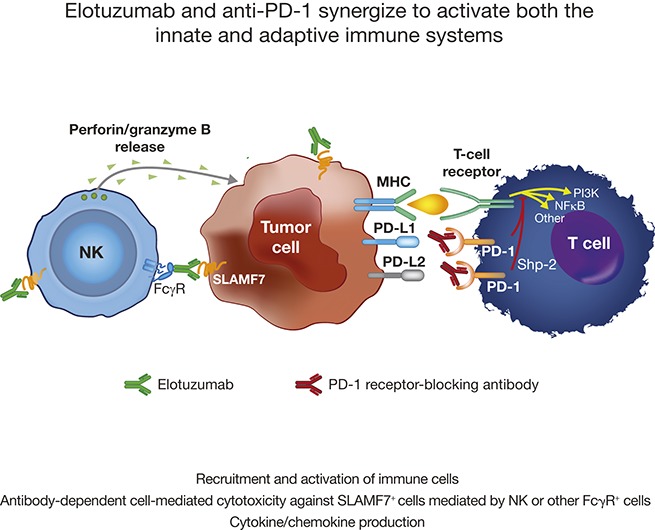
Introduction
The ability of tumor-targeted monoclonal antibodies (mAbs) to stimulate immune effector functions is a critical component of durable tumor regression.1,2 In mouse tumor models, innate effector cells expressing activating Fcγ receptors (FcγR), such as natural killer (NK) cells and myeloid cells, are required for the therapeutic efficacy of tumor-targeted mAbs.3-6 Human NK cells are activated on exposure to tumor cells coated with human immunoglobulin G1 (hIgG1) mAbs, such as rituximab.7 In lymphoma, expression of high-affinity alleles of FcγRIIIa (FcγRIIIa-158V) and FcγRIIa (FcγRIIa-131H) is associated with an improved response to rituximab therapy, likely due to enhanced antibody-dependent cellular cytotoxicity (ADCC).8,9 Similarly, benefits in progression-free survival have been reported in patients with relapsed/refractory multiple myeloma (RRMM) who were homozygous for the FcγRIIIa-158V allele and treated with elotuzumab in combination with bortezomib and dexamethasone.10 Studies in immunocompetent mice with syngeneic tumor allografts showed that the therapeutic effects of tumor-targeted mAbs decrease when CD8+ T cells are depleted.11-15 Furthermore, patients with lymphoma have developed lymphoma-specific anti-idiotype CD4+ and CD8+ T-cell responses after rituximab treatment, suggesting that tumor-targeted mAbs may initiate an antitumor adaptive immune response.14
Elotuzumab is a humanized IgG1 mAb that binds human signaling lymphocytic activation molecule F7 (hSLAMF7), a glycoprotein highly expressed on malignant plasma cells in multiple myeloma (MM), irrespective of cytogenetic abnormalities or disease stage.16-18 Elotuzumab, administered in combination with lenalidomide and dexamethasone (ELd), was shown to improve progression-free survival in a phase 3 clinical trial of RRMM and was subsequently approved in the United States, the European Union (EU), and Japan for the treatment of patients with RRMM who have received 1-3 previous therapies.19-22
Preclinical studies showed that elotuzumab induces lysis of human myeloma cells when they are incubated with peripheral blood mononuclear cells (PBMCs) or purified NK cells in vitro.16,17 The lytic effect of elotuzumab requires SLAMF7 expression on the surface of myeloma cells and depends on engagement of FcγRIIIa, demonstrating the importance of ADCC in elotuzumab-mediated myeloma cell death.17 Elotuzumab also inhibits the growth of established human myeloma xenografts in immunocompromised mice.16,17 The efficacy of elotuzumab in these models was NK cell–dependent and was enhanced by coadministration of bortezomib, lenalidomide, or mAbs that additionally stimulated NK cell activity.16,17,23-25 Furthermore, elotuzumab promotes cytotoxicity against myeloma cells through direct engagement of SLAMF7 on NK cells.26 SLAMF7 is a self-ligand that stimulates NK cell activation in the presence of the adaptor protein EWS-Fl1–activated transcript-227-29; however, elotuzumab does not activate, inhibit, or directly induce apoptosis of myeloma cells. Myeloma cells do not express EWS-Fl1–activated transcript-2 (or CD45, a phosphatase also required for SLAMF7 signaling), which may explain why elotuzumab does not directly induce MM cell apoptosis.30
The activity of tumor-targeted mAbs can be improved with mAbs that modulate adaptive immune system responses.12,31 Programmed death receptor-1 (PD-1) is an inhibitory receptor expressed on activated T cells as well as on NK cells and other immune cells.32 Binding of PD-1 to its ligands, PD-ligand 1 (PD-L1) and PD-L2, dampens antitumor immune responses,33,34 allowing tumor cells to evade immunosurveillance. PD-L1 is often expressed on myeloma cells, and expression of PD-1 on NK and CD8+ T cells is higher in patients with MM than in healthy individuals.35-37 Antibodies blocking PD-1/PD-L1 interaction enhance the functionality of hyporesponsive NK cells and CD8+ T cells from patients with MM and prolong survival in disseminated myeloma-bearing mice.36-38 Nevertheless, in a phase 1 study of 27 patients with MM, the best response to monotherapy with the anti–PD-1 mAb nivolumab was a complete response in 1 patient and stable disease in 17 patients, suggesting that PD-1 blockade may need to be combined with other modalities to achieve efficacy in patients with MM.39
In this study, we investigated the effects of elotuzumab on the phenotype and functionality of human NK cells isolated from healthy donors and patients with MM. We also assessed how an antitumor immune response is generated after treatment with elotuzumab alone or with an anti–PD-1 mAb in mouse tumor models engineered to express hSLAMF7.
Methods
Study approval
All patient samples from clinical study HuLuc63-1703, a phase 1/2 study of ELd in RRMM (#NCT00742560),40 were collected and used in accordance with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use, the Declaration of Helsinki, and all applicable national regulations. The investigator or designee was required to explain to each subject the nature of the study, the procedures, the possible side effects, and all other elements of consent as defined in US Food and Drug Administration regulation 21 CFR Part 50, EU regulations (for EU sites), and other applicable national and local regulations governing informed consent. Each subject was required to provide a signed and dated informed consent prior to enrollment.
