

Summary
ARID1A, a SWI/SNF chromatin remodeling gene, is commonly mutated in cancer and hypothesized to be tumor suppressive. In some hepatocellular carcinoma patients, ARID1A was highly expressed in primary tumors but not in metastatic lesions, suggesting that ARID1A can be lost after initiation. Mice with liver-specific homozygous or heterozygous Arid1a loss were resistant to tumor initiation while ARID1A overexpression accelerated initiation. In contrast, homozygous or heterozygous Arid1a loss in established tumors accelerated progression and metastasis. Mechanistically, gain of Arid1a function promoted initiation by increasing Cytochrome P450 mediated oxidative stress, while loss of Arid1a within tumors decreased chromatin accessibility and reduced transcription of genes associated with migration, invasion, and metastasis. In summary, ARID1A has context-dependent tumor suppressive and oncogenic roles in cancer.
In Brief
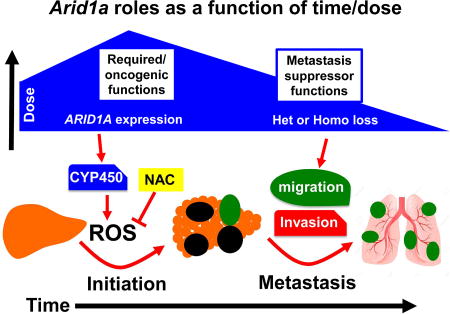
Sun el al. uncover context-specific roles for the SWI/SNF component Arid1a in liver cancer, where elevated Arid1a promotes tumor initiation through CYP450-mediated oxidative stress, whereas reduced Arid1a in established tumors increases metastasis due to reduced expression of inhibitory factors.
Introduction
The underlying principles by which key chromatin remodelers regulate cancer initiation, growth, and metastasis are unclear. Genes encoding components of the SWI/SNF chromatin-remodeling complex have been identified in genome sequencing efforts as some of the most commonly mutated genes in cancer, with ARID1A being the most frequently mutated SWI/SNF gene (Kadoch et al., 2013; Wu and Roberts, 2013). Because the majority of the mutations are loss-of-function, ARID1A is hypothesized to be a tumor suppressor (Hohmann and Vakoc, 2014; Wu and Roberts, 2013). Approximately 10–15% of hepatocellular carcinoma (HCC) harbor loss of function mutations in this gene (Fujimoto et al., 2012; Guichard et al., 2012; Kan et al., 2013; Schulze et al., 2015). Recent work showed that Arid1a has tumor suppressor functions in chemical liver cancer models (Fang et al., 2015).
Other observations suggest a more complex role for ARID1A in tumorigenesis and some SWI/SNF components are oncogenic in certain contexts (Jubierre et al., 2016; Kadoch and Crabtree, 2013). In HCC, Zhao et. al. showed that 83% of tumors have ARID1A overexpression when compared to adjacent tissues (Zhao et al., 2015). In endometrial cancer, primary tumors expressed wild-type ARID1A but metastatic subclones from the same patient harbored deleterious mutations, suggesting that perhaps ARID1A was required in early cancers (Gibson et al., 2016). Recent studies in mice showed that Arid1a loss impaired ovarian cancer formation (Zhai et al., 2015) and delayed colon cancer formation in the context of Apc mutations (Mathur et al., 2017). Given these conflicting data and the fact that epigenetic regulators often have context specific functions, we sought to delineate the roles of ARID1A in liver tumorigenesis.
Results
Homozygous Arid1a loss protected against tumor initiation in multiple mouse models
Because ARID1A is presumed to be a tumor suppressor gene, we reasoned that reduced expression would be associated with worse outcomes in patients. However, we found ARID1A expression levels negatively correlated with survival in HCC patients (Figure S1A) (Uhlen et al., 2017). While this may be due to normal variation of chromatin machinery expression associated with proliferation and other processes, these findings also suggest that there may be more complicated roles for ARID1A. To elucidate these roles, we generated liver-specific Arid1a knockout (Homo) mice using Cre driven by the Albumin promoter. As previously reported, neither wild-type (WT) nor Homo mice spontaneously developed liver tumors by one year of age (Sun et al., 2016). We excluded the possibility of truncated or alternatively spliced versions of ARID1A by probing with N- and C-terminal antibodies and six qPCR primer sets that span the entire protein (Figure S1B and S1C). Since human HCC most frequently develops in the setting of chronic liver injury, we used toxins to induce HCC in these mice. We injected the mutagen diethylnitrosamine (DEN) into WT and Homo mice at age p14, then induced chronic liver injury with biweekly carbon tetrachloride (CCl4) injections (Figure S1D). We found Homo mice formed significantly fewer and smaller tumors (p < 0.05 and 0.01, respectively; Figure S1E and S1F). Despite differences in overall tumor burden, there were no differences in histology and proliferation between genotypes (Figure S1G). To rule out the possibility that Arid1a was affecting toxin metabolism, we generated an alternative model where Arid1a was deleted after DEN mutagenesis. We injected DEN into two-week-old Arid1a+/+ and Arid1aFl/Fl mice followed two weeks later by injections of AAV8-TBG-Cre (hereafter called AAV-Cre) (Figure 1A and Figure S1H). Again, we found that Arid1aFl/Fl + AAV-Cre (KO) mice had fewer and smaller tumors on the liver surface, as well as fewer microscopic tumor nodules (all p < 0.05; Figures 1B–1G). Once formed, tumors with and without Arid1a had similar proliferation levels (Figure S1I). Thus, Arid1a loss protected against HCC initiation in two different chemically-induced models.
Figure 1. Arid1a loss protected against chemical and MYC-induced HCC initiation.

(A) Wild-type (WT) and Arid1aFl/Fl (KO) mice were injected with DEN at p14, then Arid1a was deleted with AAV-Cre at p28. 10–11 months after AAV-Cre, the mice were sacrificed.
(B) Representative gross image of WT and KO livers. Scale bar = 10 mm.
(C) H&E histology of tumor nodules. “T” denotes tumor. Scale bar = 100 µm.
(D) Number of visible tumors on liver surface (n = 6 WT and 6 KO mice).
(E) Tumor volume (n = 6 WT and 6 KO mice).
(F) Quantification of pre-malignant or malignant nodules on H&E sections (n = 6 WT and 6 KO mice, three 40x fields/per mouse).
(G) Liver to body weight ratios (n = 6 WT and 6 KO mice).
(H) Schema to generate LAP-MYC; Arid1a WT, Het, and Homo mice.
(I) Kaplan-Meier curves of LAP-MYC; Arid1a WT, Het, and Homo mice on a pure FVB background.
(J) Representative gross images of LAP-MYC; Arid1a WT and Homo livers at p49. Scale bar = 10 mm.
(K) Liver to body weight ratios (n = 7 WT and 9 Homo mice)
(L) H&E histology of tumor and normal adjacent tissue at p49. “T” denotes tumor. Scale bar = 200 µm.
(M) This genotyping protocol is used to detect Arid1a WT, floxed, and exon 8 excision mediated by Cre within LAP-MYC tumors.
(N) ARID1A IHC in LAP-MYC; Arid1a WT, Het, and Homo tumors. Scale bar = 100 µm.
Two-tailed Student’s t-tests (two-sample equal variance) were used to test the significance of differences between two groups. Kaplan-Meier method was used to estimate survival curves, which were compared using the log-rank test. All data in this figure are represented as mean ± SEM, * (p < 0.05), ** (p < 0.01), *** (p < 0.001). See also Figure S1.
To determine if genetic drivers might reveal additional insights, we employed a liver-specific tet-off cancer model based on MYC overexpression (LAP-tTA; TRE-MYC, referred hereafter as the LAP-MYC model; Figure 1H) (Shachaf et al., 2004). In human HCC, ARID1A mutations often co-exist with MYC amplification (Figure S1J). While all LAP-MYC mice with intact Arid1a succumbed to their disease by 75 days, with a median overall survival (OS) of 57 days, all LAP-MYC; Arid1a Homo mice had dramatically less tumor burden and remained alive beyond 100 days (p < 0.0001; Figures 1I–1L). Heterozygous Arid1a loss led to an intermediate phenotype with median OS of 94 days (p < 0.001; Figure 1I). Arid1a deletion was confirmed in both Het and Homo tumors on the DNA and protein levels (Figure 1M and 1N). As was the case in the chemical models, proliferation was similar after WT, Het, and Homo tumors were established (all p < 0.05; Figure S1K and S1L). To ensure that the driver oncogene in this model was not completely dependent on the presence of ARID1A and SWI/SNF, we performed ChIP-seq to quantify levels of MYC engagement with its transcriptional targets. Between WT, Het, and Homo tumors, there were no differences in genome-wide and individual gene binding by MYC (Figure S1M and S1N). Thus, we found that in this endogenous genetic model of liver carcinogenesis, Arid1a loss did not accelerate cancer initiation as would be expected if it were a tumor suppressor, but instead, was necessary for tumor initiation.
