

Metabolomics data from two independent Solanum penellii populations cast light on the genetic robustness of primary and secondary metabolite contents in tomato.
Abstract
To explore the genetic robustness (canalization) of metabolism, we examined the levels of fruit metabolites in multiple harvests of a tomato introgression line (IL) population. The IL partitions the whole genome of the wild species Solanum pennellii in the background of the cultivated tomato (Solanum lycopersicum). We identified several metabolite quantitative trait loci that reduce variability for both primary and secondary metabolites, which we named canalization metabolite quantitative trait loci (cmQTL). We validated nine cmQTL using an independent population of backcross inbred lines, derived from the same parents, which allows increased resolution in mapping the QTL previously identified in the ILs. These cmQTL showed little overlap with QTL for the metabolite levels themselves. Moreover, the intervals they mapped to harbored few metabolism-associated genes, suggesting that the canalization of metabolism is largely controlled by regulatory genes.
INTRODUCTION
The concept of canalization was originally formulated by Conrad H. Waddington (1905–1975) to define the ability of certain phenotypes to remain relatively constant in spite of environmental and genetic perturbations (Waddington, 1940, 1942). Waddington’s view of canalization encompassed developmental stability, i.e., the stability of developmental pathways, and was based on the observation that, in multicellular organisms, the formation of mature cells and organs was, to a large extent, almost invariable regardless of minor disturbances during the process. Today, the best known example of such stability of developmental pathways is the vulva fate patterning in the nematode Caenorhabditis elegans. The developmental fate of the vulva precursor cells is quasi-invariant, leading to the formation of the complete organ, despite null heterozygous mutations in the gene encoding epidermal growth factor (Félix and Barkoulas, 2012)
Since Waddington’s original conception, the phenomenon of canalization has been observed beyond narrowly defined developmental traits to encompass a broad range of phenotypes from both uni- and multicellular organisms, showing that stability of various molecular traits might well be under genetic control (Alon et al., 1999; Li et al., 2009; Lehner, 2010).
Plant breeders have recognized the relevance of canalization in identifying the best varieties adapted to specific environments: Although these studies usually adopted terms such as stability or uniformity, what was actually observed was canalization of crop yields (Finlay and Wilkinson, 1963; Becker and Leon, 1988). More recently, the analysis of stability has been extended beyond the evaluation of yields to quality-related and physiological traits (Dia et al., 2016; Kumagai et al., 2016).
It is now known that the extent to which a phenotype is canalized is under genetic control, although few studies have investigated the molecular basis of canalization in plants (Hall et al., 2007; Jimenez-Gomez et al., 2011; Lee et al., 2014). Under specific conditions, trait canalization may be beneficial to the organism, leading to the increase of the allele frequencies contributing to the appearance of the canalized phenotype; on the other hand, there might be cases where the plasticity of a specific trait could confer an adaptive advantage, especially in environments subjected to unpredictable fluctuations (Lachowiec et al., 2016).
Although canalization and plasticity may at first appear to be opposing concepts, they are actually aspects of the same, broader phenomenon, namely, the effects that external (environment) and internal (mutations) factors have on overall phenotypic variation. Whether a trait shows a canalized or plastic response ultimately depends on its underlying genetic architecture: For example, genetic redundancy of close paralogs (gene duplication) has been shown to contribute to functional compensation (Hanada et al., 2011); on the other hand, control over differential gene expression of distant paralogs is a typical feature enabling plasticity of the trait response (Zhang et al., 2017).
In this report, we use the term canalization, irrespective of its potential adaptive value, to refer to the property of those phenotypic traits showing no environmental effect when individuals of a specific genotype G are exposed to a set of different environments.
In plants, the genetics of microenvironmental canalization was studied in Arabidopsis thaliana by measuring developmental stability in genetically identical replicates of two recombinant inbred line populations and a population of accessions of wide geographic origin grown under different photoperiods (Hall et al., 2007). This study allowed the mapping of quantitative trait loci (QTL) associated with developmental stability and revealed that ERECTA likely contributes to microenvironmental canalization in rosette leaf number. It additionally provided evidence of genotypic selection both for increased and decreased canalization. Recently, another study in Boechera stricta (a close relative of Arabidopsis) included mapping of, among others, a QTL associated with genetic canalization of flowering time (Lee et al., 2014). Similarly, treatment of Arabidopsis ecotypes and recombinant inbred lines with pharmacological inhibitors of Hsp90 suggests that this protein acts as a capacitor of phenotypic variation in plants as well as in the fruit fly (Rutherford and Lindquist, 1998; Queitsch et al., 2002).
Beyond analyses of developmental traits, canalization in plants has received little attention; we thus turned our interest to the analysis of canalization of metabolic traits, given that in the past decade extensive metabolite profiling studies have been performed aimed at understanding the genetic basis of metabolite accumulation in tomato (Solanum lycopersicum; Mathieu et al., 2009; Smeda et al., 2016; Quadrana, et al., 2014; Schauer et al., 2008; Alseekh et al., 2015). In these previous studies, we investigated metabolic QTL (based on phenotypic means), their heritability, and mode of inheritance for a total of 219 metabolites of primary and secondary metabolism, including sugars, organic and amino acids, vitamins, glycoalkaloids, phenylpropanoids, hydroxycinnamates, and acyl sugars.
Here, we report the assembly of a comprehensive data set of both archival (Schauer et al., 2008; Alseekh et al., 2015) and gathered metabolite data to allow an assessment of the canalization of metabolism in tomato. The collection of novel data from the 2003 growth trial provided information from a third harvest that allowed us, on the one hand, to perform a comprehensive analysis of the variation of primary and secondary metabolite abundances across three seasons, and on the other gave us enough data sets to perform an analysis of metabolic canalization at the whole genome level.
Quantification of organismal canalization is particularly challenging since other traits may be variable or plastic to maintain stability of certain phenotypes. Studies of physiological phenotypes in plants have focused on their phenotypic means rather than on their stability or plasticity. However, a couple of recent exceptions have evaluated stability and plasticity both at the transcriptional and metabolic levels (Dal Santo et al., 2013; Joseph et al., 2015). In these studies, the coefficient of variation was used to assess the stability of transcription in grapevine (Vitis vinifera; Dal Santo et al., 2013) and of metabolite content in Arabidopsis (Joseph et al., 2015). While these pioneering studies have identified and characterized features of canalization and plasticity (Fernie and Tohge, 2013), alternative statistical approaches for estimating canalization, especially when applied to mapping populations (Dworkin, 2005a), may provide a better understanding of the molecular mechanisms underlying trait phenotypic robustness.