Mice
BALB/c or C57BL/6 female mice were purchased from Charles River (San Diego, CA), Taconic (Hudson, NY), or The Jackson Laboratory (Sacramento, CA). All mouse experimentation was carried out subsequent to proper review and approval by the Bristol-Myers Squibb Animal Care and Use Committee, accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (accreditation #001085). All mice were maintained under specific pathogen-free conditions following the “Guide for the Care and Use of Laboratory Animals” and used between 6 to 18 weeks of age. All mouse experiments were carried out after proper review and approval by MuriGenics Institutional Animal Care and Use Committee, an Office of Laboratory Animal Welfare (accreditation #A457301).
Antibody generation
To generate a mouse IgG2a variant of elotuzumab (elotuzumab-g2a), the heavy chain variable domain (VH) for the elotuzumab parental mAb (muLuc63; AbbVie Biotherapeutics, Redwood City, CA) was cloned into an expression vector containing the mouse IgG2a (mIgG2a) constant region. The light chain variable region (Vκ) was cloned into an expression vector containing the mouse light chain constant region. The 2 vectors were cotransfected into Chinese hamster ovary–S cells, and 1 stable clone was selected and scaled up for mAb production. The generation of an anti-mouse PD-1 mAb, clone 4H2, has previously been described.41 To generate non–FcγR-binding PD-1–mg1-D265A42 and elotuzumab–mg1-D265A variants, VH and Vκ sequences from the parental mAbs were grafted onto a murine IgG1 constant region containing the D265A mutation. Control antibodies included mIgG2a (clone C1.18.4, BioXCell) and mIgG1 (Bristol-Myers Squibb, Princeton, NJ). Zab098 (a surrogate mAb for daratumumab) was produced by using published sequences (GenBank: ADS96869 and ADS96865, corresponding to sequences 17 and 12 from patent publication number US7829673).
Cell lines and culturing conditions
A20 cells (ATCC, Manassas, VA) were maintained in RPMI 1640 with 10% fetal bovine serum (FBS) and 0.05 mM 2-mercaptoethanol (Life Technologies, Waltham, MA). EG7 cells (ATCC) were cultured in RPMI 1640 with 2 mM l-glutamine, 10% FBS, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 1 mM sodium pyruvate, 0.05 mM 2-mercaptoethanol, and 0.4 mg/mL G418 (Life Technologies). MC38 cells were cultured in Dulbecco's modified Eagle medium with 10% FBS. OPM-2 (DSMZ, Braunschweig, Germany) and MM1.S (Kerry Campbell, Fox Chase Cancer Center, Philadelphia, PA) cells were cultured in RPMI 1640 with 10% FBS and 10 mM HEPES. A20 or EG7 cells were retrovirally transduced with constructs encoding either GFP alone (pFB-GFP) or GFP and full-length hSLAMF7 (pFB-hSLAMF7-GFP; kindly provided by Andre Veillette, Institut de Recherches Cliniques de Montréal, Montréal, QC, Canada), selected to make stable cell lines, and sorted by flow cytometry for GFP+ cells.
Tumor cell implantation and treatment
A20-derived tumors were established via subcutaneous injection of 10 × 106 A20-hSLAMF7 cells into the hind flanks of BALB/c mice. After 11 to 14 days, mice were randomized into treatment groups when tumor volumes reached 150 to 180 mm3. EG7-derived tumors were established via subcutaneous injection of 5 × 106 EG7-hSLAMF7 cells into the hind flanks of C57BL/6 mice. After 5 to 7 days, mice were randomized into treatment groups when tumor volumes reached 80 to 120 mm3. mAbs were administered intraperitoneally at the indicated times for a total of 3 doses. For depletion experiments, 50 µg of anti–asialo-GM1 (eBioscience, San Diego, CA) or 200 µg of anti-CD8α (clone 53-6.72, BioXCell, West Lebanon, NH) were injected intraperitoneally 3 days before elotuzumab-g2a and every 7 days thereafter. For tumor rechallenge, long-term surviving mice were implanted with 10 Χ 106 of EG7-hSLAMF7 or MC38 cells 88 days after the previous tumor implantation. Tumor growth was determined biweekly by using electronic digital calipers (Fowler, Newton, MA). Mice with a tumor volume of 0 mm3 for 3 consecutive measurements were considered tumor free.
Human NK cell assays
Blood was collected from healthy donors, and PBMCs were isolated by using Ficoll-Paque Plus (GE Healthcare, Pittsburgh, PA). PBMCs were cocultured with OPM-2 target cells at a 10:1 ratio in RPMI 1640 with 10% FBS. Control hIgG1 or elotuzumab at varying concentrations was added to wells; Golgi Stop (1:1000; BD Pharmingen, San Diego, CA) was added to detect CD107a, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α). After 4 or 20 hours, cells were stained with live/dead dye, CD3, and CD56 to identify NK cells; CD107a, CD69, and CD54 to assess activation status; and intracellular IFN-γ and TNF-α (FoxP3 intracellular staining kit, eBioscience) to determine cytokine expression. For cytokine bead array (CBA) assays, NK cells were isolated from PBMCs by negative selection using the MACS NK isolation kit (Miltenyi Biotec, Auburn, CA). Purified NK cells (>95% purity by flow cytometry) were maintained at 1 × 106 cells/mL in RPMI 1640 with 10% fetal calf serum, 2 mM l-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 100 µM nonessential amino acids, 50 µM 2-mercaptoethanol, and 25 U/mL human recombinant interleukin-2 (IL-2) (National Cancer Institute, Rockville, MD) for 24 to 48 hours. NK cells were cocultured with OPM-2 at a 5:1 ratio for 5 hours, and cytokines were measured using BD CBA Flex Sets (BD Pharmingen). For ADCC assays, NK cells were isolated from PBMCs by negative selection using a magnetic bead–based separation kit (StemCell Technologies, Vancouver, BC, Canada) and cultured in MyeloCult media (StemCell Technologies) with 500 IU/mL human recombinant IL-2 for 24 hours. NK cells were cocultured in a 96-well V-bottom plate with target cells at a 10:1 ratio in the presence of elotuzumab or hIgG1 mAb. After a 3-hour incubation at 37°C, cells were washed and stained with Fixable Viability Dye (eBioscience) and the percentage of dead target cells was measured by flow cytometry.