To test if ARID1A was sufficient to exert functionally relevant, pro-cancer effects, we performed an overexpression experiment by injecting LAP-MYC mice with adenoviruses carrying CMV driven Cre (as a control) or full length human ARID1A (Figures 2A–2C). After 14 days, mice with ARID1A overexpression showed increased abdominal girth and livers that were replaced with tumor, while control mice had almost no tumor burden (p < 0.01; Figure 2D and 2E). ARID1A overexpressing LAP-MYC mice also had shorter survival (p < 0.01; Figure 2F). To mitigate the inflammatory effects of adenovirus infection and to prevent immune clearance of cells that harbored adenoviruses, we performed the same experiment in an immunodeficient Rag1−/−; LAP-tTA+/+; TRE-MYC background, and found similar results (Figures 2G and 2H). In agreement with loss-of-function studies, these data show that ARID1A can have tumor promoting roles.
Figure 2. ARID1A overexpression accelerated oncogenesis in multiple HCC models.

(A)LAP-MYC mice injected at p28 with adenovirus carrying Cre recombinase (Ad-Cre) or human ARID1A (Ad-ARID1A) under a CMV promoter and sacrificed at p42.
(B) Western blot of liver tissues from LAP-MYC mice 5 days after Ad-Cre or Ad-ARID1A injection. Antibody detects human and mouse ARID1A proteins.
(C)ARID1A IHC in LAP-MYC tumors. Scale bar = 100 µm.
(D) Girth of LAP-MYC abdomens injected with Ad-Cre or Ad-ARID1A. Box plots show the minimum, lower quartile, median, upper quartile, and maximum abdominal circumference values (n = 6 and 7 mice, respectively).
(E) Gross images of LAP-MYC livers injected with Ad-Cre or Ad-ARID1A. Liver weights and liver to body weight ratios quantified on the right (n = 6 and 6 mice). Scale bar = 10 mm.
(F) Kaplan-Meier curve of LAP-MYC mice injected with Ad-Cre or Ad-ARID1A.
(G) Kaplan-Meier curve of immunodeficient Rag1−/−; LAP-MYC mice injected with Ad-Cre or Ad-ARID1A.
(H)ARID1A expression in Rag1−/−; LAP-MYC livers 14 days after adenovirus injection, as measured by qPCR.
(I) ARID1A protein expression in H2.35 cells as measured by V5 and ARID1A antibodies. Sleeping Beauty transposase (SB2) construct and pT3-hARID1A transposon were co-transfected using standard methods.
(J) V5 Immunostaining for V5-ARID1A 7 days after HDT in WT livers. Scale bar = 50 µm.
(K) Gross images from pT3-β-Catenin + pT3-MET +/− pT3-hARID1A experiment 23 days after HDT (n = 4 pT3-empty and 10 pT3-hARID1A). Scale bar = 10 mm. V5 staining shows exogenous ARID1A expression. Scale bar = 100 µm. On the right, box plots show the minimum, lower quartile, median, upper quartile, and maximum liver vs. body weight ratios.
(L) Images from pT3-β-Catenin + pT3-YAP +/− pT3-hARID1A experiment 23 days after HDT (n =7 and 7). Scale bar = 10 mm. V5 staining shows exogenous ARID1A expression. Scale bar =100 µm. On the right, box plots show the minimum, lower quartile, median, upper quartile, and maximum liver vs. body weight ratios.
(M) Images from pT3-Myr-Akt1 + pT3-Nras(V12) +/− pT2-mArid1a experiment 28 days after HDT (n = 9 pT2-empty and 10 pT2-mArid1a). Scale bar = 10 mm. On the right, box plots show the minimum, lower quartile, median, upper quartile, and maximum liver vs. body weight ratios.
Two-tailed Student’s t-tests (two-sample equal variance) were used to test the significance of differences between two groups. Kaplan-Meier method was used to estimate survival curves, which were compared using the log-rank test. All data in this figure are represented as mean ± SEM, * (p < 0.05), ** (p < 0.01). See also Figure S2.
To corroborate this using a long-term and less immunogenic overexpression strategy, we used Sleeping Beauty transposons delivered via hydrodynamic transfection (HDT). We first validated the expression of a human pT3-ARID1A transposon (Figure 2I and 2J). Then, we induced MYC in LAP-MYC mice at 4 weeks of age, which exhibits delayed tumor initiation at around 10 weeks of age. Within one week after induction, we used HDT to deliver the SB2 transposase and the pT3-ARID1A transposon (Figure S2A) and found that ARID1A overexpression promoted tumor formation and liver/body weight ratios (Figure S2B). We also used this system to determine whether ARID1A could promote cancer initiation in the context of other oncogenic drivers. The CTNNB1 + MET, CTNNB1 + YAP, and myristoylated Akt + Nras(V12) HDT tumor models were each previously shown to drive HCC with defined latency (Chow et al., 2012; Tao et al., 2014; 2016). In all three models, the addition of human or mouse ARID1A overexpression increased tumor burden and liver/body weight ratios (Figure 2K–2M).
Arid1a promoted liver cancer initiation by increasing reactive oxygen species
To explain how ARID1A accelerated tumor initiation, we probed underlying molecular changes downstream of Arid1a perturbation. RNA-seq data from the non-malignant, MYC-expressing livers of Rag1−/−; LAP-tTA+/+; TRE-MYC mice with and without ARID1A overexpression showed the clustering of samples based on ARID1A expression, with 241 up and 190 downregulated genes (Figure 3A and Table S1). Gene Set Enrichment Analysis (GSEA) revealed that Hnf4a’s transcriptional targets were upregulated in tissues overexpressing ARID1A (Figure 3B). Hnf4a is known to drive the transcription of CYP450 genes, a superfamily of monooxygenase enzymes that oxidizes exogenous and endogenous metabolites (Tirona et al., 2003; Wiwi and Waxman, 2005). Indeed, many CYP450 genes were increased in ARID1A overexpressing livers (Figure 3C) and decreased in Arid1a deficient livers (Sun et al., 2016). qPCR confirmed that CYP450 genes were down and upregulated in Arid1a loss and gain of function livers, respectively (Figure 3D and 3E). To determine if Cyp2e1 is directly downstream of Arid1a, we examined ChIP-Seq data generated from WT liver tissues and found that ARID1A and the SWI/SNF complex bound to multiple putative enhancers and the promoter of Cyp2e1 (Figure 3F). To quantify CYP450 activity, we injected acetaminophen (APAP) into Arid1a WT and Homo mice. APAP is oxidized by CYP450 to form NAPQI, a reactive metabolite that ultimately forms APAP protein adducts, which in turn cause hepatic necrosis. APAP protein adducts were lower in Arid1a Homo livers, indicating reduced CYP450 activity (Figure S3A).
Figure 3. ARID1A drives oxidative stress via CYP450 expression, thereby promoting MYC induced liver cancers.

(A) Differentially expressed genes from an RNA-seq experiment comparing normal liver tissues from Rag1−/−; LAP-MYC + Ad-Cre and Ad-ARID1A overexpression (n = 3 and 3 mice, red is higher and blue is lower expression).
(B) GSEA for Hnf4a target gene enrichment in ARID1A overexpressing livers.
(C) Differentially expressed CYP450 family genes.
(D)CYP450 mRNA levels in LAP-MYC; Arid1a WT and Homo livers (n = 6 and 6 livers), as measured by qPCR.
(E)CYP450 mRNA levels in ARID1A overexpressing livers that are expressing MYC (n = 6 and 6 livers), as measured by qPCR.
(F) ChIP-seq tracks on the murine Cyp2e1 locus for H3K27ac (marking active enhancers and promoters), V5-ARID1A, and Cebpα (a known transcriptional activator of CYPs). Red and blue mark significant peaks over input.
(G) ROS levels in LAP-MYC + Ad-Cre vs. Ad-ARID1A livers, as measured by the DHE assay. Scale bar = 50 µm.
(H) ROS intensity fold change from DCFH-DA assays performed in normal hepatocytes from LAP-MYC; Arid1a WT, Het, and Homo mice.
(I) Representative gross images of LAP-MYC; Arid1a WT livers treated with vehicle or NAC and sacrificed at p45. Scale bar = 10 mm. Histology of LAP-MYC; Arid1a WT livers treated with vehicle or NAC. Tumor are outlined (n = 6 and 6 mice). Scale bar = 500 µm.
(J) Kaplan-Meier plot of LAP-MYC; WT and Arid1a Het mice treated with vehicle or NAC.