For this purpose, we analyzed canalization in the framework of ANOVA following the reaction norm of the variance (RxNV) definition of canalization (Dworkin, 2005b). In contrast to the more common reaction norm of the mean approach, this approach is based on measuring the variance (rather than the mean) of a trait across a set of environments. In this way, we evaluated both the robustness of the phenotypic means and the stability of the trait variances across three independent environments. The results obtained are discussed in the context both of our current understanding of metabolic regulation and of biological robustness in general.
RESULTS
Analysis of the Variation of Secondary Metabolites in an Introgression Line Population
We assembled a complete data set of the abundance and variance of secondary metabolites across three harvests. We included here newly collected metabolic profiling data resulting from a third harvest that performed in 2003 (Figure 1; see Supplemental Figure 1 for a fully annotated version; Supplemental Data Set 1). Figure 1 provides a heat map of the average metabolite fold changes across the three years such that large average increases are depicted in deep red, large average decreases are denoted in deep blue, and minor fold change variation with respect to M82 in a white hue. Hierarchical clustering has been applied on both rows and columns to group side-by-side metabolites and introgression line (IL) populations showing similar patterns of variation.
Figure 1.

Hierarchical Clustering Heat Map of the Secondary Metabolite Profiles of Three Independent Studies of the Pericarp Metabolite Content of the ILs Compared with the Parental Control S. lycopersicum (cv M82).
Data represent measurements of fruit material harvested in field trials performed in 2001, 2003, and 2004 and are presented as the log2 average of fold changes across the three seasons compared with M82. Red and blue regions indicate that the metabolite content is increased or decreased, respectively, after introgression of S. pennellii segments. A fully annotated heat map is provided in Supplemental Figure 1, and the complete data set is presented in Supplemental Data Set 1.
The relative difference in the content of any given metabolite in three harvest years was similar to that previously reported (Alseekh et al., 2015), ranging between 0 (absent) and 94-fold increase in comparison to the levels observed in the parental cultivar M82.
Next, we identified metabolite quantitative trait loci (mQTL), based on phenotypic means, using mixed effect two-way ANOVA, comparing every IL with the common control (M82). We considered only those combinations of metabolites and ILs for which we had at least three replicates per harvest (to avoid the risk that the models are overparameterized). In total, this resulted in examining 9397 statistical models out of 11,020 possible for the case of secondary metabolites and 1946 out of 6916 possible for the case of primary metabolites. For secondary metabolites, we identified 99 mQTL due to the significance of the genotype effect and 647 mQTL due to the significance of the genotype × environment interaction (P value < 0.05, Bonferroni correction for multiple hypotheses testing; Supplemental Data Sets 2 and 3). When broken down into compound classes, this corresponded to 153 mQTL for hydroxycinnamates, 161 mQTL for flavonols, 103 mQTL for glycoalkaloids, 3 mQTL for acyl-sugars, 74 mQTL for N-containing compounds, 18 mQTL for phenolics, and 135 mQTL for unclassified compounds. These numbers correspond to sharing of 57 and 89 mQTL with those reported previously (Alseekh et al., 2015) for genotype and genotype × environment interaction, respectively.
For primary metabolites, we identified 113 mQTL on the basis of the significance of the genotype effect and 129 on the basis of the significance of the genotype × environment interaction (P value < 0.05, Bonferroni correction for multiple hypotheses testing; Supplemental Data Sets 4 and 5).
We also determined the extent to which the detected mQTL are affected by excluding each one of the harvests, given that one of them (2004) followed an atypically hot and dry summer. For secondary metabolites, we identified 211, 177, and 114 mQTL due to the genotype effect for the pairs of harvests 2003 and 2004, 2001 and 2003, and 2001 and 2004, respectively (Supplemental Data Set 6). For secondary metabolites, we identified 248, 29, and 517 mQTL due to the genotype × environment interaction for the pairs of harvests 2003 and 2004, 2001 and 2003, and 2001 and 2004, respectively (Supplemental Data Set 7). For primary metabolites, we identified 121, 135, and 93 mQTL due to the genotype effect for the pairs of harvests 2003 and 2004, 2001 and 2003, and 2001 and 2004, respectively (Supplemental Data Set 8). We also found 95, 60, and 33 mQTL due to the genotype × environment interaction for the pairs of harvests 2003 and 2004, 2001 and 2003, and 2001 and 2004, respectively (Supplemental Data Set 9). The lack of robustness in some of the identified mQTL, particularly those due to the genotype × environment interaction, can be explained by the data coming from one atypical harvest in comparison to the other two. The dependence of the QTL on the season/harvest has been observed for other tomato traits (Capel et al., 2017b; Rambla et al., 2017). This underscores the need to confirm QTL across different environments and populations (Collard and Mackill, 2008; Capel et al., 2017a).
Analysis of QTL for Metabolic Canalization
Having assembled complete data sets for the abundance and variance of primary and secondary metabolites across three harvests, we next sought to identify metabolic canalization QTL (cmQTL) using the reaction norm of the variance (RxNV) approach to canalization (Dworkin, 2005a). We first assessed the significance of the cmQTL based on nominal P values (“permissive approach”), with the objective of obtaining a general overview of the type and distribution of cmQTL; we then applied Bonferroni correction for multiple hypotheses testing to retain a reduced set of significant cmQTL (“conservative approach”). The rationale for analyzing the results using also a permissive threshold is that we have found the Bonferroni correction to be too strict in removing many of the mQTL, which were actually biologically confirmed and even cloned (Fridman et al., 2004; Alseekh et al., 2015). Following our permissive approach, we identified 216 cmQTL for secondary metabolites (Supplemental Figure 2 and Supplemental Data Set 10) and 93 cmQTL for primary metabolites (Figure 2; Supplemental Data Set 11 and Supplemental Table 1). The cmQTL were distributed over 10 and 12 of the chromosomes for primary and secondary metabolites, respectively, with a preponderance of cmQTL located on chromosome 10 (Figure 2 delineates the location of these cmQTL, while Supplemental Figure 3 provides representative interaction plots of trait variability of cmQTL on IL10-1; the detailed data sets are provided in Supplemental Data Sets 10 and 11).