Proliferation and apoptosis assays
Apoptosis was measured after the incubation of platebound antibodies (10 µg/mL) with 1 × 105 cells per well in RPMI 1640 with 10% FBS for 24 hours at 37°C. Cells were stained with Annexin V-PE (BD Biosciences). Cell proliferation was measured by MTS assay (CellTiter 96 AQueous One Solution Cell Proliferation Assay; Promega, Madison, WI). Briefly, 5 × 104 cells were plated in U-bottom 96-well plates, incubated with soluble mAbs (10 µg/mL), and analyzed at 24, 48, and 72 hours.
Primary patient samples
All samples were derived from a subset of patients with paired screening and cycle 1 day 22 (C1D22) samples from clinical study HuLuc63-1703.40 Bone marrow aspirate samples were collected in sodium heparin tubes at screening and on C1D22.
Immunophenotypic and intratumoral cytokine analysis
A20-hSLAMF7 or EG7-hSLAMF7 tumor masses were resected 4 to 7 and 10 to 14 days after mAb treatments. Single-cell suspensions were prepared by dissociation and passing cells through a 70-µm filter. Cells (5 × 105) were plated in 96-well plates, treated with Fc block (clone 2.4G2; BD Biosciences), and stained with fluorochrome-conjugated antibodies against surface or intracellular markers (see supplemental Table 1 for antibody list). For intracellular staining, samples were fixed, permeabilized, and stained with antibodies (FoxP3 staining buffer set; eBioscience). For ovalbumin (OVA) tetramer (tet) staining, cells were stained with 5 µL per sample of H-2Kb OVA (SIINFEKL) PE tetramer (MBL International, Woburn, MA) in RPMI 1640 with 50 nM dasatinib (Bristol-Myers Squibb). For intratumoral cytokine analysis, tumors were harvested into 1 mL of complete media (RPMI 1640 with 3% FBS). Tumor homogenates were centrifuged and supernatants frozen at −80°C for batch sample processing. Neat supernatant (30 μL) from each sample was analyzed for concentrations of intratumoral IL-1α, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-9, IL-10, IL-12 p40, IL-12 p70, IL-13, IL-15, IL-17A, IL-17F, IL-21, IL-22, IL-23, IL-25/IL-17E, IL-27, IL-31, IL-33, IP-10, KC, granulocyte-macrophage colony-stimulating factor, granulocyte-CSF, TNF-α, IFN-γ, macrophage inflammatory protein–1α (MIP-1α), MIP-1β, MIP-2, monocyte chemoattractant protein-1, and RANTES by using bead-based assays (Millipore, Billerica, MA). MIP-1α and MIP-1β were measured by using enzyme-linked immunosorbent assay (ELM-MIP-1α, RayBiotech, Norcross GA, and MMB00, R&D Systems, Minneapolis, MN, respectively).
Statistics
Patient sample data were analyzed by using paired Student t tests. Mouse tumor volumes at specific time points were analyzed by using the Mann-Whitney U test. Flow cytometry data were analyzed by using paired Student t test, unpaired Student t test, or analysis of covariance (ANCOVA), and cytokine levels in CBA assays were analyzed by unpaired Student t test. Error bars represent standard error of the mean (SEM), with the exception of tumor volumes at randomization (standard deviation) and pre- and posttreatment patient data (median ± standard error). Binary logistic regression predictors consisted of treatment (isotype control, elotuzumab-g2a, anti–PD-1, and elotuzumab-g2a/anti–PD-1) and study (to control for possible study effects). Differences between survival curves were analyzed by using the log-rank (Mantel-Cox) test.
Results
Effect of elotuzumab on NK cell activation in vitro and in ELd-treated patients with MM
Adding elotuzumab to a mixture of human OPM-2 myeloma cells and PBMCs led to upregulation of CD107a, a marker of granule mobilization, and to production of IFN-γ and TNF-α by CD56dimCD3− NK cells (Figure 1A). This was followed by upregulation of activation markers CD69 and CD54 on CD56dimCD3− NK cells (Figure 1B). Elotuzumab treatment also increased secretion of IFN-γ, TNF-α, MIP-1α, and MIP-1β from purified NK cells when cocultured with OPM-2 cells (Figure 1C). Concomitant with the increase in NK cell activation observed in cocultures, elotuzumab reduced the number of live OPM-2 cells compared with hIgG1 treatment (data not shown), likely due to NK cell–mediated cytotoxicity.43
Figure 1.

Elotuzumab induces NK cell activation in PBMC/myeloma cell coculture in vitro and in patients with MM when combined with lenalidomide and dexamethasone. (A-B) PBMCs from healthy donors were incubated alone or with OPM-2 cells for 4 hours (A) or 20 hours (B) in the presence of either hIgG1 or increasing doses of elotuzumab (Elo). Surface expression of CD107a, CD69, and CD54 and intracellular levels of IFN-γ and TNF-α were determined by using flow cytometry with gating on live CD56dimCD3− lymphocytes. Results are representative of at least 4 donors. (C) Culture supernatant was collected at 20 hours, and NK-secreted IFN-γ, TNF-α, MIP-1α, and MIP-1β levels were measured by CBA. Data are presented as fold change comparing the cytokine levels in the presence of elotuzumab with those obtained in the presence of hIgG1. Differences were assessed by using a Student t test. (D-E) Analysis of bone marrow samples taken at time of screening (SCRN, gray box) and at C1D22 (red box) in patients with RRMM treated with ELd. (D) Median fluorescence intensity (MFI) of CD54 on CD56dimCD16+CD3− cells; n = 44 (P < .01). (E) Percentage of plasma cells (CD45dimCD138+) from bone marrow; n = 44 (P = .026). *P < .05; **P < .01; ***P < .001.
We next analyzed the frequency and phenotype of NK cells in bone marrow aspirate from patients with MM after treatment with ELd in the clinical study HuLuc63-1703.40 A significant increase in the percentage of bone marrow NK cells (CD56dimCD16+CD3−) was observed compared with pretreatment (21.8% ± 12.1% at C1D22 vs 15.3% ± 8.5% at screening; P =.001). In addition, there was a significant increase in expression of the activation marker CD54 on CD56dimCD16+CD3− bone marrow NK cells after ELd treatment (Figure 1D). Previous studies suggest that elotuzumab, but not lenalidomide, is responsible for recruiting NK cells to murine xenograft tumors.43 Along with changes in NK cell frequency and activation status, ELd treatment led to a significant reduction in the percentage of CD45dimCD138+ myeloma cells in the bone marrow (median of 0.7% at C1D22 vs 4.05% at screening, P =.026; Figure 1E).