(K) Kaplan-Meier plot of LAP-MYC; WT and Arid1a Het mice treated with AAV-GFP or AAV-Cyp2e1.
(L) Liver to body weight ratios for LAP-MYC; Arid1a WT and Het mice treated with AAV-GFP or AAV-Cyp2e1 and sacrificed at p56.
(M) Representative gross images of LAP-MYC; Arid1a Het mice + AAV-GFP or AAV-Cyp2e1, sacrificed at p56. Scale bar = 10 mm.
(N) Representative fluorescent images of livers from these mice, as measured by DHE assay. Scale bar = 50 µm.
Two-tailed Student’s t-tests (two-sample equal variance) were used to test the significance of differences between two groups. Kaplan-Meier method was used to estimate survival curves, which were compared using the log-rank test. All data in this figure are represented as mean ± SEM, * (p < 0.05), ** (p < 0.01), *** (p < 0.001). See also Figure S3 and Table S1.
Because Cyp2e1 is a generator of reactive oxygen species (ROS), which are well-known mediators of liver injury and hepatocarcinogenesis, we hypothesized that ARID1A promoted cancer initiation in part through its transcriptional control of Cyp2e1 expression, which in turn stimulated ROS production from endogenous metabolites. We quantified ROS levels in premalignant MYC overexpressing livers using a fluorescence flow cytometry assay based on the oxidation of 2’,7’-dichlorofluorescin-diacetate (DCFH-DA) and a confocal imaging assay staining for dihydroethidium (DHE). These methods showed that LAP-MYC; ARID1A overexpressing tissues had increased ROS (Figure 3G), while Arid1a Het and Homo hepatocytes had reduced ROS (p < 0.05; Figure 3H and Figure S3B–S3E). To determine if decreasing ROS mimicked Arid1a loss, we employed the antioxidant N-acetyl cysteine (NAC). NAC treatment reduced ROS (Figure S3F and S3G), tumor burden (Figure 3I) and improved median OS of LAP-MYC; Arid1a WT mice from 55 to 81 days (p < 0.0001; Figure 3J). These data are consistent with the known tumor inhibitory effects of NAC (P. Gao et al., 2007; Lin et al., 2013). In terms of effect size, the NAC curve mirrored the vehicle treated Arid1a Het curve, suggesting that NAC reproduced an antioxidant state similar to that of Arid1a haploinsufficiency.
To determine if reduced Cyp2e1 was responsible for the antioxidant and tumor protective effect, we attempted to counter the delayed tumor initiation by overexpressing Cyp2e1 in Arid1a Het mice. AAV-Cyp2e1 had little impact on LAP-MYC; Arid1a WT mice, while it accelerated death in Het mice (Figure 3K). AAV-Cyp2e1 treated Hets had increased liver to body weight ratios and tumor burden than AAV-GFP treated Hets at 56 days of age (Figure 3L and 3M). Mice injected with AAV-Cyp2e1 had increased Cyp2e1 expression (Figure S3H and S3I) as well as ROS (Figure 3N). This demonstrated that Cyp2e1 suppression, secondary to Arid1a loss, reduced oxidative stress and tumorigenesis. We also examined Cyp2c29, which is closely related to Cyp2e1, and found that it was also significantly up or downregulated in response to Arid1a overexpression or loss, respectively (Figures 3D and 3E). Cyp2c29 could also rescue the delayed tumorigenesis of LAP-MYC; Arid1a Het mice (Figure S3J and S3K).
Dynamic Arid1a expression patterns in mouse and human HCC
Given the evidence for the pro-cancer activities of ARID1A, we explored the expression pattern of ARID1A in HCC to understand when and why deleterious mutations are found in HCC samples. We reasoned that if ARID1A were a simple tumor suppressor, then protein expression should be frequently reduced in cancer samples. In chemically-induced murine HCC, there was equivalent ARID1A protein expression in tumor compared to adjacent normal tissues (Figure S4A and S4B). In the LAP-MYC model, ARID1A was modestly overexpressed in tumor as compared to adjacent normal tissues (Figure S4C) and the absence of expression was confirmed in Homo livers (Figure S4D). We then performed ARID1A immunostaining in 34 human HCC samples whose intensities were graded as “none”, “weak”, “moderate”, or “strong” (Figure 4A). All adjacent non-cancerous background tissues had either weak or moderate staining (Figure 4B). 86% (29/34) of the tumor samples had strong or moderate expression and 15% (5/34) had no expression (Figure 4C). ARID1A overexpression in HCC was also seen in other HCC databases (Figures S4E and S4F). These data were consistent with a previous report showing that 101/115 (88%) of HCC have higher or equal amounts of ARID1A expression as compared to adjacent liver (Zhao et al., 2015). Thus, ARID1A expression was frequently maintained or overexpressed in both mouse and human liver tumors, supporting the possibility that ARID1A has a promotional role in cancer development.
Figure 4. ARID1A expression in human HCC.

(A) Representative H&E histology and ARID1A staining in human HCC samples, scored as 0 (none), 1+ (weak), 2+ (moderate), and 3+ (strong). Scale bar = 100 µm.
(B) Representative picture of adjacent non-malignant liver showing 1 to 2+ ARID1A staining. Scale bar = 100 µm.
(C) Proportion of human HCC with each level of ARID1A staining.
(D) ARID1A staining in human primary HCC and metastatic samples. 21 paired samples were analyzed, and four representative pairs with loss of expression in metastases are shown. Scale bar = 100 µm.
One explanation to rectify the mouse and human data is the possibility that ARID1A is required early in tumorigenesis but is then mutated, lost, or suppressed at later time points. This phenomenon was observed for ARID1A in endometrial cancer, where primary tumors expressed wild-type ARID1A but metastatic subclones harbored deleterious mutations (Gibson et al., 2016). Among common cancer driver genes, ARID1A was the only gene mutated in this subclonal fashion. To assess this scenario in human HCC, we screened over 1500 patients and found 21 with paired primary and metastatic HCC samples. 7/21 (33%) showed substantial loss of ARID1A in the metastatic versus the primary lesion (Figure 4D). This indicated that late ARID1A loss was an observed event in the context of human HCC metastatic progression.
We examined the possibility that a haploinsufficient dose is an alternative mechanism for retaining ARID1A during initiation that also allowed for dose reduction during the course of progression. When we re-analyzed all previously identified ARID1A mutations from four HCC sequencing studies, we found only 4 of 65 tumor samples (6.1%) had biallelic ARID1A gene lesions (Fujimoto et al., 2012; Guichard et al., 2012; Kan et al., 2013; Schulze et al., 2015) (Table S2). Furthermore, while copy number variation analysis of gene gain and loss in Schulze et al. identified recurrent homozygous deletions of CFH, IRF2, CDKN2A, PTPN3, PTEN, AXIN1 and RPS6KA3, only one of 30 samples (3.3%) with ARID1A mutations showed homozygous deletion (Schulze et al., 2015). A majority of HCC samples that had ARID1A mutations carried monoallelic alterations, thus suggesting that the ARID1A haploinsufficient state is a phenotypically relevant one.
Homozygous and heterozygous Arid1a loss in tumors promoted growth and metastasis
This encouraged us to ask if the impact of Arid1a loss is dependent either on timing or dose. Although LAP-MYC; Arid1a Homo mice had improved survival (Figure 1I), we hypothesized that this was due to differences in tumor initiation. To interrogate progression in isolation, we injected Ad-Cre into LAP-MYC; Arid1aFl/Fl and LAP-MYC; Arid1a+/+ mice with pre-existing tumors and 48 hours later, tumor fragments were harvested and transplanted into NSG mice, where Arid1a deleted tumors grew more rapidly (Figure 5A and 5B). Similar results were obtained with pre-formed LAP-MYC; Arid1a WT and Homo tumors (Figure 5A and 5B). This showed that Arid1a deletion accelerated tumor once tumors had formed, indicating that Arid1a promoted tumor initiation but its presence in later tumor stages impaired progression.
Figure 5. Homozygous and heterozygous loss of Arid1a in established liver cancers promoted progression and metastasis.

(A) Schema for assessing the influence of Arid1a loss in established tumors. LAP-MYC; Arid1a+/+ or Arid1aFl/Fl mice were given Ad-Cre (2 × 109 PFU/mouse delivered IV) after tumors were established in the endogenous model. Tumor fragments were transplanted into NSG flanks and growth was measured. A second approach involved transplanting established LAP-MYC; Arid1a WT or Arid1a Homo (Alb-Cre mediated) tumors.
(B) Growth rate of tumor fragments (n = 10 tumors in each group were measured and each line represents one tumor). Tumor genotyping for Arid1a locus shown below for the Ad-Cre experiment.