Figure 2.

Putative cmQTL.
Chromosome mapping of putative canalized primary metabolite QTL based on the genetic map of the S. pennellii introgression lines (http://www.sgn.cornell.edu). The figure shows 84 major cmQTL primary metabolites using the reaction norm of the trait (RxNV) approach. cmQTL highlighted in blue are significant at a level of α = 0.05 after Bonferroni correction for multiple hypotheses testing. For more detailed analysis, see Supplemental Data Sets 11 and 12.
When assessing the significance of the ANOVA terms for each individual metabolite (at a significance level of α = 0.05 after Bonferroni correction for multiple hypotheses testing), we found that the  interaction was significant for two primary metabolites: maltitol and phenylalanine (Figure 3; Supplemental Data Set 12). For the secondary metabolites, the
interaction was significant for two primary metabolites: maltitol and phenylalanine (Figure 3; Supplemental Data Set 12). For the secondary metabolites, the  interaction was significant after the conservative Bonferroni correction for seven metabolites (i.e., F009, F045, F074, F080, F401, F413, and F620; Figure 3; Supplemental Data Set 13).
interaction was significant after the conservative Bonferroni correction for seven metabolites (i.e., F009, F045, F074, F080, F401, F413, and F620; Figure 3; Supplemental Data Set 13).
Figure 3.

Variability of Canalized Metabolites.
Interaction plots of canalized metabolites after Levene’s transformation and Bonferroni correction for multiple hypotheses testing. Data represent the relative abundance of metabolites from M82 and ILs after Levene’s transformation from three independent harvest seasons (2001, 2003, and 2004). Data are represented as mean ± sd (n > 6).
The significance of the genotype effect demonstrates that there is a difference between the genotypes over all considered environments with respect to the trait (i.e., the variation of the metabolite level per Levene’s transformation). This effect points further at the respective bin harboring QTL for variation. The statistical significance of the  interaction was detected in the following introgression lines: IL-10-1 for maltitol and phenylalanine, 10-2-2 for phenylalanine, IL-12-2 for F009, F045, F401, and F413, IL-4-4 for F074, IL-7-4 for F080, and IL-12-1 for F620. Visualization via interaction line graphs illustrates that M82 differs from the particular ILs with respect to change of variability in different years (Figure 3).
interaction was detected in the following introgression lines: IL-10-1 for maltitol and phenylalanine, 10-2-2 for phenylalanine, IL-12-2 for F009, F045, F401, and F413, IL-4-4 for F074, IL-7-4 for F080, and IL-12-1 for F620. Visualization via interaction line graphs illustrates that M82 differs from the particular ILs with respect to change of variability in different years (Figure 3).
Similarly to the analysis of mQTL, we also determined the extent to which the detected cmQTL were affected by excluding each one of the harvests. We identified 188, 24, and 119 cmQTL for secondary metabolites and 36, 35, and 33 cmQTL for primary metabolites before Bonferroni correction for the pairs of harvests 2003 and 2004, 2001 and 2003, and 2001 and 2004, respectively. For secondary and primary metabolites, we found 6, 1, and 1 cmQTL and 0, 0, and 1 cmQTL after Bonferroni correction for the respective pairs of harvests. Given that, in the explorative analysis, we detected cmQTL under field conditions in all pairs of conditions, we concluded that the revealed cmQTL over the three harvests captured a robust signal for QTL underlying metabolic canalization.
Validation of Identified cmQTL with the Solanum pennellii BIL Population
We next sought to validate these putative canalization QTL by analyzing a subset of the independently derived S. pennellii backcrossed introgression lines (BILs) (Ofner et al., 2016), which provide increased resolution of the regions of interest. For this purpose, we focused on the putative canalized primary metabolite QTL on chromosome 10, specifically the hot spot in IL10-1, and were able to confirm 9 (out of 25) cmQTL mapped to chromosome 10, using the BIL population. The validated canalized primary metabolite QTL were phenylalanine on both arms of chromosome 10 and Fru-6-P, Glc-6-P, and maltose on the upper arm of chromosome 10 (Figures 4, 5, 6 and 7). It is important to note that the higher resolution afforded by the BIL population allowed us to distinguish the QTL for Fru-6-P, Glc-6-P, and maltose from those for phenylalanine, malate, myo-inositol, and erythritol. Intriguingly, there is almost no overlap between the cmQTL identified here (and validated by the BILs) and those for the metabolite levels themselves (Schauer et al., 2008; Alseekh et al., 2015), with only one of them being mapped to the same region, namely, phenylalanine on chromosome 10 (IL10-3).
Figure 4.

Validation of Putative cmQTL Using the BIL Population.
(A) Schematic representation of chromosome 10 and BILs that have the S. pennellii genomic segments (represented as blue bars).
(B) Box plots of relative abundance of metabolites after Levene’s transformation for the validated cmQTL on IL10-1. The left panel represents the analyzed overlapping BIL genotypes. The right panel shows relative metabolite content in the IL and M82 genotypes from three environments. Data are represented as box plots and display the full range of variation from minimum to maximum (n > 6).
(C) Schematic representation of the genomic interval harboring the cmQTL for Phe on IL10-1 (corrected P value = 4.01E-6, nominal P value = 5.8E-10). The corresponding region in S. pennellii is characterized by multiple presence-absence variants.
E, environment followed by the harvest year; 01, from year 2001; 03, from year 2003; 04, from year 2004; BILs-M-E1, backcross inbred lines that have the M82 genomic segments in this region and grew in environment (harvest) 1; BILs_P E1, backcross inbred lines that have S. pennellii segments in this region and grew in environment (harvest) 1.
Figure 5.

Validation of Putative cmQTL for Glc-6-P, Fru-6-P, and Maltose Using the BIL Population.