Antitumor effects of elotuzumab against hSLAMF7-expressing mouse tumor lines in vivo
Previous models demonstrated that elotuzumab inhibited the growth of hSLAMF7-expressing myeloma xenografts in immunocompromised mice.43 To evaluate whether elotuzumab mediates antitumor efficacy in immunocompetent mice, we generated mouse tumor cell lines expressing hSLAMF7. A20-hSLAMF7 and EG7-hSLAMF7 cells showed stable expression of hSLAMF7 at levels similar to OPM-2 cells (supplemental Figure 1). Elotuzumab bound to and promoted NK cell–mediated cytotoxicity toward hSLAMF7-expressing cells, but not toward control A20 or EG7 cells (supplemental Figures 2 and 3).
The in vivo efficacy of elotuzumab against established hSLAMF7-expressing tumors was evaluated (Figure 2A-D). Treatment of BALB/c mice bearing A20-hSLAMF7 tumors or C57BL/6 mice bearing EG7-hSLAMF7 tumors with elotuzumab-g2a (an mIgG2a isotype variant of elotuzumab) reduced tumor growth in both models vs controls (A20-hSLAMF7 volume at day 21: 377 mm3 ± 157 mm3 vs 1062 mm3 ± 240 mm3, P =.022; EG7-hSLAMF7 volume at day 27: 293 mm3 ± 137 mm3 vs 1273 mm3 ± 338 mm3, P =.03; Figure 2A,D).
Figure 2.

Maximal antitumor effect of elotuzumab requires both FcR-expressing NK cells and CD8+T cells in mouse tumor models. (A) BALB/c mice were injected subcutaneously with A20-hSLAMF7 tumor cells (10 × 106 cells) and randomized at day 11 when their tumors reached an average size of 180 ± 60 mm3. Mice were injected intraperitoneally with 10 mg/kg of either control mIgG2a (circles) or elotuzumab-g2a (triangles) on days 11, 14, and 18. n = 12 mice per group. *P = .022 vs IgG2a, day 21. (B-C) A20-hSLAMF7–bearing BALB/c mice were first randomized on day 7 (average tumor size: 117 ± 44 mm3) to receive anti-CD8α or anti–asialo-GM1 (days 7, 14, and 21) and then randomized again on day 10 (average tumor size: 166 ± 59 mm3) to receive 10 mg/kg of either control IgG2a or elotuzumab-g2a (days 10, 13, and 17) mAb. n = 10 mice per group. **P = .002 for elotuzumab-g2a/anti–asialo-GM1 vs elotuzumab-g2a; ***P = .0008 for elotuzumab-g2a/anti-CD8α vs elotuzumab-g2a, day 20. (D) C57BL/6 mice were injected subcutaneously with EG7-hSLAMF7 tumor cells (5 × 106 cells) and randomized on day 7 when their tumors reached an average size of 98.9 ± 9.6 mm3. Mice were injected intraperitoneally with 10 mg/kg of either control IgG2a (circles) or elotuzumab-g2a (triangles) on days 7, 11, and 14. n = 7-8 mice per group. *P = .04 vs IgG2a, day 25; and P = .03 vs IgG2a, day 27. (E) EG7-hSLAMF7–bearing C57BL/6 mice were randomized on day 7 (average tumor size: 82 ± 42 mm3) to receive 10 mg/kg of control IgG2a (circles), elotuzumab-g2a (triangles), or elotuzumab–mg1-D265A (inverted triangles) on days 7, 11, and 14. n = 10-11 mice per group. *P = .04 elotuzumab-g2a vs control IgG2a, day 18. Data are mean ± SEM. Statistical analyses were performed at the indicated time point using the Mann-Whitney U test.
Mice depleted of NK cells or treated with a non–FcγR-binding variant of elotuzumab (elotuzumab–mg1-D265A) had higher tumor volumes than mice in the control group (Figure 2B,E), suggesting that the antitumor effects of elotuzumab-g2a were likely due to FcγR-dependent functions, such as ADCC. Consistent with this, soluble or platebound elotuzumab-g2a had no direct effect on the apoptosis or growth of hSLAMF7-expressing tumor cells in vitro (supplemental Figure 4). To assess whether adaptive immunity was involved in elotuzumab-g2a antitumor activity, we treated mice with a CD8α-depleting mAb prior to elotuzumab-g2a treatment. Tumors in CD8α-depleted mice were larger than those in nondepleted mice (Figure 2C). Thus, adaptive as well as innate immune responses appear to be necessary for optimal elotuzumab-g2a–mediated antitumor efficacy, which is consistent with observations for other tumor-targeted mAbs.11,14,15
Effect of the elotuzumab-g2a and anti–PD-1 combination on in vivo tumor growth
In both models, PD-1 expression was higher on tumor-infiltrating T cells compared with splenic T cells (Figure 3A). Additionally, both A20-hSLAMF7 and EG7-hSLAMF7 tumors expressed high levels of mouse PD-L1 (Figure 3B). Mice with established A20-hSLAMF7 tumors were treated with control mAbs, anti–PD-1, elotuzumab-g2a, or the elotuzumab-g2a/anti–PD-1 combination. Although anti–PD-1 and elotuzumab-g2a each demonstrated antitumor activity alone, their combination significantly enhanced tumor regression and extended survival compared with single-agent treatment (Figure 3C-D). In the binary logistic regression analysis of complete tumor regression, both the elotuzumab-g2a– and anti–PD-1–treated groups were significantly more likely to be tumor free (Wald χ2 [1] = 7.30, P =.007, odds ratio [OR]: 18.48; 95% confidence interval [CI]: 2.23-153.37; Wald χ2 [1] = 10.06, P =.002; OR: 30.26; 95% CI: 3.68-248.85, respectively) than the control-treated group (supplemental Figure 5A). Treatment with the elotuzumab-g2a/anti–PD-1 combination resulted in the greatest number of tumor-free mice (Wald χ2 [1] = 22.51, P < .0001; OR: 206.84; 95% CI: 22.86-1871.88).
Figure 3.