(C) Kaplan-Meier plot of LAP-MYC; WT and Arid1a Het mice in the mixed strain background.
(D)LAP-MYC; Arid1a Het mice developed metastases to the peritoneum, mesenteric lymph nodes, and lungs (yellow arrowheads). Scale bar = 10 mm.
(E) Histology of gut (40x, scale bar = 500 µm) and lung (200x, scale bar = 100 µm) metastases from LAP-MYC; Arid1a Het mice.
(F) Mice with distant metastases in the LAP-MYC model. Intrahepatic metastases were not included.
(G) Gross (Scale bar = 5 mm) and H&E images (Scale bar = 200 µm) of lung metastases from NSG mice injected IV with LAP-MYC; Arid1a WT or Het cancer cells.
(H) Tumor burden in livers and lungs of Arid1a+/+ and Arid1aFl/+ mice given DEN at p14 and AAV-Cre at p28. Mice were examined at 11 months of age. Scale bar = 10 mm for livers and scale bar = 5 mm for lungs.
(I) Mice with distant metastases in the DEN model. Intrahepatic metastases were not included.
Kaplan-Meier method was used to estimate survival curves, which were compared using the log-rank test. All data in this figure are represented as mean ± SEM, ** (p < 0.01). See also Figure S5.
We also examined Ubc-CreERT and Mx-Cre; Arid1aFl/Fl inducible knockout mice to confirm that Arid1a loss after tumorigenesis promoted growth (Figure S5A). We gave DEN at p15 then performed magnetic resonance imaging (MRI) 7 months later to confirm the presence of tumors (Figure S5B). We then deleted Arid1a with either tamoxifen or poly I:C and waited another 4 months. Both inducible Cre models showed larger tumors after induced Arid1a deletion (Figure S5C–S5E), consistent with the LAP-MYC transplant data.
Next, we assessed the possibility that heterozygous ARID1A loss was sufficient to drive progression after the initiation of tumors. As mentioned earlier, LAP-MYC; Arid1a Het mice had improved survival as compared to WT mice in a pure FVB strain background (Figure 1I). In a mixed B6/129/FVB cohort with longer latency and incomplete penetrance, all Arid1a Het mice succumbed to disease while 22% of WT mice achieved long-term survival, suggesting unfavorable tumor biology in Arid1a Het mice (Figure 5C). In support of this, we observed metastatic disease in 8 of 32 LAP-MYC; Arid1a Het mice, while none of 25 Arid1a WT mice had metastatic spread (Figures 5D–F). To test the metastatic potential of established tumors, we isolated WT and Het tumor cells and injected them into circulation, after which Het cells caused higher rates of lung metastasis (Figure 5G).
We also asked if Arid1a suppression was associated with metastasis in the DEN model, so we injected AAV-Cre into Arid1aFl/+ mice two weeks after DEN mutagenesis (same schema as Figure 1A). After 11 months, we identified reduced primary tumor burden but increased distant metastatic spread in Arid1a Het mice (Figure 5H and 5I). In sum, these data showed that heterozygous loss of Arid1a potentiated metastasis in established tumors.
Arid1a haploinsufficiency alters global chromatin occupancy and metastasis genes
We next sought to determine how both heterozygous and homozygous Arid1a loss were sufficient to drive more aggressive cancer phenotypes. Arid1a heterozygosity led to protein levels that were intermediate between WT and Homo samples (Figure 6A). Given the biochemical hypothesis that SWI/SNF remodels chromatin by using ATP to evict nucleosomes, we asked if both partial and complete Arid1a loss were sufficient to reduce chromatin density by performing ATAC-seq, a technique to assess chromatin accessibility. Compared to WT tumors, there were fewer ATAC-seq peaks in Het and Homo tumors (14489 vs. 11181 and 9609, respectively), indicating decreased chromatin accessibility across the genome upon Arid1a reduction (Figure 6B). When we examined the 3256 peaks uniquely called in WT tumors, we found similar levels of reduced accessibility in both Het and Homo tumors (Figures 6C and 6D). RNA-seq expression profiling was also performed to assess transcription associated with these epigenetic changes. Het and Homo tumor expression profiles were divergent from WT but clustered together, indicating that the transcriptional impact of partial or complete Arid1a loss was similar (Figure 6E and (Table S3). More genes were downregulated (442) than upregulated (193), an observation that was consistent with reduced chromatin access for transcription factors. Indeed, reduced chromatin accessibility is closely associated with a subset of these differentially regulated genes (Figures 6F). These data showed that the haploinsufficient dose of Arid1a was sufficient to exert a genome-wide impact on chromatin accessibility and gene expression to a similar extent as homozygous loss.
Figure 6. Partial and complete Arid1a loss increased chromatin occupancy and altered gene expression to similar extents.

(A) ARID1A protein expression in LAP-MYC tumors.
(B) ATAC-seq peaks in LAP-MYC; Arid1a WT (n = 4), Het (n = 4), and Homo (n = 3) tumors. Total peaks called are in parentheses.
(C) 3256 ATAC-seq peaks uniquely called in WT tumors are visualized here.
(D) The global view of chromatin accessibility for these 3256 tracks.
(E) RNA-seq of LAP-MYC; Arid1a WT (n = 4), Het (n = 4), and Homo (n = 4) tumors from pure FVB mice. Of 634 differentially expressed genes, 193 were up and 442 were downregulated.
(F) ATAC-seq peaks for 153 genes that are differentially expressed after Arid1a loss. See also Table S3.
To identify specific molecular mechanisms of increased aggressiveness associated with Arid1a heterozygosity, we examined another set of RNA-seq expression profiles from mixed strain LAP-MYC; Arid1a WT and Het primary tumors prone to metastasis (Figure S6A–S6C, Table S4). GSEA showed that cell cycle and E2F target genes were upregulated and liver-specific genes were downregulated, suggesting a more proliferative, less differentiated state (Figure 7A). GSEA also showed that programs associated with increased cell motility and metastasis were upregulated in Arid1a Het cancers (Figure 7B). In agreement with this analysis, we found that a high fraction of differentially expressed genes was associated with metastasis (73 of 383 differentially expressed genes (19%); Figure 7C).
Figure 7. Arid1a haploinsufficiency promoted gene expression programs that supported cell migration, invasion, and distant lung colonization.

(A) GSEA for upregulation of cell cycle and E2F target gene pathways and downregulation of liver-specific genes.
(B) GSEA showed upregulation of metastasis and cell migration pathways.
(C) Differentially expressed genes associated with metastasis from mixed strain background LAP-MYC; Arid1a WT and Het tumors (n = 8 and 8 mice, red is higher and blue is lower expression).
(D) Knockdown of ARID1A protein with shRNAs in human Huh7 HCC cells.
(E) In vitro migration assay for shGFP and shARID1A in Huh7, with empty vector and EMILIN1 cDNA lentiviral overexpression. 40x images show wound size at time 0 and 48 hr after scratch. Scale bar = 500 µm.
(F) Relative change in area covered by cells between time 0 and 48 hr.
(G) ChIP-Seq data showing binding of EMILIN1 MAT1A, LCN2, and IL1R1 loci by ARID1A and SNF5 over input in human HepG2 hepatoma cells (data from Raab et al.).
(H) Schema for evaluation of metastasis suppressor genes. Murine Hepa1c1c7 cells with shARID1A knockdown and lentiviral rescue were transplanted IV into NSG mice, then organs were harvested and luciferase imaged after 6 weeks.
(I) Gross images of tumor burden in the livers and lungs of NSG mice 6 weeks after Hepa1c1c7 cells were injected IV. On right, number of gross tumors on organ surfaces (n = 5 mice for shGFP, n = 8 mice for shARID1A). Scale bar = 10 mm for livers and scale bar = 5 mm for lungs.
(J) Quantification of lung macromets from n = 5 (shGFP) and 7 (shARID1A) mice per group.
Two-tailed Student’s t-tests (two-sample equal variance) were used to test the significance of differences between two groups. All data in this figure are represented as mean ± SEM, * (p < 0.05), ** (p < 0.01), *** (p < 0.001). See also Figure S6 and Table S4.
To screen for functionally relevant metastasis genes, we first used shRNA to determine if ARID1A loss was sufficient to promote migration and/or invasion in HCC cells. We observed that ~70% ARID1A knockdown in human Huh7 cells (Figure 7D) resulted in greater migration (Figure 7E). To ensure that shRNAs were causing a relevant dose reduction of ARID1A, we examined Huh7 cells treated with distinct doses of ARID1A siRNA as well as ARID1A wild-type, heterozygous, and homozygous mouse H2.35 cells generated using Cas9 gene-editing (Figure S6D and S6E). Both sets of cells replicated the shRNA results in migration assays, indicating that shRNA knockdown faithfully modeled the dose dependent loss of ARID1A.