Box plots of relative abundance of Glc-6-P, Fru-6-P, and maltose after Levene’s transformation (left panel) and validation using BILs (right panel). On the y axis, the left panel represents the relative abundance of metabolites after Levene’s transformation; data from three independent harvest seasons (2001, 2003, and 2004). The right panel represents the analyzed overlapping BIL genotypes from three independent environments. Data are represented as box plots and display the full range of variation from minimum to maximum (n > 6). E, environment followed by the harvest year; 01, from year 2001; 03, from year 2003; 04, from year 2004; BILs-M-E1, backcross inbred lines that have the M82 genomic segments in this region and grew in environment (harvest) 1; BILs_P E1, backcross inbred lines that have S. pennellii segments in this region and grew in environment (harvest) 1.
Figure 6.

Validation of Putative cmQTL for Phenylalanine in IL10-3 Using the BIL Population.
Box plots of relative abundance of phenylalanine in IL10-3 after Levene’s transformation (left panel) and the validation using BILs (right panel). On the y axis, the left panel represents the relative abundance of metabolites after Levene’s transformation. Data from three independent harvest seasons (2001, 2003, and 2004) are shown. The right panel represents the analyzed overlapping BIL genotypes from three independent environments. Data are represented as box plots and display the full range of variation from minimum to maximum (n > 6). E, environment followed by the harvest year; 01, from year 2001; 03, from year 2003; 04, from year 2004; BILs-M-E1, backcross inbred lines that have the M82 genomic segments in this region and grew in environment (harvest) 1; BILs_P E1, backcross inbred lines that have S. pennellii segments in this region and grew in environment (harvest) 1.
Figure 7.

Validation of Putative cmQTL Using the BIL Population.
Box plots of relative abundance of erythritol (A), malate (B), glycerate (C), and myo-inositol (D) after Levene’s transformation (left panel) and their validation using BILs (right panel). The y axis in the left panel represents the relative abundance of metabolites after Levene’s transformation. Data from three independent harvest seasons (2001, 2003, and 2004) are shown. The right panel represents the analyzed overlapping BIL genotypes from three independent environments. Data are represented as box plots and display the full range of variation from minimum to maximum (n > 6). E, environment followed by the harvest year; 01, from year 2001; 03, from year 2003; 04, from year 2004; BILs-M-E1, backcross inbred lines that have the M82 genomic segments in this region and grew in environment (harvest) 1; BILs_P E1, backcross inbred lines that have S. pennellii segments in this region and grew in environment (harvest) 1.
However, while perusal of the genes in the vicinity of these cmQTL revealed three glucosyltransferases in the subregion of IL10-2 and phenylalanine ammonia lyase on IL10-3, there were very few metabolism-associated genes in any of the other intervals; with the exception of those mentioned above, none of the cmQTL contained genes that were directly involved in synthesis or degradation of the metabolite in question (Supplemental Data 2). The high-resolution nature of the BILs, along with the fact that the genomes of both parental lines have been sequenced (Bolger et al., 2014), rendered it possible to obtain relatively short gene lists for the nine regions that we used for validation via the independent population (Supplemental Data Set 14). Interestingly, several of the genes contained within the cross-validated intervals are associated with regulatory proteins, including those encoding chaperones, receptor-like kinases, and transcription factors (see Supplemental Files 1 and 2 for a detailed discussion) and as such are similar to previously described genes associated with canalization of developmental phenotypes in both plants (Queitsch et al., 2002; Hall et al., 2007; Jimenez-Gomez et al., 2011) and other biological systems (Rutherford and Lindquist, 1998; Specchia et al., 2010).
To further investigate the genetic basis of canalization, we focused on the cmQTL for phenylalanine detected in IL10-1, which represents a significant cmQTL under both the permissive and conservative approaches (uncorrected P value = 5.8E-10, Bonferroni-corrected P value = 4.01E-6). The genomic interval for this cmQTL was further reduced with a subset of overlapping BILs to a 0.5-Mb segment containing 32 genes (Figure 4C, map of cmQTL for phenylalanine on IL10-1). A genomic survey of the corresponding region in S. pennellii revealed long stretches of nonorthologous genes and a general lack of colinearity between the two species. The main genomic polymorphisms observed in S. pennellii are the absence of the 10 FAD binding domain containing genes and a higher number of retrotransposon-related genes (e.g., those encoding gag/pol, reverse transcriptase, and Ribonuclease H).
DISCUSSION
Here, we report a genome-wide analysis for canalization of metabolic traits in tomato fruits. To our knowledge, this represents one of the first experimental approaches attempted in plants on the subject; as such, it extends our previous mQTL mapping efforts in IL populations, which were based solely on phenotypic data. A previous study has examined the canalization of metabolic fluxes in Escherichia coli (Ho and Zhang, 2016): On the basis of computed fluxes in a genome-scale model, the authors identified six capacitor reactions they believed to be important in conferring robustness of primary metabolism. However, it is important to note that these data were not validated experimentally.
Our findings indicate that in tomato fruit, the levels of only some metabolites are canalized and that the tomato genes encoding the proposed capacitor reactions for E. coli are not among the candidate genes we report here. This is not surprising, given the notable difference between multi- and unicellular organisms based on the structure of the underlying metabolic networks (Rosenfeld and Alon, 2003; Milo et al., 2004). Intriguingly, several putative canalized primary metabolite QTL map to the same, albeit large, region of chromosome 10. This finding suggests that trait variation of these metabolites may be buffered by a common regulatory locus within this region. In the case of the phenylalanine cmQTL in IL10-1, however, the genetic basis of trait canalization seems more complex: The comparison of the genomic sequence between the two species highlights a series of presence-absence variants, with the S. pennellii region also differing, with respect to M82, for the presence of several additional retrotransposon-related genes. In this cmQTL, the genetic difference between S. lycopersicum and S. pennellii does not appear to reside in a single candidate gene, but instead extends over 0.5 Mb and includes long stretches of noncolinearity between the two species. However, while most of the putative canalized metabolite QTL represent relatively large genomic regions, one of these corresponding to the 15 secondary metabolites mapped to the same region of chromosome 10, consisted of only 58 genes (Supplemental File 1).