Elotuzumab-g2a and anti–PD-1 mAbs show synergistic antitumor activity. (A) Splenocytes and tumor-infiltrating lymphocytes from A20-hSLAMF7– or EG7-hSLAMF7–bearing mice were harvested. The proportion of splenic or tumor-infiltrating CD4+CD3+, CD8+CD3+, or NKp46+CD3− cells that expressed PD-1 was quantified. Results are a composite of 3 independent experiments with a total of 5 to 22 mice per group; error bars show SEM. Frequencies of tumor-infiltrating vs splenic PD-1+ cells were compared by using the paired Student t test. (B) A20-hSLAMF7 and EG7-hSLAMF7 cells prior to implantation were stained with either isotype control (filled red histogram) or anti-mouse PD-L1 (mPD-L1; blue histogram). A flow cytometry histogram representative of PD-L1 expression is shown. (C-D) A20-hSLAMF7–bearing mice were randomized to different treatment groups on day 10 when their tumors reached an average size of 157 ± 63 mm3 and were treated with 10 mg/kg elotuzumab-g2a or control mIgG2a and/or 3 mg/kg anti–PD-1 injected intraperitoneally on days 10, 14, and 17. Tumor volumes for individual mice (C) and percent survival (D) are shown. n = 9 per group. Differences between survival curves were analyzed by using the Mantel-Cox test. (E) EG7-hSLAMF7–bearing mice were randomized to different treatment groups on day 7 when their tumors reached an average size of 120 ± 51 mm3 and were treated with 10 mg/kg elotuzumab-g2a or control mIgG2a and/or 10 mg/kg anti–PD-1 injected intraperitoneally on days 7, 10, and 14. Tumor volumes for individual mice and the number of tumor-free mice per group are shown. n = 9 per group. **P < .01; ***P < .0001.
Enhanced efficacy of the elotuzumab-g2a/anti–PD-1 combination was also observed in the EG7-hSLAMF7 model. Coadministration of elotuzumab-g2a and anti–PD-1 significantly inhibited tumor growth compared with the control group (tumor volume at day 21: 289 ± 164 mm3 vs 1323 ± 244 mm3, P =.004; Figure 3E) and extended survival (P < .05). Binary logistic regression analyses demonstrated that elotuzumab-g2a or anti–PD-1 alone did not yield statistically significant increases in the proportion of tumor-free mice vs the control group (supplemental Figure 5B). By contrast, the elotuzumab-g2a/anti–PD-1 combination resulted in a significant increase in the number of tumor-free mice (Wald χ2 [1] = 8.41, P =.004; OR: 24.05; 95% CI: 2.80-206.38) compared with the control group. Therefore, in both models, the elotuzumab-g2a/anti–PD-1 combination showed superior efficacy to either agent alone, even when no single-agent antitumor activity was observed. Importantly, mice cured of EG7-hSLAMF7 tumors after elotuzumab-g2a/anti–PD-1 combination treatment were protected against a secondary tumor challenge, suggesting that mice developed immunological memory against EG7-hSLAMF7 tumor cells (Figure 4).
Figure 4.

Long-term surviving mice treated with the elotuzumab-g2a/anti–PD-1 combination are protected against subsequent tumor rechallenge. (A-B) At day 88 after initial EG7-hSLAMF7 inoculation, a cohort of long-term surviving mice originally treated with 3 doses of the elotuzumab-g2a/PD-1 combination were rechallenged with either EG7-hSLAMF7 (A) or MC38 (B) cells. (C-D) Naive C57BL/6 mice were implanted with the same tumor cells to confirm tumor growth. Groups contained 3 to 4 mice and are from a single experiment. Symbols represent individual data points from individual mice.
NK cell activation and cytokine and chemokine release after elotuzumab-g2a and anti–PD-1 combination treatment
The antitumor efficacy of the elotuzumab-g2a/anti–PD-1 combination could be due to innate and/or adaptive immune system activation within the tumor. A greater proportion of tumor-infiltrating NK cells from A20-hSLAMF7 tumor-bearing mice treated with the elotuzumab-g2a/anti–PD-1 combination expressed IFN-γ (118% increase in IFN-γ+ cells, P < .0001) and TNF-α (85% increase in TNF-α+ cells, P = .011), compared with control mice (Figure 5A-B). MIP-1α and MIP-1β levels were consistently elevated intratumorally after elotuzumab-g2a or elotuzumab-g2a/anti–PD-1 treatment (Figure 5C, data not shown). Unlike cytokine and chemokine increases that occurred 4 to 7 days after elotuzumab-g2a/anti–PD-1 combination treatment, changes in activation marker expression on tumor-infiltrating NK cells occurred 10 to 14 days after treatment. With the elotuzumab-g2a/anti–PD-1 combination, tumor-infiltrating NK cells degranulated more (167% increase in CD107a+ cells, P < .0001) and expressed higher levels of CD69 (32% increase in CD69+ cells, P = .045) relative to the control-treated group (Figure 5D-E). Tumor-infiltrating (but not splenic) NK cells from A20-hSLAMF7–bearing mice treated with the combination regimen showed a significant increase in cytokine and activation marker expression, consistent with the requirement for the antigen (hSLAMF7)-expressing tumor cells for NK cell activation (Figure 5A-B,D-E). Similar results were seen in EG7-hSLAMF7 tumor-infiltrating NK cells (supplemental Figure 6).
Figure 5.

Elotuzumab-g2a and anti–PD-1 combination promotes NK cell activation and cytokine and chemokine release within A20-hSLAMF7 tumors. A20-hSLAMF7–bearing mice were treated with control, 3 mg/kg anti–PD-1, 10 mg/kg elotuzumab-g2a (Elo), or elotuzumab-g2a plus anti–PD-1. Tumor-infiltrating and splenic NK cells were analyzed by flow cytometry. Results are a composite of 3 independent experiments with a total of 9 to 15 mice per group unless indicated otherwise. NK cells were gated as live, GFP−CD19−CD3−NKp46+. (A-B) Quantification of (A) IFN-γ–producing and (B) TNF-α–producing tumor-infiltrating and splenic NK cells as percentages of total NK cells isolated 4 to 7 days after the start of treatment. (C) Intratumoral concentration of MIP-1β in tumors isolated 5 days after the start of treatment, measured by enzyme-linked immunosorbent assay. n = 5 per group. (D-E) Quantification of (D) CD107a+ and (E) CD69+ tumor-infiltrating and splenic NK cells as percentages of total NK cells isolated 10 to 14 days after the start of treatment. Results are shown as mean ± SEM. Statistical analyses were performed by using ANCOVA (A-B,D-E) and unpaired Student t test (C). *P < .05; ** P < .01; ***P < .0001.
