

Key Points
First study of cognitive functioning in adult patients with SCD genotypes other than homozygous for hemoglobin S.
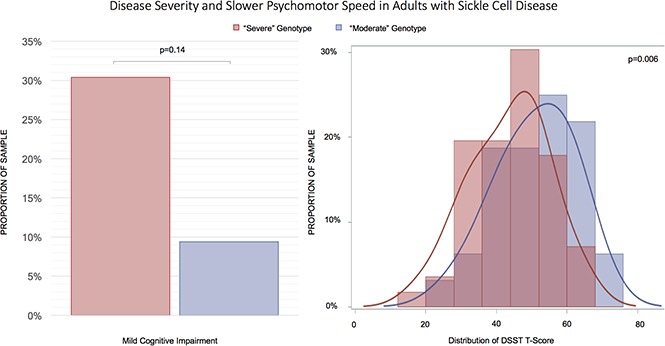
Patients with “severe” genotypes had significantly poorer speed of processing when compared with those with “moderate” genotypes.
Abstract
Psychomotor slowing is common in children with sickle cell disease (SCD), but little is known about its severity in adults. We conducted a cross-sectional study to quantify psychomotor speed, measured with the digit symbol substitution test (DSST), in relationship with disease severity in adults with SCD attending an outpatient clinic (n = 88, age 36.3 years). Genotype was used to group patients in “severe” (homozygous for hemoglobin S or compound heterozygous with β0 thalassemia) or “moderate” groups (compound heterozygous for HbS, with either HbC or β+ thalassemia). Analyses were repeated after exclusion of patients with a history of stroke (n = 11). Mild impairment in processing speed was detectable in both the “severe” and the “moderate” group (30% and 9%, respectively; age-adjusted P = .14). Compared with the “moderate” group, those in the “severe” group had significantly lower standardized DSST scores (P = .004), independent of adjustment for factors that differed between the groups: hemoglobin, ferritin, hydroxyurea use, blood pressure parameters, and stroke history. Results were similar after excluding patients with stroke. Psychomotor slowing in SCD differs in relationship to genotype; this difference appears unrelated to history of stroke or severity of anemia and other risk factors examined cross-sectionally. Although less prevalent, mild cognitive impairment was also detectable in patients with a less severe genotype. Longitudinal studies of SCD should include all diseases genotypes and examine factors that would reduce the risk of slow processing speed and perhaps more general cognitive impairment in each subgroup.
Visual Abstract
Introduction
Sickle cell disease (SCD) is a term used to define syndromes characterized by the inheritance of a mutated hemoglobin (HbS) that polymerizes under hypoxic conditions leading to dysfunctional and abnormally shaped (sickled) red blood cells. Both patients with homozygous HbS (HbSS disease or sickle-cell anemia) and patients with HbS coinherited with other hemoglobin mutations (HbC [HbSC], D, O, E, and β-thalassemia [HbS/β thalassemia]) suffer from red blood cell sickling and its downstream effects.1 On an organ level, ischemia and infarction are common and lead to loss of function. The central nervous system is highly affected in SCD, with young children with HbSS disease suffering from a very high prevalence of overt and silent ischemic stroke and all patients from a rising, high incidence of both ischemic and hemorrhagic stroke as they age.2-5 Psychomotor slowing, an indicator of brain dysfunction and cognitive impairment, has been amply documented in children with SCD,6-9 but there are limited data on the prevalence of this complication in adult patients. Although the primary risk factor for psychomotor slowing is stroke,10 there has been mounting evidence that cognitive impairment also occurs in patients without a history of overt or silent stroke.11-13 Risk factors for cognitive impairment in patients with SCD without stroke are, however, not completely known,14 particularly in relationship to the SCD genotype. To date, there is only 1 major study characterizing cognitive impairment in adult patients with SCD in the absence of stroke. It was conducted on a selected cohort of 149 patients with HbSS disease, without a history of recent transfusion or major chronic complications including liver and kidney disease. The results of this study showed that cognitive impairment was common, predominantly affected psychomotor speed, and was associated with anemia in the older patients.13 Studies of cognition in other SCD genotypes are needed, particularly because approximately one-third of patients with SCD in the United States and millions worldwide have SCD genotypes other than HbSS.15 As care for SCD improves, a larger number of patients are surviving to an older age16,17 and may be exposed to currently unknown risk factors of cognitive impairment. Thus, it is extremely important to identify modifiable risk factors to prevent the onset or slow the progression of cognitive impairment within this population.