Importantly, however, several of the putative primary metabolite QTL and two of the BIL-validated primary metabolite QTL that we reported share a common metabolite. An evaluation of the metabolic roles of these metabolites and of the validated canalized metabolite QTL for which only a single locus was observed suggest that the pathways of starch degradation (reflected by maltose, isomaltose, Fru-6-P, and Glc-6-P content), which play a key role in ripening (Carrari et al., 2006), alongside the metabolism of phenylpropanoids, volatile organic compounds, and the amino acids asparagine (an important component of foliar nitrate assimilation) and phenylalanine (a precursor of lignin) (Dal Cin et al., 2011), are maintained at constant levels by highly robust genetic mechanisms within the tomato pericarp. Indeed, the phenylalanine QTL was even maintained following the application of the highly stringent Bonferroni correction. Since the pathway of starch degradation (which yields sugars) and those related to phenylalanine-derived volatiles contribute to fruit nutritional quality and flavor (Zhang et al., 2015; Tieman et al., 2017), it is possible that these pathways are related to the production of enticing fruits that aid in seed dispersal.
In conclusion, by analyzing two independent populations derived from crossing cultivated tomato with one of its wild species relatives, we have demonstrated that genetic canalization extends beyond developmental phenotypes and additionally encompasses metabolite levels as traits. We have shown that, at least in some instances, some of these loci for metabolite canalization could be mapped in different genomic locations from those mQTL for phenotypic means. This finding opens the possibility of breeding for stability of metabolic traits independently of the amount of metabolites present. Our analysis of genomic segments harboring the cmQTL also indicated that canalization is likely achieved through the action of regulatory genes, rather than being embedded in the structural genes of metabolic pathways. As with other QTL studies of metabolic traits, our results may be confined to the tissue and developmental stages from which the material was obtained. Therefore, establishing the extent to which the same QTL can be identified in other settings provides an exciting new research perspective. These findings thus not only open up a novel fundamental research avenue, given that they identify genomic regions that suppress variability in several traits, but also have important implications for commercial applications, particularly with regard to the renewed interest in breeding high quality crops that are both tasty and nutritious.
METHODS
Growth Conditions
The metabolite data set presented is based on field-grown introgression lines (and in 2004 their respective heterozygous counterparts; Semel et al., 2006) over three harvests (2001, 2003, and 2004). The field trials were conducted in the Western Galilee Experimental Station in Akko, Israel. Plants were grown in a completely randomized design with one plant per m2. Seedlings were grown in greenhouses for 35 to 40 d and then transferred to the field. Twelve seedlings of each homozygous IL and heterozygous ILH (IL*M82) were transplanted as well as 70 seedlings of M82. Eight ILs were not included in the analysis of 2004 because of poor germination (ILH2-4, IL3-1, ILH3-4, ILH6-2, ILH6-2-2, ILH6-4, ILH7-2, and ILH9-3-2). Fruit was harvested when 80 to 100% of the tomatoes were red (Eshed and Zamir, 1995). The field was irrigated with 320 m3 of water per 1000 m2 of field area throughout the season.
Three to five red ripe fruits were collected from each biological replicate (single plant) and three to six independent plants were used for each IL. For the cultivated variety M82, we had at least 80 independent biological replicates in each harvest season. Fruit pericarp materials were frozen in liquid nitrogen and stored at −80°C until further analysis. From the frozen tomato powder, an aliquot of fresh weight was weighed and extracted for both primary and secondary metabolites as described previously (Schauer et al., 2006; Tohge and Fernie, 2010).
Morphological and reproductive traits have previously been described for the 2001 and 2003 harvests (Schauer et al., 2006, 2008) and for the 2004 harvest (Semel et al., 2006) and variance in primary metabolite traits have also been recorded for three harvests (Alseekh et al., 2015).
Secondary Metabolite Profiling
Secondary metabolites were profiled using the Waters Acquity UPLC system coupled to an Exactive Orbitrap mass detector according to the previously published protocol (Giavalisco et al., 2009). UPLC was equipped with a HSS T3 C18 reversed phase column (100 × 2.1-mm i.d. 1.8-µm particle size; Waters) which was operated at a temperature of 40°C. The mobile phases consisted of 0.1% formic acid in water (Solvent A) and 0.1% formic acid in acetonitrile (Solvent B). The flow rate of the mobile phase was 400 µL/min, and 2 μL sample was loaded per injection. The UPLC was connected to an Exactive Orbitrap (Thermo Fisher Scientific) via a heated electro spray source (Thermo Fisher Scientific). The spectra were recorded using full scan mode in negative ion detection, covering a mass range from m/z 100 to 1500. The resolution was set to 25,000 and the maximum scan time was set to 250 ms. The sheath gas was set to a value of 60, while the auxiliary gas was set to 35. The transfer capillary temperature was set to 150°C, while the heater temperature was adjusted to 300°C. The spray voltage was fixed at 3 kV, with a capillary voltage and a skimmer voltage of 25 and 15 V, respectively. MS spectra were recorded from min 0 to 19 of the UPLC gradient. Molecular masses, retention time, and associated peak intensities were extracted from the raw files using RefinerMS software (version 5.3; GeneData), Metalign (Lommen, 2012), and Xcalibur software (Thermo Fisher Scientific). Metabolite identification and annotation were performed using standard compounds, literature, and tomato metabolomics databases (Moco et al., 2006; Iijima et al., 2008; Tohge and Fernie, 2009, 2010; Rohrmann et al., 2011). Data are reported in a manner compliant with the standards suggested by Fernie et al. (2011).
Heat Maps
Heat maps were created by MeV software (http://mev.tm4.org), using the log2 of the metabolite fold changes with respect to the recurrent parent M82. Each cell in the heat map represents the log2 average fold change value from the independent seasons. Hierarchical clustering was applied to rows and columns using Pearson’s correlation coefficient.
mQTL Mapping in the IL Population
To identify mQTL, a two-way mixed effect ANOVA (see subsection below) with genotype (IL) was used as a fixed effect and environment and genotype × environment interactions were treated as random effects. mQTL were detected if the genotype effect or the genotype × environment effect was statistically significant at a level of 0.05. To provide conservative results, all P values were Bonferroni corrected for multiple hypotheses testing. Correlation analysis was also performed across the entire population by means of the Pearson correlation coefficient to determine possible technical artifacts. A gas chromatography-mass spectrometry analysis was also performed on fruit materials harvested in three independent field trials from selected Solanum pennellii BILs (Ofner et al. 2016).