T-cell activation after elotuzumab-g2a/anti–PD-1 combination treatment
We next investigated how combination treatment modulates the activation and effector function of T cells. A20-hSLAMF7 tumor-infiltrating CD8+ T cells expressed lower levels of CD107a than splenic CD8+ T cells. However, upon treatment with either anti–PD-1 alone or the elotuzumab-g2a/anti–PD-1 combination, tumor-infiltrating (but not splenic) CD8+ T cells exhibited enhanced CD107a expression (95% increase in CD107a+ cells with anti–PD-1, P < .0001; 106% increase with combination treatment, P < .0001 vs control; Figure 6A). The proportion of CD8+ T cells positive for CD69 and PD-1 followed a similar pattern (80% increase, P = .001 and 73% increase, P = .001, vs control, respectively; Figure 6B-C). Only treatment with elotuzumab-g2a alone or in combination with anti–PD-1 led to a significant increase in the proportion of tumor-infiltrating CD8+ T cells expressing granzyme B (GrzB; 62% increase, P = .008 and 68% increase, P = .006 vs control, respectively; Figure 6D). Effector CD4+ T-cell activation was also greater with the elotuzumab-g2a/anti–PD-1 combination than with control treatment (supplemental Figure 7). Similar changes in tumor-infiltrating CD8+ T-cell phenotypes were observed in the EG7-hSLAMF7 model (supplemental Figure 8).
Figure 6.

Elotuzumab-g2a/anti–PD-1 combination promotes infiltration and effector function of CD8+T cells. (A-E) A20-hSLAMF7–bearing mice were treated with control, 3 mg/kg anti–PD-1, 10 mg/kg elotuzumab-g2a, or elotuzumab-g2a plus anti–PD-1. Tumor-infiltrating and splenic CD8+ T cells were analyzed by flow cytometry (gated as live, GFP−CD19−CD3+CD8+). Quantification of (A) CD107a, (B) CD69, (C) PD-1, and (D) intracellular GrzB tumor-infiltrating and splenic CD8+ T cells as percentages of total CD8+ T cells isolated 10 to 14 days after the start of treatment. Results are a composite of 3 independent experiments with a total of 8 to 18 mice per group. (E) EG7-hSLAMF7–bearing mice were treated with control, 10 mg/kg elotuzumab-g2a, 10 mg/kg anti–PD-1, or elotuzumab-g2a plus anti–PD-1, injected intraperitoneally. Tumor-infiltrating and splenic OVA-tet+ CD8+ T cells were analyzed by flow cytometry (gated as live, GFP−CD19−CD3+CD8+OVA-tet+) and enumerated. Results are shown as mean ± SEM and are from a representative experiment with 7 to 8 mice per group. Statistical analyses were performed by using ANCOVA (A-D) and unpaired Student t test (E). *P < .05; **P < .01; ***P < .0001.
To determine treatment effects on tumor-specific T-cell responses, we enumerated tumor-infiltrating and splenic OVA-specific CD8+ T cells from EG7-hSLAMF7–bearing mice. The OVA-tet+CD8+ T-cell population was increased 3.6-fold and 3.5-fold in tumors in the anti–PD-1 and elotuzumab-g2a/anti–PD-1 combination groups, respectively, vs control (Figure 6E). Together, these data suggest that the elotuzumab-g2a/anti–PD-1 combination enhanced CD8+ T-cell infiltration and effector function.
Discussion
Combined antibody regimens that stimulate both innate and adaptive immunity may afford greater treatment benefits than those that primarily target 1 pathway.12 In this study, we demonstrated that anti-hSLAMF7 (elotuzumab-g2a) and anti–PD-1 mAbs exhibit enhanced antitumor activity when used in combination. Flow cytometric and cytokine analyses of mouse tumors demonstrated that the potency of elotuzumab-g2a/anti–PD-1 treatment may be due to the combined effects of these mAbs on different cell populations within the tumor. Anti–PD-1 treatment, alone or together with elotuzumab-g2a, resulted in increased numbers of tumor-infiltrating T cells expressing high levels of activation markers. Elotuzumab-g2a/anti–PD-1 treatment also promoted NK cell activation and led to the production of inflammatory cytokines IFN-γ and TNF-α in the tumor microenvironment. Our data support the hypothesis that elotuzumab-g2a mediates the initial destruction of tumors by ADCC and promotes NK cell activation. Stimulation of NK cells may lead to both the direct killing of tumor cells and the modulation of other FcγR-expressing cells through the release of chemoattractants (eg, MIP-1α/β) or cytokines (eg, IFN-γ) that promote recruitment and activation of macrophages and/or neutrophils.44,45 Cumulative ADCC/antibody-dependent cellular phagocytosis mediated by FcγR-expressing cells may then lead to enhanced antigen presentation and initiation of downstream CD8+ T-cell–mediated adaptive immune responses. Such a response is likely to be augmented by anti–PD-1 mAbs blocking the effect of PD-1/PD-L1–mediated inhibitory signals on T cells.
Tumor rechallenge experiments demonstrated that mice cured with the elotuzumab-g2a/anti–PD-1 combination generated a memory immune response and subsequently rejected rechallenge with EG7-hSLAMF7 cells. DiLillo et al46 demonstrated that mice primed with an anti-hCD20 mAb and hCD20+ EL4 lymphoma cells reject lymphoma rechallenge with hCD20+ EL4 but not wild-type EL4 cells, suggesting that the T-cell response was directed at the hCD20 neoantigen. Whether the elotuzumab-g2a–mediated immune response was solely directed at the hSLAMF7 antigen or whether the combination with the PD-1 blockade enhanced both epitope spreading and antineoantigen T-cell responses remains unknown. Memory NK cells have also been described47 and additional studies are needed to evaluate their role in the generation of immune memory to the elotuzumab-g2a/anti–PD-1 combination.
Notably, anti–PD-1 also appears to synergize with elotuzumab-g2a–mediated activation of NK cells: an activating effect of PD-1 blockade is supported by previous studies demonstrating the expression of PD-1 on NK cells in MM.35,36 Additional studies are needed to clarify the capacity of PD-1 to signal in NK cells and to explore the effects of this pathway on NK cell responses. In this study, T-cell activation appeared to be driven primarily by anti–PD-1. Activation of T cells by anti–PD-1 is consistent with previous reports that T cells from patients with MM express high levels of PD-1.35 Although it is not yet clear whether T cells in patients with MM have fully “exhausted” or senescent phenotypes,48,49 these data highlight the importance of additional mechanistic studies of PD-1 blockade in MM.