The objective of this analysis was to assess psychomotor speed in relationship to the SCD genotype. Because little is known about genotypic differences in cognition, we examined several factors related to SCD severity and determined whether any of these factors would attenuate the association between genotype and psychomotor speed in our cohort. We hypothesized that if differences in cognition by genotype were seen, they could be explained, at least partly, by differences in demographic, disease-related, or treatment factors between the 2 groups.
Methods
Study population
Patients with SCD were recruited from the University of Pittsburgh Medical Center Adult Sickle Cell Program outpatient clinic under the University of Pittsburgh Institutional Review Board protocols PRO12040139 and PRO08110422. All patients with HbSS, HbSC, and HbS/β, the 3 most prevalent genotypes of SCD, older than 18 and able to provide informed consent were informed about the study by staff members during their routine clinic visit and offered entry into the study if they were in steady-state SCD.18 Eligibility criteria also included whether they were (1) English speaking and (2) currently receiving routine follow-up care at the University of Pittsburgh Medical Center Adult Sickle Cell Program. Exclusion criteria included the following: (1) pregnancy as determined by a positive urine human chorionic gonadotropin test at the time of informed consent and (2) acute medical problem including acute vaso-occlusive crisis. Of 182 patients who visited the clinic during this time, 118 were interviewed, 113 provided informed consent, and 88 completed testing. Four were ineligible after providing consent, 2 refused to consent, 1 died before completing testing, and 18 were unable to schedule testing during the study period. Upon obtaining informed consent, patients received a blood draw and the digit symbol substitution test (DSST). For the analysis, patients with HbSS disease were clustered with patients with HbS/β0 thalassemia (n = 56 for both genotypes combined) as these syndromes share similar disease features and patients with HbSC disease with patients with HbS/β+ thalassemia (n = 32 for both genotypes combined). For brevity, SCD genotypes were defined as “severe” (HbSS and HbS/β0 thalassemia) and “moderate” (HbSC and HbS/β+ thalassemia).
DSST
The DSST is a pencil and paper test of information processing speed.19 In this decoding test, participants are given 90 seconds to match and write as many digits to symbols as possible from a key consisting of the correct digit-symbol pairs. Scores reflect the number of correct digit-symbol matches made in 90 seconds, such that higher scores indicate higher function. The digit-symbol matching task assesses encoding, retrieval, visuomotor coordination and speed, learning and memory, and selective attention and thus was chosen as the primary outcome of interest, as cognitive impairment among patients with SCD affects primarily working memory and psychomotor speed.13 Furthermore, the DSST does not suffer from a ceiling effect and can identify changes at the highest levels of cognition.20 The raw DSST scores were standardized based on age, race, sex, and education-based norms21; analyzed as a continuous variable; and will be referred to as standardized DSST scores. Standardized DSST scores were used to define mild cognitive impairment (MCI), defined as ≤1.5 standard deviations (SDs) below the DSST T-score (T-scores had a mean of 50 and SD of 10). In our sample, a T-score of 40 was 1 SD below the mean (17th percentile); a T-score of 35 was 1.5 SDs below the mean (10th percentile).21
Other variables of interest