Statistical Analysis of Canalization
Starting from the entire metabolite data sets for the wild-type M82 and each introgression line in three seasons, canalization was analyzed using ANOVA following the reaction norm of the trait (RxNT) definition of canalization (Dworkin, 2005a). According to this definition, canalization is the opposite of phenotypic plasticity (Nijhout and Davidowitz, 2003): Thus, a genotype would be considered canalized with respect to a set of environments if there is no environmental effect on the trait. By this definition, the more canalized a genotype is, the less its trait should vary across environments. For the metabolite trait  we consider its variance, resulting in the reaction norm of the variance (RxNV) approach.
we consider its variance, resulting in the reaction norm of the variance (RxNV) approach.
In this approach, canalization is inferred by decanalizing the system via multiple environments to induce a change in the trait away from its normal manifestation. In our case, the RxNT applied to metabolite 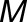 can be captured by a two-way ANOVA model:
can be captured by a two-way ANOVA model:
 , where
, where 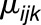 is the
is the 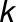 th replicated measurement of metabolite
th replicated measurement of metabolite 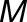 ’s trait in genotype
’s trait in genotype  under environment (season)
under environment (season)  ,
,  is the grand mean for metabolite
is the grand mean for metabolite  ’s trait,
’s trait,  corresponds to the effect of genotype
corresponds to the effect of genotype 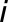 (with
(with  ),
),  corresponds to the season
corresponds to the season 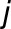 (with
(with  ),
), 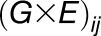 is the interaction between genotype
is the interaction between genotype  and season
and season 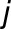 , and
, and 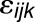 is a normally distributed error term. We treated
is a normally distributed error term. We treated  and
and 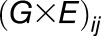 as random effects, while
as random effects, while 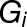 was considered a fixed effect. The model was implemented in the R programming environment using the package lme4. The significance of the fixed effects was determined with the ANOVA function from the car package. The significance of the random effect
was considered a fixed effect. The model was implemented in the R programming environment using the package lme4. The significance of the fixed effects was determined with the ANOVA function from the car package. The significance of the random effect  was determined by comparing the fits of the models with and without inclusion of the interaction (Pinheiro and Bates, 2000). In RxNV, the model is based on the Levene’s transformation of
was determined by comparing the fits of the models with and without inclusion of the interaction (Pinheiro and Bates, 2000). In RxNV, the model is based on the Levene’s transformation of 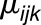 given by
given by  , where
, where 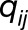 is the median of the replicates in genotype
is the median of the replicates in genotype  under environment
under environment 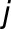 .
.
The following is a code snippet which allows replication of the findings:
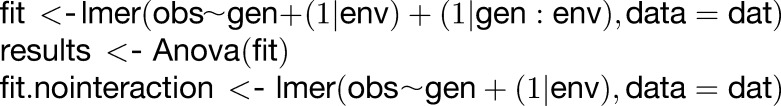 |
results.compare <- anova(fit, fit.nointeraction) where dat contains the ANOVA table (see Supplemental File 2). The same lines with the nominal levels of the metabolites (i.e., without Levene’s transformation) were used to determine the mQTL.
A significant  term then indicates genetic variation for plasticity of metabolite
term then indicates genetic variation for plasticity of metabolite  ’s trait.
’s trait.
Since the two-way ANOVA model is tested for each metabolite and introgression line, Bonferroni adjustment of P values was performed for multiple hypotheses testing (i.e., the obtained P values were multiplied by  , where
, where  denotes the number of metabolites and
denotes the number of metabolites and  represents the number of introgression lines).
represents the number of introgression lines).
Selection of Candidate Genes in the cmQTL
To detect functional candidate genes in the identified cmQTL, the full list of the genes contained in each introgression/cmQTL was retrieved from the Sol Genomics network (https://solgenomics.net/, genome version: tomato Sl2.50, ITAG2.4). For each introgression, the gene lists were then filtered to retain only those genes which, on the basis of previous studies, have shown to possess a role in the genetics of trait canalization (in plants or other organisms). These functional candidate genes were subjected to a combination of sequence analyses and annotation tools (see Supplemental File 1).
Supplemental Data
Supplemental Figure 1. Fully annotated heat map of secondary metabolite profiles of three independent studies of the pericarp metabolite content of the ILs compared with M82.
Supplemental Figure 2. Putative canalized secondary metabolite quantitative trait loci.
Supplemental Figure 3. Interaction plots showing the variability of putative canalized primary metabolites in the hotspot IL10-1.
Supplemental Table 1. Summary of number of mQTL and cmQTL identified in this study.
Supplemental Data Set 1. Average fold changes of secondary metabolites in the S. pennellii introgression lines over the 3-year observation period.
Supplemental Data Set 2. Significant mQTL for secondary metabolites due to the genotype interaction effect at a level of α = 0.05 after Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 3. Significant mQTL for secondary metabolites due to genotype by environment interaction effect at a level of α = 0.05 after Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 4. Significant mQTL for primary metabolites due to genotype effect at a level of α = 0.05 after Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 5. Significant mQTL for primary metabolites due to genotype by environment interaction effect at a level of α = 0.05 after Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 6. Significant mQTL for secondary metabolites due to genotype interaction effect at a level of α = 0.05 without the conservative Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 7. Significant mQTL for secondary metabolites due to genotype by environment interaction effect at a level of α = 0.05 without the conservative Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 8. Significant mQTL for primary metabolites due to genotype effect at a level of α = 0.05 without the conservative Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 9. Significant mQTL for primary metabolites due to genotype by environment interaction effect at a level of α = 0.05 without the conservative Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 10. Significant cmQTL for secondary metabolites at a level of α = 0.05 without the conservative Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 11. Significant cmQTL for primary metabolites at a level of α = 0.05 without the conservative Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 12. Significant cmQTL for primary metabolites at a level of α = 0.05 after Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 13. Significant cmQTL for secondary metabolites at a level of α = 0.05 after Bonferroni correction for multiple hypotheses testing.
Supplemental Data Set 14. List of candidate genes of each validated canalized mQTL using the backcross inbred lines.
Supplemental File 1. Detailed dissection of genes within validated cmQTL.
Supplemental File 2. R data R script.codes file.
Acknowledgments
Work in the Zamir and Fernie laboratories is funded by the Deutsche Forschungsgemeinschaft in the framework of Deutsche Israeli Project FE 552/12-1. The Zamir lab was also supported by the Israel Science Foundation (653/15).