Next, we generated a lentiviral overexpression library of 52 candidate genes corresponding to genes implicated in progression/metastasis and downregulated in Het tumors (genes from Figure 7C). We confirmed that many of these genes were also downregulated in the setting of Arid1a knockdown in Huh7 (Figure S6F), and that lentiviral overexpression was effective (Figure S6G and S6H). We reasoned that the overexpression of some of these candidate metastasis suppressing genes would block the pro-migratory state of ARID1A deficient cells. This screen identified genes that satisfied three criteria: 1) significantly reduced the migration of ARID1A knockdown cells, 2) had little to no effect on WT cells, and 3) abrogated the migratory differences between WT and ARID1A knockdown cells (Figures 7E and 7F). Among these genes, EMILIN1, MAT1A, IL1R1, and LCN2 were previously associated with metastasis suppression (Dagenais et al., 2017; Danussi et al., 2012; Wang et al., 2013; J. Zhang et al., 2013). To determine if these genes have direct interactions with ARID1A and SWI/SNF components in human HCC, we examined ChIP-Seq experiments from HepG2 cells performed by Raab et al. Both ARID1A and SNF5 showed physical interactions with the promoters of EMILIN1, MAT1A, IL1R1, LCN2, suggesting direct regulation (Figure 7G).
To more fully validate these candidate genes, we performed additional in vitro and in vivo metastasis assays (Figure S6I–L and Figure 7H–J). Migration and transwell invasion assays in Huh7, Hepa1c1c7, and HepG2 HCC cells showed that ARID1A knockdown promoted invasion and migration, and that EMILIN1, MAT1A, IL1R1, and LCN2 suppressed these activities (Figure S6I–K). To evaluate these phenomena in vivo, we injected Hepa1c1c7 cells with and without shARID1A into the circulation of NSG mice and found that shARID1A promoted lung colonization (Figure 7H and 7I). Overexpression of LCN2, IL1R1, MAT1A, or EMILIN1 abrogated the increased lung colonization seen with ARID1A downregulation (Figure 7J and Figures S6L). Altogether, our data showed that Arid1a suppression resulted in impaired terminal differentiation, increased metastatic potential, and provide a molecular basis for the observed increased aggressiveness in Arid1a Het tumors (Figure 8).
Figure 8. Model for oncogenic and tumor suppressive activities of ARID1A.

Discussion
ARID1A is presumed to be a tumor suppressor based on loss-of-function mutational profiles observed in a broad variety of cancers (Hohmann and Vakoc, 2014; Wu and Roberts, 2013). Our data confirmed tumor suppressive functionality for Arid1a within established DEN-induced tumors, corroborating the findings of Fang et al. We extended these findings to the MYC liver cancer model. Since Fang et al. evaluated DEN-induced tumor burden at relatively late stages, it is possible that there were differences in tumor initiation at much earlier stages. In addition, differences such as the use of chronic CCl4, AAV-Cre, or the microbiome in our models may have also contributed to potential differences. The advance in our study is that in multiple in vivo liver cancer models, ARID1A clearly has tumor promoting functions during the early phases of transformation. It is likely that contrasting results found in different cancer models underscores the context-dependency of Arid1a function.
By examining how ARID1A overexpression promoted carcinogenesis, we identified a mechanism that linked ARID1A, oxidative stress, and carcinogenesis through the CYP450 system. We previously showed that Arid1a loss resulted in a hyper-regenerative state associated with increased proliferation and decreased maturation without spontaneous cancer formation (Sun et al., 2016). Reduced oxidative stress reconciles the observations of lower tissue damage and less HCC initiation in Arid1a null livers. Consistent with our findings, antioxidants have been shown to both antagonize tumor initiation and promote tumor progression (Lin et al., 2013; Piskounova et al., 2015).
Clinical observations from human cancer samples also suggest complex roles for ARID1A. Human mRNA expression and mutation analyses suggest the possibility that there may be a role for wild-type ARID1A expression at some point in tumorigenesis. The pro-cancer functions identified here in tandem with human loss-of-function mutations could be reconciled by the fact that ARID1A activity changes depending on contextual variables such as timing and dose. Though late stage ARID1A mutations do occur, as we have shown, it is unlikely that ARID1A loss only occurs during metastasis. It is possible that ARID1A is lost immediately after tumor initiation, but it would be challenging to pinpoint the timing of such a mutational event.
Our work also showed that Arid1a haploinsufficiency was sufficient to drive key aspects of tumor progression and will be a relevant state to investigate in future mouse or human models. The fact that Arid1a heterozygosity promoted metastasis is consistent with previous studies that showed partial ARID1A loss in human gastric and liver cancer samples was associated with metastasis and that incomplete ARID1A knockdown in cancer cell lines promoted migration and invasion (He et al., 2015; Huang et al., 2012; Yan et al., 2014). While we provided in vivo evidence that this also occurred in genetically engineered mouse models and offered mechanisms by which this occurs, it is likely that the downregulation of many metastasis or growth suppressing genes collectively contribute to the phenotypic effects of ARID1A loss in tumors, and our study identified only a subset of such genes. Other possibilities are that ARID1A loss may be tumorigenic by protecting tumor cells from lowered ROS levels or by decreasing HNF4A expression.
It is not surprising that epigenetic machines such as ARID1A have highly context-specific functions. Aberrant SWI/SNF dependent chromatin-remodeling could in principle support the actions of both oncogenic and tumor suppressive networks, resulting in directionally opposite effects. The net carcinogenic effect likely stems from the context in which ARID1A mutations are found, such as tissue, cooperative mutations, timing, or dose. Previous studies of Brg1, the ATPase component of the SWI/SNF complex, showed that it has stage dependent functions in lung and pancreatic cancers (Glaros et al., 2008; Roy et al., 2015). Our study identified unexpected and potent roles for ARID1A in a series of liver cancer models, but cannot be used to stereotype ARID1A functions for all settings. These findings underscore the importance of considering stage, dose, and tissue context when evaluating the function of ARID1A and when contemplating therapeutic strategies to modulate epigenetic machinery in cancer.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Hao Zhu (hao.zhu@utsouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
H2.35 cells (Source: mouse, female, strain BALB/c) were cultured in Dulbecco’s modified Eagle Medium (Life Technologies) supplemented with 4% fetal bovine serum and 200 nM dexamethasone. Hepa1c1c7 (Source: mouse, strain C57L) , Huh7 (Source: human, male) and 293T (Source: human, male) cells were cultured in Dulbecco’s modified Eagle Medium (Life Technologies) supplemented with 10% fetal bovine serum. All cells were cultured at 37 °C in a humidified atmosphere with 5% CO2.
Mice
All mice were approved by and handled in accordance with the guidelines of the Institutional Animal Care and Use Committee at UTSW. In Arid1a floxed mice, induced deletion between the two loxP sites produced cells lacking exon 8 of Arid1a (see Table S1 for primers), which resulted in a frameshift mutation and nonsense-mediated decay in the resulting transcript (X. Gao et al., 2008). The strain backgrounds of these mice were a mix of C57/B6, 129, and FVB, unless otherwise noted. All experiments were done in an age and sex controlled fashion unless otherwise noted in the figure legends.
DEN and CCl4 cancer model
Diethylnitrosamine (DEN, Sigma) was injected intraperitoneally (IP) at age p14 then chronic liver injury was induced with biweekly carbon tetrachloride (CCl4, Sigma) injections.
DEN and AAV-Cre cancer model
DEN was given to Arid1aFlFl and Arid1a WT mice at age p14 by IP injection. Two weeks later, 5×1010 genomic particles AAV8-Cre was injected to delete Arid1a.
LAP-MYC liver cancer model
LAP-MYC liver cancer model was driven by liver-specific MYC overexpression. Tetracycline transactivator (tTA) expression was under the control of the liver activator protein (LAP). The c-MYC transgene was under the control of the tetracycline response element (TRE). Conditional deletion of Arid1a was induced with Alb-Cre. Doxycycline (Dox; 1 g/liter in water) was removed at birth to induce MYC.
Transposon liver cancer model
HCC inducing oncogenes (pT3-β-Catenin/pT3-MET/pT3-YAP) were introduced with and without pT3-hARID1A or pT2-mArid1a along with SB2 into wild-type FVB mice. Livers were harvested at a fixed time after hydrodynamic transfection (HDT) to determine tumor burden. LAP-MYC mice were induced at 4 weeks of age, then between 4–5 weeks of age, HDT with either pT3-empty or pT3-hARID1A along with SB2 was performed.