Variables related to severity of SCD genotype and factors known to be related to cognitive function8,9,22 were considered for inclusion in the analyses. Self-reported participant age, race, sex, and completed level of education were recorded. Blood pressure was collected by trained clinic personnel at the time of cognitive testing and later abstracted from the participant’s medical records. Mean arterial pressure (MAP) was calculated from diastolic blood pressure (DBP) and systolic blood pressure (SBP) values: MAP = [(2 × DBP) + SBP]/3. Blood samples were used to determine a variety of clinical measures of interest, including serum creatinine (mg/dL), reticulocyte count, albumin (g/dL), lactate dehydrogenase (LDH, IU/L), ferritin (ng/mL), white blood cell (WBC) count, and platelet count. Hydroxyurea and opiate use history were collected because use of these medications may differ between genotypes and potentially be associated with cognitive function.22,23 The variables were defined as holding a current, active prescription for hydroxyurea and any opiate, as determined at the time of testing, by abstracting prescription records from the patients’ medical records. History of stroke, silent cerebral infarct (SCI) diagnosed by magnetic resonance imaging (MRI), and transfusion within the last 90 days were abstracted from the patients’ medical records. In addition, a study neuroradiologist reviewed any clinically available brain MRIs for all patients participating in the study. The neuroradiologist’s read superseded medical history in cases of discrepancy as follows: patients with cerebral infarct by MRI were categorized as having a history of stroke, whereas those with findings of small vessel disease were categorized as having a history of SCI. In one case, the MRI of the brain of a patient with a history of stroke was not available for review; that participant was included in the history of stroke group.
Statistical analyses
Descriptive statistics were calculated first for the entire analytic sample, and then by genotype, comparing the “severe” with “moderate” group to help identify potential confounders of the association between genotype and standardized DSST. Linear regression was used to assess the association between genotype and continuous demographic and disease-related characteristics. Logistic regression was used to assess the associations between genotype and categorical disease-related characteristics. All models were adjusted for age, except the model of age and genotype, and standardized DSST with genotype. Linear regression models were then used to assess the relationship between standardized DSST and genotype, individually adjusted by potential confounders that were associated with genotype severity as identified in the previous descriptive analyses, followed by a model fully adjusted for all potential confounders. We adjusted for individual factors first because we sought to understand the influence of each variable on the association between DSST and genotype. All assumptions of normality and independence of predictors were met. As some covariates were not collected for all 88 participants, and given the lack of prior research in this field, we ran each model with the maximum sample size possible. We repeated these methods for those without a history of stroke. Additionally, we conducted sensitivity analyses fitting these models among those people who had complete data on all variables in order to directly compare the model results. All analyses were conducted with SAS, version 9.4 (SAS Institute Inc., Cary, NC).
Results
Main characteristics of the study participants
A greater proportion of participants in this study had a “severe” genotype (63.6%) as opposed to a “moderate” genotype (36.4%). Because patients with a “severe” genotype were younger compared with those in the “moderate” group (33.7 years old vs 40.9 years old, P = .006), all between-group comparisons were adjusted for age. There were no significant differences in education (13.1 vs 13.2 years, P = .78) or the proportion of males (37.5% male vs 37.5% male, P = .97) between the genotype groups. When compared with participants with “moderate” genotype, participants with “severe” genotype had lower hemoglobin levels (9.2 g/dL vs 11.5 g/dL, P < .0001), higher ferritin (1141.6 ng/mL vs 403.4 ng/mL, P = .02), lower DBP (68.8 mm/Hg vs 73.5 mm/Hg, P = .02) and MAP (83.1 mm/Hg vs 88.6 mm/Hg, P = .01), and a higher proportion of hydroxyurea use (57.1% vs 31.2%, P = .04). There was not a significant difference in platelet count, LDH, SBP, oxygen saturation, WBC count, reticulocyte count, creatinine, opiate use, and history of transfusion, stroke, or SCI (based on MRI) between the groups (Table 1). Although there was a higher proportion of patients with MCI in the “severe” group (30.4% vs 9.4%), the difference was not statistically significant (P = .14).
Table 1.