AUTHOR CONTRIBUTIONS
S.A., D.Z., Z.N., and A.R.F. conceived the work. I.O. and D.Z. provided genetic materials and managed their growth and harvest, while S.A., F.S., Y.B., F.V., and T.T. conducted the analysis. H.T. and Z.N. designed and developed the bioinformatic analyses. S.A., Z.N., and A.R.F. interpreted the data and wrote the article.
Footnotes
Articles can be viewed without a subscription.
References
- Alon U., Surette M.G., Barkai N., Leibler S. (1999). Robustness in bacterial chemotaxis. Nature 397: 168–171. [DOI] [PubMed] [Google Scholar]
- Alseekh S., et al. (2015). Identification and mode of inheritance of quantitative trait loci for secondary metabolite abundance in tomato. Plant Cell 27: 485–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker H.C., Leon J. (1988). Stability analysis in plant-breeding. Plant Breed. 101: 1–23. [Google Scholar]
- Bolger A., et al. (2014). The genome of the stress-tolerant wild tomato species Solanum pennellii. Nat. Genet. 46: 1034–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel C., Yuste-Lisbona F.J., López-Casado G., Angosto T., Cuartero J., Lozano R., Capel J. (2017a). Multi-environment QTL mapping reveals genetic architecture of fruit cracking in a tomato RIL Solanum lycopersicum × S. pimpinellifolium population. Theor. Appl. Genet. 130: 213–222. [DOI] [PubMed] [Google Scholar]
- Capel C., Yuste-Lisbona F.J., López-Casado G., Angosto T., Heredia A., Cuartero J., Fernández-Muñoz R., Lozano R., Capel J. (2017b). QTL mapping of fruit mineral contents provides new chances for molecular breeding of tomato nutritional traits. Theor. Appl. Genet. 130: 903–913. [DOI] [PubMed] [Google Scholar]
- Carrari F., Baxter C., Usadel B., Urbanczyk-Wochniak E., Zanor M.I., Nunes-Nesi A., Nikiforova V., Centero D., Ratzka A., Pauly M., Sweetlove L.J., Fernie A.R. (2006). Integrated analysis of metabolite and transcript levels reveals the metabolic shifts that underlie tomato fruit development and highlight regulatory aspects of metabolic network behavior. Plant Physiol. 142: 1380–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collard B.C.Y., Mackill D.J. (2008). Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363: 557–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Cin V., et al. (2011). Identification of genes in the phenylalanine metabolic pathway by ectopic expression of a MYB transcription factor in tomato fruit. Plant Cell 23: 2738–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Santo S., Tornielli G.B., Zenoni S., Fasoli M., Farina L., Anesi A., Guzzo F., Delledonne M., Pezzotti M. (2013). The plasticity of the grapevine berry transcriptome. Genome Biol. 14: r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dia M., Wehner T.C., Perkins-Veazie P., Hassell R., Price D.S., Boyhan G.E., Olson S.M., King S.R., Davis A.R., Tolla G.E., Bernier J., Juarez B. (2016). Stability of fruit quality traits in diverse watermelon cultivars tested in multiple environments. Hortic. Res. 3: 16066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin I. (2005a). Canalization, cryptic variation, and developmental buffering: A critical examination and analytical perspective. In Variation: A Central Concept in Biology, B. Hallgrímsson and B.K. Hall, eds (New York: Academic Press; ), pp. 131–158. [Google Scholar]
- Dworkin I. (2005b). A study of canalization and developmental stability in the sternopleural bristle system of Drosophila melanogaster. Evolution 59: 1500–1509. [PubMed] [Google Scholar]
- Eshed Y., Zamir D. (1995). An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141: 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Félix M.A., Barkoulas M. (2012). Robustness and flexibility in nematode vulva development. Trends Genet. 28: 185–195. [DOI] [PubMed] [Google Scholar]
- Fernie A.R., Aharoni A., Willmitzer L., Stitt M., Tohge T., Kopka J., Carroll A.J., Saito K., Fraser P.D., DeLuca V. (2011). Recommendations for reporting metabolite data. Plant Cell 23: 2477–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernie A.R., Tohge T. (2013). Plastic, fantastic! Phenotypic variance in the transcriptional landscape of the grape berry. Genome Biol. 14: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay K.W., Wilkinson G.N. (1963). The analysis of adaptation in a plant-breeding programme. Aust. J. Agric. Res. 14: 742–754. [Google Scholar]
- Fridman E., Carrari F., Liu Y.S., Fernie A.R., Zamir D. (2004). Zooming in on a quantitative trait for tomato yield using interspecific introgressions. Science 305: 1786–1789. [DOI] [PubMed] [Google Scholar]
- Giavalisco P., Köhl K., Hummel J., Seiwert B., Willmitzer L. (2009). 13C isotope-labeled metabolomes allowing for improved compound annotation and relative quantification in liquid chromatography-mass spectrometry-based metabolomic research. Anal. Chem. 81: 6546–6551. [DOI] [PubMed] [Google Scholar]
- Hall M.C., Dworkin I., Ungerer M.C., Purugganan M. (2007). Genetics of microenvironmental canalization in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 104: 13717–13722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K., Sawada Y., Kuromori T., Klausnitzer R., Saito K., Toyoda T., Shinozaki K., Li W.-H., Hirai M.Y. (2011). Functional compensation of primary and secondary metabolites by duplicate genes in Arabidopsis thaliana. Mol. Biol. Evol. 28: 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho W.C., Zhang J. (2016). Adaptive genetic robustness of Escherichia coli metabolic fluxes. Mol. Biol. Evol. 33: 1164–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima Y., et al. (2008). Metabolite annotations based on the integration of mass spectral information. Plant J. 54: 949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Gomez J.M., Corwin J.A., Joseph B., Maloof J.N., Kliebenstein D.J. (2011). Genomic analysis of QTLs and genes altering natural variation in stochastic noise. PLoS Genet. 7: e1002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B., Corwin J.A., Kliebenstein D.J. (2015). Genetic variation in the nuclear and organellar genomes modulates stochastic variation in the metabolome, growth, and defense. PLoS Genet. 11: e1004779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai E., Homma K., Kuroda E., Shimono H. (2016). Finlay-Wilkinson’s regression coefficient as a pre-screening criterion for yield responsiveness to elevated atmospheric CO2 concentration in crops. Physiol. Plant. 158: 312–317. [DOI] [PubMed] [Google Scholar]