Mx-Cre and Ubc-CreERT models
DEN was given IP to Arid1aFl/Fl and Mx-Cre; Arid1aFl/Fl mice or Ubc-CreERT; Arid1aFl/Fl mice at p15 then MRI was performed 7 months later to confirm the presence of tumors. Arid1a was deleted with either tamoxifen or poly I:C and 4 months later mice were sacrificed to determine tumor burden.
Human samples
34 HCC samples from Figure 4A were obtained from percutaneous biopsies from patients who underwent informed consent. HCC diagnosis and ARID1A staining intensity were confirmed by a board-certified pathologist specializing in gastrointestinal oncology (P.G.) in a blinded fashion. The study was performed under IRB #STU 062013-063. 21 paired primary and metastatic HCC from Figure 4D were obtained from the Department of Pathology at Zhongshan Hospital, Fudan University. All samples were re-evaluated independently by two pathologists (Y. Ji and F. Tian) before further analysis. For all of the HCC samples used in this study, the mean age range for HCC patients was 57 years (range: 25–86) and 91% of the samples were male. The Ethics Committee of Zhongshan Hospital approved the collection and use of human samples.
METHOD DETAILS
Histology
Tissue samples were fixed in 4% paraformaldehyde (PFA) and embedded in paraffin. Primary antibodies used were Ki-67 (Abcam ab15580), p-H3 (Cell Signaling #9706), ARID1A (Sigma HPA005456), V5 (Cell Signaling 13202). Detection was performed with the Elite ABC Kit and DAB Substrate (Vector Laboratories), followed by hematoxylin counterstaining (Sigma).
Chemical injury experiments
CCl4 was diluted 1:10 in corn oil (Sigma) and administered with IP injections at 0.5 mL/kg (Beer et al., 2008). Mice were injected and monitored twice per week. Diethylnitrosamine was given IP at a dose of 25 mg/kg at two weeks of age. Mice were given 500 µl of 20 mg/ml tamoxifen by oral gavage for 2 consecutive days to induce whole body Cre excision mediated by Ubc-CreERT.
N-acetyl cysteine
Mice were injected with N-acetyl cysteine (NAC; Sigma) at 200 mg/kg mixed in PBS at 28 and 29 days of age. The mice were then maintained on NAC water (1 g/L), which was replaced every two to three days. The pH of the water was adjusted to 7.0–7.4.
siRNA transfection
siRNA oligonucleotides were synthesized from Life Technology (s15785). 3×105 Huh7 cells were plated in 6 well plate, cells were transfected with scramble or ARID1A siRNA using Lipofectamine RNAiMAX Transfection reagent (Life Technologies).
APAP adduct measurements
Mice were given 300 mg/kg by IP injections. 24 hr later, liver samples were collected, snap frozen, crushed, and powdered under liquid nitrogen. APAP-Cys adducts in a portion of the liver powder were quantified by high pressure liquid chromatography and electrochemical detection (HPLC-ECD) as described previously (Heard et al., 2011).
ROS detection
2,7-Dichlorofluorescin-diacetate (DCFH-DA)) (Life Technologies) is oxidized by ROS to form fluorescent 2,7-dichlorofluorescein (DCF) (Rosenkranz et al., 1992). Primary hepatocytes were isolated as previously described (W.-C. Li et al., 2010). Cells were incubated with 10 µM DCFH-DA for 30 min in 37 °C , transferred to ice, an d underwent flow cytometry. The average intensity of DCF was used as a marker for intracellular ROS levels. For ROS detection in fresh tissues, dihydroethidium(DHE)(Sigma) staining for superoxide was performed (Owusu-Ansah et al., 2008). Cryosections (30 µm) were freshly cut and incubated with 10 µM DHE at 37°C for 30 min. Ethidium staining was visualized using laser confocal microscopy (LSM 780, Zeiss) at the UTSW Live Cell Imaging Facility. The red signal corresponded to levels of cellular superoxide anion and the intensities were quantified in five fields for each mouse by ImageJ.
Adenoviruses and adeno associated viruses
5×1010 genomic particles of AAV8.TBG.PI.Cre.rBG (University of Pennsylvania Vector Core), AAV8.TBG.PI.eGFP.WPRE.bGH or AAV8.TBG.PI.Cyp2e1.rBG or AAV8.TBG.PI.Cyp2c29.rBG in 100µL of buffer were injected retro-orbitally. For ARID1A overexpression experiments, full-length human ARID1A (NM_006015.4) was cloned into an adenovirus serotype 5 construct containing a CMV promoter. Adenovirus carrying Cre and ARID1A (Vector Biolabs) were injected at a dose of 1×109 viral PFU/mouse.
Lentivirus production
Gateway donor vectors containing cDNA of candidate metastasis suppressor genes were obtained from The UTSW McDermott Sequencing Core. cDNAs of target genes were inserted from donor vectors into the plenti7.3 destination vector (Invitrogen) using Gateway LR Clonase II Plus enzyme mix (Invitrogen) as described in the manufacturer’s instructions. The integrity and fidelity of all constructs were verified by DNA sequencing. 293T cells were cultured in DMEM supplemented with 10% fetal bovine serum (Sigma) and were maintained at 37 °C in a humidified atmosphere with 5% CO2. For virus production, 3.5 µg of the appropriate plasmid and 3.2 µg of helper plasmids (2.7 µg of gag-pol and 0.5 µg of VSVg) were transfected into 293T cells cultured at 50% confluence in a 10 cm dish using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. Viral supernatants were collected 48 hr after transfection and filtered through a 0.45 µm filter.
Cell proliferation, migration and invasion assays
For proliferation, 2.5×104 cells were plated in triplicates in 12-well plates and cell numbers were counted for 3 days. For migration, 2×105 cells stably expressing either shRNA against Arid1a or GFP were plated into 6well plates the day before infection. When the cells reached ~70% confluence, 1 mL of lentiviral supernatant was added and removed 24 hr later. The cells were starved in DMEM supplemented with 2% FBS to inhibit proliferation. After at least 18 hr of starvation, a wound was created by scraping dry cells with a 200 µL pipette tip and the media was replaced with fresh DMEM with 2% FBS. Images were taken immediately after the scratch and 48 hr later. The wound sizes of were assessed using MRI Wound Healing Tool (http://dev.mri.cnrs.fr/projects/imagej-macros/wiki/Wound_Healing_Tool) in ImageJ. The rate of migration was calculated as the change in wound areas. Invasion assays were performed using transwell filter chambers coated with matrigel (Corning). According to the manufacturer’s instructions, 5×104 cells in 500 µL serum-free DMEM were seeded in the upper chamber of a transwell and 750 µL medium supplemented with 20% FBS was added to the lower chamber. After 24 hr incubation, cells migrated through the membrane were fixed, stained, and counted with a light microscope.
Xenograft models
Hep1c1c7 cells were infected with lentivirus expressing shRNA against GFP or Arid1a and selected with 1 µg/mL puromycin. Then cells were infected with lentivirus overexpressing candidate genes and after 24 hr, cells were sorted for GFP to obtain transduced cells. The cells were then infected with a luciferase lentivirus which is used for in vivo bioluminescence imaging. Viability was confirmed before inoculation into NSG mice. For tail vein injections, 0.5×106 cells were used. Six weeks after tail vein injection, tumor burden in mice was evaluated with bioluminescence imaging. Primary LAP-MYC tumors were dissociated as previously described (Piskounova et al., 2015). Briefly, the tumors were harvested and dissociated sterilely using a closed system tissue grinders (SKS Science) followed by enzymatic digestion using 200 U/mL collagenase IV (Worthington) for 20 min at 37°C. DNase (50–100 U/mL) was added to reduce clumping of cells during digestion. Cells were filtered with a 70 mm cell strainer to obtain single cell suspensions. 1×106 cells were injected into NSG mice through the tail vein. For xenograft experiments, LAP-MYC; Arid1a+/+ or LAP-MYC; Arid1aFl/Fl mice were given IV Ad-Cre (2 × 109 PFU/mouse delivered IV) after tumors were clearly established. 48 hr later, 30 mm3 tumor fragments were implanted in the subcutaneous tissue of the flanks of NSG mice. The tumor volumes were measured starting one week post-transplant and established as the baseline. The growth of each tumor was calculated relative to its own baseline.
RNA extraction and RT-qPCR
Total RNA was isolated using Trizol reagent (Invitrogen). cDNA synthesis was performed with 1µg of total RNA using iScript II Reverse Transcription Kit (Biorad). See Table S5 for primers used. Gene expression levels were measured using the DDCt method as described previously (H. Zhu et al., 2010).