Characteristics of the entire analytic sample and by genotype severity
| Entire analytic sample | “Severe” genotype | “Moderate” genotype | P* | |
|---|---|---|---|---|
| Mean (SD)* (n = 88) | Mean (SD)* (n = 56) | Mean (SD)* (n = 32) | ||
| Age, y | 36.3 (12.0) | 33.7 (10.8) | 40.9 (12.8) | .006 |
| Male sex, n (%)* | 33 (37.5) | 21 (37.5) | 12 (37.5) | .97 |
| Education, y | 13.1 (1.7) | 13.1 (1.8) | 13.2 (1.7) | .78 |
| MCI, n (%) | 20 (22.7) | 17 (30.4) | 3 (9.4) | .14 |
| DSST T-score | 46.3 (11.7) | 47.6 (14.5) | 51.0 (13.4) | .004 |
| O2 saturation, % | 97.8 (1.8) | 97.5 (1.8) | 98.1 (1.7) | .15 |
| WBC count, ×109/L | 9.5 (3.7) | 9.7 (3.8) | 9.2 (3.7) | .86 |
| Hemoglobin, g/dL | 10.0 (1.9) | 9.2 (1.5) | 11.5 (1.5) | <.0001 |
| Platelet count, ×109/L | 314.0 (162.8) | 344.1 (179.8) | 263.3 (115.1) | .17 |
| Reticulocytes, % | 1.5 (3.4) | 1.7 (4.0) | 1.1 (1.9) | .49 |
| LDH, IU/L | 302.8 (146.0) | 321.2 (142.3) | 269.2 (149.1) | .16 |
| Ferritin, ng/mL | 892.6 (1664.3) | 1141.6 (1864.4) | 403.4 (1042.2) | .02 |
| Creatinine, mg/dL | 0.7 (0.2) | 0.7 (0.3) | 0.8 (0.2) | .91 |
| SBP, mm/Hg | 114.2 (13.9) | 111.3 (13.4) | 118.9 (13.6) | .08 |
| DBP, mm/Hg | 70.6 (8.8) | 68.8 (7.7) | 73.5 (9.8) | .02 |
| MAP, mm/Hg | 85.2 (9.4) | 83.1 (8.4) | 88.6 (10.0) | .01 |
| Hydroxyurea use, n (%) | 42 (47.7) | 32 (57.1) | 10 (31.2) | .04 |
| Opiate use, n (%) | 25 (28.4) | 15 (26.8) | 10 (31.2) | .57 |
| Transfusion history, n (%) | 22 (25.9) | 17 (31.5) | 5 (16.1) | .15 |
| Stroke history, n (%) | 11 (12.5) | 9 (16.1) | 2 (6.3) | .13 |
| SCI history, n (%) | 4 (4.5) | 3 (5.4) | 1 (3.1) | .65 |
Age-adjusted P value calculated with linear regression for continuous variables and logistic regression for categorical variables; the models with age were unadjusted, and the models with standardized DSST T-score were unadjusted. The bold font indicates P < .05.
Associations with DSST
Participants with a “severe” genotype had lower DSST scores (47.6 vs 51.0, P = .004), indicating poorer psychomotor speed (Table 1); after excluding the 11 patients with stroke, participants with a “severe” genotype still had lower standardized DSST scores, compared with those with a “moderate” genotype (45.1 ± 10.6 vs 52.1 ± 10.2, P = .01). Adjustment for hemoglobin, ferritin, DBP, MAP, and history of taking hydroxyurea did not attenuate the association between genotype severity and DSST (Table 2). Results were similar when excluding the patients with stroke (supplemental Tables 1 and 2). Sensitivity analyses excluding participants with a history of stroke, and analyses limited to the sample of participants with complete data for all models, yielded similar results (supplemental Table 3).
Table 2.