- Lachowiec J., Queitsch C., Kliebenstein D.J. (2016). Molecular mechanisms governing differential robustness of development and environmental responses in plants. Ann. Bot. 117: 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.R., Anderson J.T., Mitchell-Olds T. (2014). Unifying genetic canalization, genetic constraint, and genotype-by-environment interaction: QTL by genomic background by environment interaction of flowering time in Boechera stricta. PLoS Genet. 10: e1004727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B. (2010). Genes confer similar robustness to environmental, stochastic, and genetic perturbations in yeast. PLoS One 5: e9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Cassidy J.J., Reinke C.A., Fischboeck S., Carthew R.W. (2009). A microRNA imparts robustness against environmental fluctuation during development. Cell 137: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommen A. (2012). Data (pre-)processing of nominal and accurate mass LC-MS or GC-MS data using MetAlign. Methods Mol. Biol. 860: 229–253. [DOI] [PubMed] [Google Scholar]
- Mathieu S., Cin V.D., Fei Z., Li H., Bliss P., Taylor M.G., Klee H.J., Tieman D.M. (2009). Flavour compounds in tomato fruits: identification of loci and potential pathways affecting volatile composition. J. Exp. Bot. 60: 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo R., Itzkovitz S., Kashtan N., Levitt R., Alon U. (2004). Response to comment on “network motifs: Simple building blocks of complex networks” and “superfamilies of evolved and designed networks”. Science 305: 1107. [Google Scholar]
- Moco S., Bino R.J., Vorst O., Verhoeven H.A., de Groot J., van Beek T.A., Vervoort J., de Vos C.H.R. (2006). A liquid chromatography-mass spectrometry-based metabolome database for tomato. Plant Physiol. 141: 1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhout H.F., Davidowitz G. (2003). Developmental Perspectives on Phenotypic Instability, Canalization, and Fluctuating Asymmetry. (New York: Wiley; ). [Google Scholar]
- Ofner I., Lashbrooke J., Pleban T., Aharoni A., Zamir D. (2016). Solanum pennellii backcross inbred lines (BILs) link small genomic bins with tomato traits. Plant J. 87: 151–160. [DOI] [PubMed] [Google Scholar]
- Quadrana L., et al. (2014). Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 5: 3027. [DOI] [PubMed] [Google Scholar]
- Queitsch C., Sangster T.A., Lindquist S. (2002). Hsp90 as a capacitor of phenotypic variation. Nature 417: 618–624. [DOI] [PubMed] [Google Scholar]
- Pinheiro J.C., Bates D.M. (2000). Mixed-Effects Models in S and S-PLUS. (New York: Springer).
- Rambla J.L., et al. (2017). Identification, introgression, and validation of fruit volatile QTLs from a red-fruited wild tomato species. J. Exp. Bot. 68: 429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrmann J., et al. (2011). Combined transcription factor profiling, microarray analysis and metabolite profiling reveals the transcriptional control of metabolic shifts occurring during tomato fruit development. Plant J. 68: 999–1013. [DOI] [PubMed] [Google Scholar]
- Rosenfeld N., Alon U. (2003). Response delays and the structure of transcription networks. J. Mol. Biol. 329: 645–654. [DOI] [PubMed] [Google Scholar]
- Rutherford S.L., Lindquist S. (1998). Hsp90 as a capacitor for morphological evolution. Nature 396: 336–342. [DOI] [PubMed] [Google Scholar]
- Schauer N., Semel Y., Balbo I., Steinfath M., Repsilber D., Selbig J., Pleban T., Zamir D., Fernie A.R. (2008). Mode of inheritance of primary metabolic traits in tomato. Plant Cell 20: 509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer N., et al. (2006). Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 24: 447–454. [DOI] [PubMed] [Google Scholar]
- Semel Y., Nissenbaum J., Menda N., Zinder M., Krieger U., Issman N., Pleban T., Lippman Z., Gur A., Zamir D. (2006). Overdominant quantitative trait loci for yield and fitness in tomato. Proc. Natl. Acad. Sci. USA 103: 12981–12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeda J.R., Schilmiller A.L., Last R.L., Mutschler M.A. (2016). Introgression of acylsugar chemistry QTL modifies the composition and structure of acylsugars produced by high-accumulating tomato lines. Mol. Breed. 36: 160. [Google Scholar]
- Specchia V., Piacentini L., Tritto P., Fanti L., D’Alessandro R., Palumbo G., Pimpinelli S., Bozzetti M.P. (2010). Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 463: 662–665. [DOI] [PubMed] [Google Scholar]
- Tieman D., et al. (2017). A chemical genetic roadmap to improved tomato flavor. Science 355: 391–394. [DOI] [PubMed] [Google Scholar]
- Tohge T., Fernie A.R. (2009). Web-based resources for mass-spectrometry-based metabolomics: a user’s guide. Phytochemistry 70: 450–456. [DOI] [PubMed] [Google Scholar]
- Tohge T., Fernie A.R. (2010). Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nat. Protoc. 5: 1210–1227. [DOI] [PubMed] [Google Scholar]
- Waddington C.H. (1940). Organisers and Genes. (Cambridge, UK: Cambridge University Press; ). [Google Scholar]
- Waddington C.H. (1942). Canalization of development and the inheritance of acquired characters. Nature 150: 563–565. [DOI] [PubMed] [Google Scholar]
- Zhang D., Zhao M., Li S., Sun L., Wang W., Cai C., Dierking E.C., Ma J. (2017). Plasticity and innovation of regulatory mechanisms underlying seed oil content mediated by duplicated genes in the palaeopolyploid soybean. Plant J. 90: 1120–1133. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Butelli E., Alseekh S., Tohge T., Rallapalli G., Luo J., Kawar P.G., Hill L., Santino A., Fernie A.R., Martin C. (2015). Multi-level engineering facilitates the production of phenylpropanoid compounds in tomato. Nat. Commun. 6: 8635. [DOI] [PMC free article] [PubMed] [Google Scholar]