Western blot
Tissue was lysed in Tissue Protein Extraction Reagent (Thermo Fisher). Western blots were performed in the standard fashion. The following antibodies were used: ARID1A (Sigma HPA005456 and LifeSpan Biosciences LS-C287870), Cyp2e1 (Abcam ab28146), β-Actin (Cell Signaling 4970), Tubulin (Cell Signaling 3873), Vinculin (Cell Signaling 13901), V5 (Cell signal 13202), anti-rabbit IgG, HRP-linked antibody (Cell Signaling 7074) and anti-mouse IgG, HRP-linked antibody (Cell Signaling 7076).
RNA-Seq
RNA was extracted from LAP-MYC; Arid1a WT, Het and Homo liver tumor tissues or normal tissues from Rag1−/−; LAP-MYC mice injected with Ad-Cre or Ad-ARID1A with the Qiagen miRNeasy Mini kit. RNA-seq libraries were prepared with the Ovation RNA-Seq Systems 1–16 (Nugen) and indexed libraries were multiplexed in a single flow cell and underwent 75 base pair single-end sequencing on an Illumina NextSeq500 using the High Output kit v2 (75 cycles) at the UTSW Children’s Research Institute Sequencing Facility.
ATAC-Seq
ATAC-Seq libraries were constructed using 10 mg of frozen tumor tissue. Frozen tissues were crushed into powder in liquid nitrogen. Tissue was suspended in 1 ml ice-cold PBS, then centrifuged at 2000 g for 5 min at 4°C. The superna tant was removed and the pellet was resuspended in 1 ml LB1 buffer (50 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM EDTA, pH 8.0, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100). Tubes were rocked at 4 °C for 10 min. Sample were transferred to a 2 ml glass douncer, and tissue was dounced with a loose pestle and transfer to a 1.5 ml tube, which was then centrifuged at 2000 g for 5 min at 4 °C . The supernatant was resuspended in 1 ml cold PBS and filtered using a 40 µm cell strainer. Nuclei were counted with a cell counter and 5×104 nuclei were used for the transposition reaction. Transposed DNA fragments were purified using a MinElute Kit (Qiagen) and PCR-amplified using PCR primer1 and a barcoded PCR primer 2 (Nextera DNA Sample Preparation Kits - Index Kit). PCR conditions: 72 °C for 5 min, 98 °C for 30 s, followed by 11 cycles of 98 °C for 10 s, 63 °C for 30 s and 72 °C for 60 s. Amplified libraries were purified with a MinElute Kit and library quality was assessed using the TapeStation system (Agilent). All libraries were sequenced by 75 bp paired-end reads using the NextSeq500 system at the Children’s Research Institute at UTSW Sequencing Facility.
ChIP-Seq
Chromatin Immunoprecipitation (ChIP) was performed as described previously with some modifications (Xu et al., 2010). Briefly, 0.5–1 g frozen liver tissue was minced quickly with a razorblade into small pieces < 0.5 cm3. Tissue was crosslinked in 1% formaldehyde for 20 min at room temperature in PBS and quenched using 125 mM glycine for 10 min at RT. Tissue was dounced in ice-cold PBS first with the loose and later with the tight pestle. This was filtered using a 40 µm cell strainer and then centrifuged at 2000 rpm for 5 min. Cold PBS was used to wash 2x and suspended to count nuclei. 1×107 nuclei were used for the ChIP assay. Chromatin was sonicated to around 500 bp in buffer 0 (10 mM Tris-HCl, 1 mM EDTA, 0.1% sodium deoxycholate, 0.1% SDS, 1% Triton X-100, 0.25% sarkosyl, pH 8.0) with 0.3 M NaCl. Sonicated chromatin was incubated with 5 µg antibody at 4 °C. Antibody against c-MYC (Santa Cruz, SC764X) was used. After overnight incubation, protein A/G Dynabeads (Invitrogen) were added to the ChIP reactions and incubated for 4 additional hr at 4 °C to collect the immunoprecipitated chromatin. Subsequently, Dynabeads were washed 2x with 1 ml of buffer 0, 2x with 1 ml of buffer 0.3 (buffer 0 with 0.3 M NaCl), 2x with 1 ml of LiCl buffer (10 mM Tris-HCl, 1 mM EDTA, 0.5% sodium deoxycholate, 0.5% NP-40, 250 mM LiCl, pH 8.0), and 2x with 1 ml of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). The chromatin was eluted in SDS elution buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.0) followed by reverse crosslinking at 65 °C overnight. ChIP DNA was treated with RNaseA (5 g/ml) and protease K (0.2 mg/ml), and purified using QIAquick Spin Columns (Qiagen). The purified ChIP DNA was used for library preparation. ChIP-seq libraries were prepared using NEBNext ChIP-Seq Library Prep Master Mix Set for Illumina Kit. And library quality was assessed using the TapeStation system (Agilent). All libraries were sequenced with 75 bp paired-end reads using the NextSeq500 Systerm at the Children’s Research Institute at UTSW Sequencing Facility.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-Sequencing Data Analysis
Raw sequencing reads were trimmed to remove adaptors and low quality sequences (Phred score < 20) using trim galore package (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). The sequence reads were aligned to the GRCm38/mm10 with HiSAT2 (Kim et al., 2015). After duplicates removal by SAMtools (H. Li et al., 2009), read counts were generated for the annotated genes based GENCODE V20, using featureCounts (Harrow et al., 2012; Liao et al., 2014). Differential gene analysis was performed use edgeR, using FDR < 0.05 as cutoff.(McCarthy et al., 2012; Robinson et al., 2010) Heatmaps to visualize the data were generated by using GENE-E (Robinson et al., 2010). GSEA analysis was performed with a pre-ranked gene list by log fold change (Subramanian et al., 2005).
ATAC-Seq Analysis
Sequences were aligned with Bowtie2 (Langmead and Salzberg, 2012). Using 3 to 4 replicates for each genotypic condition, we obtained an average of 27 uniquely mapped fragments. Peaks were called using MACS2 (Y. Zhang et al., 2008) with p-value = 1e-5 cuttoff in 200 bps smoothing window. Replicate concordance for each condition were measured by calculating IDR (Irreproducibility Discovery Rate) by IDR package (Q. Li et al., 2011). Overlapping peaks were identified and annotated using ChIPpeakAnno package (L. J. Zhu, 2013; L. J. Zhu et al., 2010). Normalized bigwig files for browser tracks visualization and heatmaps were generated by deepTools (Ramírez et al., 2016).
ChIP-Seq Analysis
Sequences were aligned to mouse genome (mm10) by bowtie2 (version 2.2.3, see Langmead and Salzberg, 2012) with parameter “–sensitive”, we perform filtering by (1) removing alignments with mapping quality less than 10 and (2) removing duplicate reads identified by Picard MarkDuplicates (version 1.92, http://broadinstitute.github.io/picard). Then enriched regions (peaks) were identified using MACS2 (Zhang et al., 2008) (version 2.0.10.20131216), with a q-value cutoff of 0.05. Peak regions were annotated by HOMER (Heinz et al., 2010).
Statistical Analysis
The data in most figure panels reflect multiple experiments performed on different days using mice derived from different litters. Two-tailed Student’s t-tests (two-sample equal variance) were used to test the significance of differences between two groups. Kaplan-Meier method was used to estimate survival curves, which were compared using the log-rank test. In all figures, statistical significance is represented as mean ± SEM, * (p < 0.05), ** (p < 0.01), *** (p < 0.001), ****(p < 0.0001).
DATA AND SOFTWARE AVAILABILITY
The raw sequencing data reported in this paper has been deposited into the NCBI GEO database under accession number GSE95541 and includes the RNA-seq data (GSE95537) and ATAC-seq data (GSE95530) and ChIP-seq data (GSE102607).
Supplementary Material
Highlights.
ARID1A promotes cancer initiation by increasing CYP450 mediated oxidative stress.
ARID1A was frequently overexpressed in primary HCC but lost in metastatic clones.
In established tumors, Het and Homo loss promoted progression and metastasis.
Arid1a loss increased chromatin occupancy, downregulated metastasis suppressors.
Significance.
ARID1A is one of the most commonly mutated genes in human cancer, including liver malignancies. As it is altered in a loss of function manner, it is widely presumed that ARID1A is a tumor suppressor gene. However, there is data suggesting that the gene has tumor promoting effects in certain contexts. Using a broad panel of genetic and chemical models of liver cancer, we show that ARID1A has both oncogenic and tumor suppressive functions. Our findings provide a more nuanced understanding of the carcinogenic function of this frequently altered gene in cancer.