Association of genotype and DSST T-score, adjusted for factors that were associated with genotype severity
| Covariates (X) | Model adjusted for X | VIF* |
|---|---|---|
| β (SE); P | ||
| Unadjusted | −7.42 (2.48); .004 | 1.00 |
| Hemoglobin | −7.10 (3.05); .02 | 1.50 |
| Ferritin | −7.62 (2.67); .006 | 1.05 |
| DBP | −7.97 (2.63); .003 | 1.07 |
| MAP | −8.14 (2.68); .003 | 1.09 |
| Hydroxyurea use | −7.54 (2.58); .004 | 1.07 |
| Stroke/SCI history | −6.63 (2.42); .008 | 1.02 |
| Fully adjusted† | −8.70 (3.47); .01 | 7.76 |
DSST T-scores are adjusted for age, race, sex, and education. The β and P values are for the variable “genotype” (severe vs moderate-severe), for the model of “genotype” predicting DSST score, adjusted for the covariate reported in the corresponding row of the table.
SE, standard error.
VIF indicates variance inflation factor of genotype in unadjusted and adjusted models.
Fully adjusted model includes genotype, hemoglobin, ferritin, DBP, MAP, hydroxyurea use, and stroke/SCI history as predictors.
Discussion
To our knowledge, this is the first study to examine cognitive performance in compound heterozygotes (HbSC and HbS/β0 thalassemia). We found differences in psychomotor speed measured by the DSST between patients classified based on genotype. This difference is remarkable, considering the relatively small sample size and the finding that the “moderate” group was nearly 10 years older than the “severe” group. Processing speed decreases with age in both normal adults and other populations, and older age was found to be associated with worse cognitive function in a subanalysis of the largest study of cognitive function in adult patients with SCD.13 Thus, our results suggest genotype is a stronger risk factor for poorer cognitive function than age in our cohort. This is important because although cognitive function has been well characterized in children with SCD,8,24 few studies have examined factors that influence cognitive function during adulthood13,25 or in milder forms of the disease.
In the entire analytic sample, ∼22% of participants met the criteria for MCI as they performed at or below the 10th percentile of the general population (accounting for age, education, race, and gender). This finding is of potential clinical relevance; information processing slowing negatively impacts numerous functional outcomes and disease management, and hence impairment in this domain deserves immediate attention.26 Importantly, poor disease self-management and medication adherence are major concerns in SCD27-29 and could be ameliorated via interventions targeting information processing and executive function domains. The 9% prevalence of MCI in the “moderate” group, although lower than the 30% observed in the “severe” group, is also notable. Patients with a moderate genotype are generally considered at lower risk of vascular complications (eg, pediatric stroke, kidney disease, and pulmonary hypertension) and have a longer life expectancy as compared with those in the “severe” group.30 Based on our findings, however, these patients are likely to experience accelerated cognitive decline with older chronological age and may benefit from cognitive impairment screening early in life.
We then explored whether factors that have been associated with other disease complications in SCD could individually explain the difference in cognitive function between the 2 groups. In particular, we focused on the factors that were significantly different between the 2 groups in bivariate comparisons (Table 1). We found that the 2 groups differed significantly with respect to demographic and laboratory characteristics and clinical factors that are expected to impact cognitive function. The association of genotype with DSST, however, remained statistically significant after individual adjustment for hemoglobin, ferritin, LDH, blood pressure parameters (DBP and MAP), history of taking hydroxyurea or stroke (P < .05), and a model adjusted for all of these factors collectively (P < .05), suggesting that the impact of genotype is independent of these potential risk factors. Although the number of patients with a history of stroke was not significantly different between the groups, there were, as expected, a higher number of patients with stroke in the “severe” genotype group. We found, however, that the results of our analyses did not differ significantly when we excluded patients with stroke (supplemental Tables 1 and 2). It was also surprising that adjusting for this complication did not attenuate the difference in DSST, considering its strong association with decreased cognitive function in other studies (Table 2; supplemental Table 2). It is possible that patients with “severe” SCD may also have a higher burden of cerebrovascular disease not detectable by conventional MRI methods but associated with cognitive impairment.31
Establishing trajectories of cognitive performance is important to understanding how SCD may impact cognitive function with aging. Differences in cognition between patients with SCD and control groups are apparent at an early age.9,32 Additionally, it was found that general domains of attention and executive functions had the largest impairments in pediatric populations.9 These differences in cognitive function in patients with SCD compared with controls are thought to increase with age,9,33 but a lack of prospective cognitive evaluations in patients with SCD makes it difficult to predict the trajectories of cognitive decline within this population. Studies examining relationships between SCD and several measures of cognitive function in older populations have yielded mixed results, some finding no difference compared with controls,34,35 and others finding poorer processing speed,13,36 worse memory,13 and lower IQ and perceptual organization.13 Thus, the question of whether cognitive function deteriorates with aging in SCD remains unanswered.