We noted several differences between the EG7-SLAMF7 and A20-SLAMF7 tumor models. Although the elotuzumab-g2a/anti–PD-1 combination was highly efficacious in both models, the A20-hSLAMF7 model was more responsive to elotuzumab-g2a and anti–PD-1 both as single and combined agents. These results may be due to differences in the tumor microenvironment of each model, higher immunogenicity of A20-SLAMF7 tumors in BALB/c mice, or differences in the expression of inhibitory or costimulatory molecules and their ligands between the models. We found fewer NK cells and CD4+FoxP3− effector cells and an increased frequency of CD11b+GR1+ myeloid-derived suppressor cells in EG7-hSLAMF7 tumors. We also observed higher PD-1 expression on lymphocyte subsets (particularly NK cells) in the EG7-hSLAMF7 vs A20-hSLAMF7 models.
We also noted several differences between our observations and current literature. Upregulation of TNF family receptors on human NK cells has been reported following ADCC.50 However, we found no significant change in CD137, GITR, or OX40 surface expression on mouse tumor-infiltrating NK cells after exposure to elotuzumab-g2a (data not shown). We did observe increased GrzB expression by CD8+ T cells after treatment with elotuzumab-g2a, but not after anti–PD-1 treatment. This contrasts with previous studies in which increased GrzB expression was observed on tumor CD8+ T cells from anti–PD-1–treated patients with metastatic melanoma.51
In addition to mediating ADCC, elotuzumab directly activates NK cells by engaging hSLAMF7.26 Elotuzumab, however, does not bind mouse SLAMF7 and, as such, any direct effects of elotuzumab through SLAMF7 on SLAMF7-expressing mouse NK or other SLAMF7-expressing leukocytes could not be captured in this study. Future studies that use knock-in mice expressing hSLAMF7 throughout the hematopoietic compartment, or that use elotuzumab surrogate antibodies that can bind mouse SLAMF7, will be helpful in establishing the contribution of SLAMF7 activation to elotuzumab-mediated antitumor activity.
Acknowledgments
The authors thank the Bristol-Myers Squibb Animal Facility and MuriGenics, where the tumor studies were conducted. The authors also thank Mary Ellen Cvijic, Elizabeth Saravia, David Walker, Xiang Shao, and David Nakamura of Bristol-Myers Squibb, Audie Rice, Steve Keller, and James Sheridan of AbbVie for assistance with the studies and data analysis, and John Engelhardt, Nathan Siemers, and Catherine Bolger of Bristol-Myers Squibb for critical reading of the manuscript. Editorial assistance was provided by Matthew Thomas of Caudex (Oxford, United Kingdom).
This work was supported by Bristol-Myers Squibb.
Supplementary Material
The full-text version of this article contains a data supplement.
Authorship
Contribution: N.A.B., A.J.K., M.D.R., and R.F.G. conceived and designed the studies; N.A.B., A.J., T.B., M.M., P.J.N., M.D.R., and R.F.G. developed the methodology; N.A.B., A.J., A.Y.K., T.B., M.M., M.R.J., A.J.R., P.J.N., A.J.K., M.D.R., and R.F.G. analyzed and interpreted the data; N.A.B., A.J.K., M.D.R., and R.F.G. wrote, reviewed, and revised the manuscript; and K.H. generated critical reagents.
Conflict-of-interest disclosure: N.A.B., K.H., A.J.K., M.D.R., and R.F.G. are employees of and/or have ownership interest in Bristol-Myers Squibb. A.J. and A.Y.K. are employees of Bristol-Myers Squibb. T.B. and M.M. were employees of Bristol-Myers Squibb at the time of the study. P.J.N. received funding from Bristol-Myers Squibb to perform aspects of this study. The remaining authors declare no competing financial interests.
Correspondence: Natalie A. Bezman, Bristol-Myers Squibb, 700 Bay Rd, Redwood City, CA 94063; e-mail: natalie.bezman@bms.com.
References
- 1.Weiner LM, Dhodapkar MV, Ferrone S. Monoclonal antibodies for cancer immunotherapy. Lancet. 2009;373(9668):1033-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28(28):4390-4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6(4):443-446. [DOI] [PubMed] [Google Scholar]
- 4.Taylor RP, Lindorfer MA. Immunotherapeutic mechanisms of anti-CD20 monoclonal antibodies. Curr Opin Immunol. 2008;20(4):444-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchida J, Hamaguchi Y, Oliver JA, et al. . The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med. 2004;199(12):1659-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Albanesi M, Mancardi DA, Jönsson F, et al. . Neutrophils mediate antibody-induced antitumor effects in mice. Blood. 2013;122(18):3160-3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowles JA, Weiner GJ. CD16 polymorphisms and NK activation induced by monoclonal antibody-coated target cells. J Immunol Methods. 2005;304(1-2):88-99. [DOI] [PubMed] [Google Scholar]
- 8.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21(21):3940-3947. [DOI] [PubMed] [Google Scholar]
- 9.Cartron G, Dacheux L, Salles G, et al. . Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99(3):754-758. [DOI] [PubMed] [Google Scholar]
- 10.Jakubowiak A, Offidani M, Pégourie B, et al. . Randomized phase 2 study: elotuzumab plus bortezomib/dexamethasone vs bortezomib/dexamethasone for relapsed/refractory MM. Blood. 2016;127(23):2833-2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dyall R, Vasovic LV, Clynes RA, Nikolić-Zugić J. Cellular requirements for the monoclonal antibody-mediated eradication of an established solid tumor. Eur J Immunol. 1999;29(1):30-37. [DOI] [PubMed] [Google Scholar]
- 12.Stagg J, Loi S, Divisekera U, et al. . Anti-ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti-PD-1 or anti-CD137 mAb therapy. Proc Natl Acad Sci USA. 2011;108(17):7142-7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park S, Jiang Z, Mortenson ED, et al. . The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell. 2010;18(2):160-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abès R, Gélizé E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood. 2010;116(6):926-934. [DOI] [PubMed] [Google Scholar]
- 15.Vasović LV, Dyall R, Clynes RA, Ravetch JV, Nikolić-Zugic J. Synergy between an antibody and CD8+ cells in eliminating an established tumor. Eur J Immunol. 1997;27(2):374-382. [DOI] [PubMed] [Google Scholar]
- 16.Hsi ED, Steinle R, Balasa B, et al. . CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14(9):2775-2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tai YT, Dillon M, Song W, et al. . Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112(4):1329-1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veillette A, Guo H. CS1, a SLAM family receptor involved in immune regulation, is a therapeutic target in multiple myeloma. Crit Rev Oncol Hematol. 2013;88(1):168-177. [DOI] [PubMed] [Google Scholar]
- 19.Bristol-Myers Squibb. Empliciti (elotuzumab) prescribing information. Available at: http://packageinserts.bms.com/pi/pi_empliciti.pdf. Accessed 5 May 2016.