Processing speed is of particular interest given that deficits in this area have been previously associated with increased markers of cerebrovascular damage and may represent evidence of accelerated brain damage in populations with SCD.35,37 Furthermore, deficits in processing speed are also detectable in pediatric populations across multiple studies and similarly correlate with the burden of cerebrovascular damage.8,9,38 Thus, it can be hypothesized that this domain may be particularly susceptible to brain aging in SCD, although direct comparisons between pediatric and adult populations are complicated by the heterogeneity of the study populations and tests used. Interestingly, Crawford and Jonassaint found that statistically adjusting for differences in processing speed attenuated other deficits in cognitive performance in SCD.36 These results suggest that processing speed may be impacted to a larger degree than other cognitive domains and that it may represent the “core” cognitive impairment in SCD, leading to a downstream effect in other cognitive processes. Here we found a significant difference in processing speed between the “moderate” and “severe” genotypes in fully adjusted models that was not explained by history of stroke/SCI. Unfortunately, the results cannot be directly compared with those of Vichinsky et al13 because comparison groups differed between these 2 studies; however, both studies support the notion that this deficit also occurs in the absence of ischemic lesions detectable by conventional MRI methods. In order to determine the impact of age, future studies should explore cognitive function prospectively, with a particular emphasis on processing speed, in patients with SCD followed from childhood into adulthood so that trajectories can be determined. Standardized scores along with raw or comparison scores should be presented.
Our study has several limitations. Although we explored the role of the most important factors responsible for disease severity in SCD, our relatively small sample size did not permit us to conduct an exhaustive analysis of the multitude of genetic, epigenetic, and environmental factors that affect the SCD phenotype and could have an impact on cognitive function. This variability could only be comprehensively assessed in a much larger epidemiological study. Furthermore, the cross-sectional design did not allow us to assess how longitudinal changes in risk factors could affect cognitive function. Finally, our study is limited by its focus on 1 specific cognitive task assessed by the DSST. Although this test is particularly advantageous in SCD because it has good test-retest variability and is suitable for longitudinal testing, is easy and quick to administer, is sensitive to MCI, and probes one domain preferentially affected in patients with SCD, further studies should more thoroughly explore differences in other neurocognitive domains.
This was the first study of cognitive functioning in adult patients with SCD genotypes other than HbSS; future longitudinal studies should assess the risk factors for cognitive impairment in these patients so that preventive and therapeutic strategies based on surveillance and early detection may be developed.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors would like to provide a special recognition to the individuals who participated in this study.
This work was supported by a grant from the National Heart, Lung and Blood Institute, National Institutes of Health (1R01HL127107-01A1) (E.M.N.) and by a Pittsburgh Heart, Lung and Blood Vascular Medicine Institute P3HVBI Award (C.R.).
Authorship
Contribution: D.R.J. collected and analyzed data and wrote the manuscript; A.M. analyzed data and edited the manuscript; M.A.B. contributed important intellectual insight into data interpretation and edited the manuscript; J.M.M. interpreted the MRIs of the participants and edited the manuscript; C.R. designed the study and edited the manuscript; and E.M.N. designed the study, directed the project, and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Enrico M. Novelli, UPMC Adult Sickle Cell Program, Division of Hematology/Oncology, University of Pittsburgh, 200 Lothrop St, BST E1240, Pittsburgh, PA 15261; e-mail: novellie@upmc.edu.
References
- 1.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644. [DOI] [PubMed] [Google Scholar]
- 2.Bernaudin F, Verlhac S, Arnaud C, et al. Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood. 2015;125(10):1653-1661. [DOI] [PubMed] [Google Scholar]
- 3.DeBaun MR, Sarnaik SA, Rodeghier MJ, et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood. 2012;119(16):3684-3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwiatkowski JL, Zimmerman RA, Pollock AN, et al. Silent infarcts in young children with sickle cell disease. Br J Haematol. 2009;146(3):300-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91(1):288-294. [PubMed] [Google Scholar]
- 6.Edwards CL, Raynor RD, Feliu M, et al. Neuropsychological assessment, neuroimaging, and neuropsychiatric evaluation in pediatric and adult patients with sickle cell disease (SCD). Neuropsychiatr Dis Treat. 2007;3(6):705-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hariman LM, Griffith ER, Hurtig AL, Keehn MT. Functional outcomes of children with sickle-cell disease affected by stroke. Arch Phys Med Rehabil. 1991;72(7):498-502. [PubMed] [Google Scholar]
- 8.Oluwole OB, Noll RB, Winger DG, Akinyanju O, Novelli EM. Cognitive functioning in children from Nigeria with sickle cell anemia. Pediatr Blood Cancer. 2016;63(11):1990-1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schatz J, Finke RL, Kellett JM, Kramer JH. Cognitive functioning in children with sickle cell disease: a meta-analysis. J Pediatr Psychol. 2002;27(8):739-748. [DOI] [PubMed] [Google Scholar]
- 10.Armstrong FD, Thompson RJ Jr, Wang W, et al. ; Neuropsychology Committee of the Cooperative Study of Sickle Cell Disease. Cognitive functioning and brain magnetic resonance imaging in children with sickle Cell disease. Pediatrics. 1996;97(6, pt 1):864-870. [PubMed] [Google Scholar]
- 11.Noll RB, Stith L, Gartstein MA, et al. Neuropsychological functioning of youths with sickle cell disease: comparison with non-chronically ill peers. J Pediatr Psychol. 2001;26(2):69-78. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann LL, Goodwin GM, Ebmeier KP. The cognitive neuropsychology of depression in the elderly. Psychol Med. 2007;37(12):1693-1702. [DOI] [PubMed] [Google Scholar]
- 13.Vichinsky EP, Neumayr LD, Gold JI, et al. ; Neuropsychological Dysfunction and Neuroimaging Adult Sickle Cell Anemia Study Group. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. 2010;303(18):1823-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeBaun MR, Kirkham FJ. Central nervous system complications and management in sickle cell disease. Blood. 2016;127(7):829-838. [DOI] [PubMed] [Google Scholar]
- 15.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lanzkron S, Carroll CP, Haywood C Jr. Mortality rates and age at death from sickle cell disease: U.S., 1979-2005. Public Health Rep. 2013;128(2):110-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4, suppl):S512-S521. [DOI] [PubMed] [Google Scholar]
- 18.Ballas SK. More definitions in sickle cell disease: steady state v base line data. Am J Hematol. 2012;87(3):338. [DOI] [PubMed] [Google Scholar]
- 19.Wechsler D. WAIS-R Manual: Wechsler Adult Intelligence Scale-Revised. San Antonio, TX: Psychological Corporation;1981. [Google Scholar]
- 20.Proust-Lima C, Amieva H, Dartigues J-F, Jacqmin-Gadda H. Sensitivity of four psychometric tests to measure cognitive changes in brain aging-population-based studies. Am J Epidemiol. 2007;165(3):344-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heaton R, Miller SW, Taylor MJ, Grant I. Revised Comprehensive Norms for an Expanded Halstead-Reitan Battery: Demographically Adjusted Neuropsychological Norms for African American and Caucasian Adults. Lutz, FL: Psychological Assessment Resources;2004. [Google Scholar]