- 20.Lonial S, Dimopoulos M, Palumbo A, et al. ; ELOQUENT-2 Investigators. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373(7):621-631. [DOI] [PubMed] [Google Scholar]
- 21.European Medicines Agency. Elotuzumab: summary of product characteristics. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003967/WC500206673.pdf. Accessed 20 June 2016.
- 22.National Institute of Pharmaceutical Medical Devices. New drugs approved in May 2016. Available at: http://www.pmda.go.jp/files/000216006.pdf. Accessed 8 March 2017.
- 23.van Rhee F, Szmania SM, Dillon M, et al. . Combinatorial efficacy of anti-CS1 monoclonal antibody elotuzumab (HuLuc63) and bortezomib against multiple myeloma. Mol Cancer Ther. 2009;8(9):2616-2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sola C, Blery M, Bonnafous C, et al. . Lirilumab enhances anti-tumor efficacy of elotuzumab [abstract]. Blood. 2014;124(21). Abstract 4711. [Google Scholar]
- 25.Robbins M, Jure-Kunkel M, Dito G, Andre P, Zhang H, Bezman N. Effects of IL-21, KIR blockade, and CD137 agonism on the non-clinical activity of elotuzumab [abstract]. Blood. 2014;124(21). Abstract 4717. [Google Scholar]
- 26.Collins SM, Bakan CE, Swartzel GD, et al. . Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother. 2013;62(12):1841-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouchon A, Cella M, Grierson HL, Cohen JI, Colonna M. Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J Immunol. 2001;167(10):5517-5521. [DOI] [PubMed] [Google Scholar]
- 28.Cruz-Munoz ME, Dong Z, Shi X, Zhang S, Veillette A. Influence of CRACC, a SLAM family receptor coupled to the adaptor EAT-2, on natural killer cell function. Nat Immunol. 2009;10(3):297-305. [DOI] [PubMed] [Google Scholar]
- 29.Kumaresan PR, Lai WC, Chuang SS, Bennett M, Mathew PA. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol Immunol. 2002;39(1-2):1-8. [DOI] [PubMed] [Google Scholar]
- 30.Guo H, Cruz-Munoz ME, Wu N, Robbins M, Veillette A. Immune cell inhibition by SLAMF7 is mediated by a mechanism requiring src kinases, CD45, and SHIP-1 that is defective in multiple myeloma cells. Mol Cell Biol. 2015;35(1):41-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohrt HE, Houot R, Marabelle A, et al. . Combination strategies to enhance antitumor ADCC. Immunotherapy. 2012;4(5):511-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDermott DF, Atkins MB. PD-1 as a potential target in cancer therapy. Cancer Med. 2013;2(5):662-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173(2):945-954. [DOI] [PubMed] [Google Scholar]
- 34.Dong H, Strome SE, Salomao DR, et al. . Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793-800. [DOI] [PubMed] [Google Scholar]
- 35.Benson DM Jr, Bakan CE, Mishra A, et al. . The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116(13):2286-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Görgün G, Samur MK, Cowens KB, et al. . Lenalidomide enhances immune checkpoint blockade-induced immune response in multiple myeloma. Clin Cancer Res. 2015;21(20):4607-4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paiva B, Azpilikueta A, Puig N, et al. . PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia. 2015;29(10):2110-2113. [DOI] [PubMed] [Google Scholar]
- 38.Chung DJ, Pronschinske KB, Shyer JA, et al. . T-cell exhaustion in multiple myeloma relapse after autotransplant: optimal timing of immunotherapy. Cancer Immunol Res. 2016;4(1):61-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesokhin AM, Ansell SM, Armand P, et al. . Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34(23):2698-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson PG, Jagannath S, Moreau P, et al. ; 1703 study investigators. Elotuzumab in combination with lenalidomide and dexamethasone in patients with relapsed multiple myeloma: final phase 2 results from the randomised, open-label, phase 1b-2 dose-escalation study. Lancet Haematol. 2015;2(12):e516-e527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li B, VanRoey M, Wang C, Chen TH, Korman A, Jooss K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor--secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res. 2009;15(5):1623-1634. [DOI] [PubMed] [Google Scholar]
- 42.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis [published correction appears in Cancer Cell. 2015;28(4):543]. Cancer Cell. 2015;28(3):285-295. [DOI] [PubMed] [Google Scholar]
- 43.Balasa B, Yun R, Belmar NA, et al. . Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol Immunother. 2015;64(1):61-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelletier M, Micheletti A, Cassatella MA. Modulation of human neutrophil survival and antigen expression by activated CD4+ and CD8+ T cells. J Leukoc Biol. 2010;88(6):1163-1170. [DOI] [PubMed] [Google Scholar]
- 45.Robertson MJ. Role of chemokines in the biology of natural killer cells. J Leukoc Biol. 2002;71(2):173-183. [PubMed] [Google Scholar]
- 46.DiLillo DJ, Ravetch JV. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell. 2015;161(5):1035-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol. 2016;16(2):112-123. [DOI] [PubMed] [Google Scholar]
- 48.Suen H, Brown R, Yang S, et al. . Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia. 2016;30(8):1716-1724. [DOI] [PubMed] [Google Scholar]
- 49.Hallett WH, Jing W, Drobyski WR, Johnson BD. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol Blood Marrow Transplant. 2011;17(8):1133-1145. [DOI] [PubMed] [Google Scholar]
- 50.Kohrt HE, Houot R, Goldstein MJ, et al. . CD137 stimulation enhances the antilymphoma activity of anti-CD20 antibodies. Blood. 2011;117(8):2423-2432. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Tumeh PC, Harview CL, Yearley JH, et al. . PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.