- 22.Puffer E, Schatz J, Roberts CW. The association of oral hydroxyurea therapy with improved cognitive functioning in sickle cell disease. Child Neuropsychol. 2007;13(2):142-154. [DOI] [PubMed] [Google Scholar]
- 23.Merkhofer C, Sylvester S, Zmuda M, et al. The impact of cognitive function on adherence to hydroxyurea therapy in patients with sickle cell disease [abstract]. Blood. 2016;128(22). Abstract 2493. [Google Scholar]
- 24.Schatz J, McClellan CB. Sickle cell disease as a neurodevelopmental disorder. Ment Retard Dev Disabil Res Rev. 2006;12(3):200-207. [DOI] [PubMed] [Google Scholar]
- 25.Jorgensen DR, Rosano C, Novelli EM. Can neuroimaging markers of vascular pathology explain cognitive performance in adults with sickle cell anemia? A review of the literature. Hemoglobin. 2016;40(6):381-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salthouse TA. The role of memory in the age decline in digit-symbol substitution performance. J Gerontol. 1978;33(2):232-238. [DOI] [PubMed] [Google Scholar]
- 27.Baskin ML, Collins MH, Brown F, et al. Psychosocial considerations in sickle cell disease (SCD): the transition from adolescence to young adulthood. J Clin Psychol Med Settings. 1998;5(3):315-341. [Google Scholar]
- 28.Brandow AM, Jirovec DL, Panepinto JA. Hydroxyurea in children with sickle cell disease: practice patterns and barriers to utilization. Am J Hematol. 2010;85(8):611-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makubi A, Sasi P, Ngaeje M, et al. Rationale and design of mDOT-HuA study: a randomized trial to assess the effect of mobile-directly observed therapy on adherence to hydroxyurea in adults with sickle cell anemia in Tanzania. BMC Med Res Methodol. 2016;16(1):140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore). 2005;84(6):363-376. [DOI] [PubMed] [Google Scholar]
- 31.Novelli EM, Elizabeth Sarles C, Jay Aizenstein H, et al. Brain venular pattern by 7T MRI correlates with memory and haemoglobin in sickle cell anaemia. Psychiatry Res. 2015;233(1):18-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hogan AM, Pit-ten Cate IM, Vargha-Khadem F, Prengler M, Kirkham FJ. Physiological correlates of intellectual function in children with sickle cell disease: hypoxaemia, hyperaemia and brain infarction. Dev Sci. 2006;9(4):379-387. [DOI] [PubMed] [Google Scholar]
- 33.Wang W, Enos L, Gallagher D, et al. ; Cooperative Study of Sickle Cell Disease. Neuropsychologic performance in school-aged children with sickle cell disease: a report from the Cooperative Study of Sickle Cell Disease. J Pediatr. 2001;139(3):391-397. [DOI] [PubMed] [Google Scholar]
- 34.Kugler S, Anderson B, Cross D, et al. Abnormal cranial magnetic resonance imaging scans in sickle-cell disease. Neurological correlates and clinical implications. Arch Neurol. 1993;50(6):629-635. [DOI] [PubMed] [Google Scholar]
- 35.van der Land V, Mutsaerts HJ, Engelen M, et al. Risk factor analysis of cerebral white matter hyperintensities in children with sickle cell disease. Br J Haematol. 2016;172(2):274-284. [DOI] [PubMed] [Google Scholar]
- 36.Crawford RD, Jonassaint CR. Adults with sickle cell disease may perform cognitive tests as well as controls when processing speed is taken into account: a preliminary case-control study. J Adv Nurs. 2016;72(6):1409-1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mackin RS, Insel P, Truran D, et al. ; Neuropsychological Dysfunction and Neuroimaging Adult Sickle Cell Anemia Study Group. Neuroimaging abnormalities in adults with sickle cell anemia: associations with cognition. Neurology. 2014;82(10):835-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stotesbury H, Kawadler JM, Koelbel M, Balfour P, Kirkham F. Processing speed index in paediatric sickle cell disease: a systematic review and meta-analysis. In: British Paediatric Neurology Association Annual Meeting; 2017; Cambridge, United Kingdom. Abstract 50. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.