Acknowledgments
We thank J. Shelton and C. Lewis for histology, the UTSW Bioinformatics and the CRI Sequencing Core for sequencing. X.S. was supported by a Hamon Center for Regenerative Science and Medicine Award. S.C.W. is a UTSW Disease-Oriented Clinical Scholar, an American College of Surgeon Faculty Research Fellow, and supported by an American Cancer Society Institutional Research Grant. X.L. is funded by Cancer Prevention and Research Institute of Texas (RP150596). P.G. was supported by the UTSW Department of Pathology Intramural Research Program. L.N. is a HHMI International Fellow. H.Z. was supported by the Pollack Foundation, a NIH/NIDDK R01 grant (R01DK111588), a Burroughs Wellcome Career Medical Award, a CPRIT New Investigator Grant (R1209), a CPRIT Early Translation Grant (DP150077), a DOD Team Science Award (CA150245), and a SUC/AACR Innovative Research Grant (IRG-10–16).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
X.S., S.C.W. and H.Z. designed and performed the experiments and wrote the manuscript. X.S., T.M., M.S., T.H.H., and X.L. generated and analyzed the bioinformatic data. Y.W., Y.J., L.L., M.Z., I.N., J.C.C., C.C., S.Z., L.H.N., L.W., Z.W., assisted with experiments. P.G. analyzed human pathology. L.L. and L.J. performed APAP metabolite analysis. S.F., W.L., L.H., F.T., Y.J., AY., A.S., and H.Z. obtained samples and clinical data.
References
- Beer S, Komatsubara K, Bellovin DI, Kurobe M, Sylvester K, Felsher DW. Hepatotoxin-induced changes in the adult murine liver promote MYC-induced tumorigenesis. PLoS ONE. 2008;3:e2493. doi: 10.1371/journal.pone.0002493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow EK-H, Fan L-L, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012;56:1331–1341. doi: 10.1002/hep.25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagenais M, Dupaul-Chicoine J, Douglas T, Champagne C, Morizot A, Saleh M. The Interleukin (IL)-1R1 pathway is a critical negative regulator of PyMT-mediated mammary tumorigenesis and pulmonary metastasis. Oncoimmunology. 2017;6:e1287247. doi: 10.1080/2162402X.2017.1287247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danussi C, Petrucco A, Wassermann B, Modica TME, Pivetta E, Del Bel Belluz L, Colombatti A, Spessotto P. An EMILIN1-negative microenvironment promotes tumor cell proliferation and lymph node invasion. Cancer Prev Res (Phila) 2012;5:1131–1143. doi: 10.1158/1940-6207.CAPR-12-0076-T. [DOI] [PubMed] [Google Scholar]
- Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, Arai Y, Takahashi H, Shirakihara T, Nagasaki M, Shibuya T, Nakano K, Watanabe-Makino K, Tanaka H, Nakamura H, Kusuda J, Ojima H, Shimada K, Okusaka T, Ueno M, Shigekawa Y, Kawakami Y, Arihiro K, Ohdan H, Gotoh K, Ishikawa O, Ariizumi S-I, Yamamoto M, Yamada T, Chayama K, Kosuge T, Yamaue H, Kamatani N, Miyano S, Nakagama H, Nakamura Y, Tsunoda T, Shibata T, Nakagawa H. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nature Genetics. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, Bhujwalla ZM, Felsher DW, Cheng L, Pevsner J, Lee LA, Semenza GL, Dang CV. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12:230–238. doi: 10.1016/j.ccr.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci USA. 2008;105:6656–6661. doi: 10.1073/pnas.0801802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson WJ, Hoivik EA, Halle MK, Taylor-Weiner A, Cherniack AD, Berg A, Holst F, Zack TI, Werner HMJ, Staby KM, Rosenberg M, Stefansson IM, Kusonmano K, Chevalier A, Mauland KK, Trovik J, Krakstad C, Giannakis M, Hodis E, Woie K, Bjorge L, Vintermyr OK, Wala JA, Lawrence MS, Getz G, Carter SL, Beroukhim R, Salvesen HB. The genomic landscape and evolution of endometrial carcinoma progression and abdominopelvic metastasis. Nature Genetics. 2016:1–11. doi: 10.1038/ng.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaros S, Cirrincione GM, Palanca A, Metzger D, Reisman D. Targeted knockout of BRG1 potentiates lung cancer development. Cancer Research. 2008;68:3689–3696. doi: 10.1158/0008-5472.CAN-07-6652. [DOI] [PubMed] [Google Scholar]
- Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clément B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouzé E, Calvo F, Zucman-Rossi J. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nature Genetics. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, Barnes I, Bignell A, Boychenko V, Hunt T, Kay M, Mukherjee G, Rajan J, Despacio-Reyes G, Saunders G, Steward C, Harte R, Lin M, Howald C, Tanzer A, Derrien T, Chrast J, Walters N, Balasubramanian S, Pei B, Tress M, Rodriguez JM, Ezkurdia I, van Baren J, Brent M, Haussler D, Kellis M, Valencia A, Reymond A, Gerstein M, Guigó R, Hubbard TJ. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Li J, Xu J, Zhang S, Xu Y, Zhao W, Yin Z, Wang X. Decreased expression of ARID1A associates with poor prognosis and promotes metastases of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2015;34:47. doi: 10.1186/s13046-015-0164-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard KJ, Green JL, James LP, Judge BS, Zolot L, Rhyee S, Dart RC. Acetaminophen-cysteine adducts during therapeutic dosing and following overdose. BMC Gastroenterol. 2011;11:20. doi: 10.1186/1471-230X-11-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends in Genetics. 2014;30:356–363. doi: 10.1016/j.tig.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Deng Q, Wang Q, Li K-Y, Dai J-H, Li N, Zhu Z-D, Zhou B, Liu X-Y, Liu R-F, Fei Q-L, Chen H, Cai B, Zhou B, Xiao H-S, Qin L-X, Han Z-G. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nature Genetics. 2012;44:1117–1121. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- Jubierre L, Soriano A, Planells-Ferrer L, París-Coderch L, Tenbaum SP, Romero OA, Moubarak RS, Almazán-Moga A, Molist C, Roma J, Navarro S, Noguera R, Sanchez-Cespedes M, Comella JX, Palmer HG, Sánchez de Toledo J, Gallego S, Segura MF. BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene. 2016;35:5179–5190. doi: 10.1038/onc.2016.50. [DOI] [PubMed] [Google Scholar]
- Kadoch C, Crabtree GR. Reversible Disruption of mSWI/SNF (BAF) Complexes by the SS18-SSX Oncogenic Fusion in Synovial Sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature Genetics. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, Hauptschein R, Rejto PA, Fernandez J, Wang G, Zhang Q, Wang B, Chen R, Wang J, Lee NP, Zhou W, Lin Z, Peng Z, Yi K, Chen S, Li L, Fan X, Yang J, Ye R, Ju J, Wang K, Estrella H, Deng S, Wei P, Qiu M, Wulur IH, Liu J, Ehsani ME, Zhang C, Loboda A, Sung W-K, Aggarwal A, Poon RT, Fan ST, Wang J, Hardwick J, Reinhard C, Dai H, Li Y, Luk JM, Mao M. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015 doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. Annals of Applied Statistics. 2011;5:1752–1779. doi: 10.1214/11-AOAS466. [DOI] [Google Scholar]
- Li W-C, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol. Biol. 2010;633:185–196. doi: 10.1007/978-1-59745-019-5_13. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lin H, Liu X-B, Yu J-J, Hua F, Hu Z-W. Antioxidant N-acetylcysteine attenuates hepatocarcinogenesis by inhibiting ROS/ER stress in TLR2 deficient mouse. PLoS ONE. 2013;8:e74130. doi: 10.1371/journal.pone.0074130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, Park PJ, Shivdasani RA, Roberts CWM. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nature Genetics. 2017;49:296–302. doi: 10.1038/ng.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Yavari A, Banerjee U. A protocol for in vivo detection of reactive oxygen species. Protocol Exchange 2008 [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–5. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz AR, Schmaldienst S, Stuhlmeier KM, Chen W, Knapp W, Zlabinger GJ. A microplate assay for the detection of oxidative products using 2′,7′-dichlorofluorescin-diacetate. Journal of Immunological Methods. 1992;156:39–45. doi: 10.1016/0022-1759(92)90008-H. [DOI] [PubMed] [Google Scholar]
- Roy N, Malik S, Villanueva KE, Urano A, Lu X, Figura von G, Seeley ES, Dawson DW, Collisson EA, Hebrok M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes & Development. 2015;29:658–671. doi: 10.1101/gad.256628.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze K, Imbeaud S, eacute EL, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller CEM, Shinde J, Soysouvanh F, Calatayud A-L, Pinyol R, Pelletier L, Balabaud C, Laurent A, Blanc J-F, Mazzaferro V, Calvo F, Villanueva A, Nault J-C, Bioulac-Sage P, Stratton MR, Llovet JM, Zucman-Rossi J. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature Publishing Group. 2015;47:1–10. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH, Felsher DW. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Chuang J-C, Kanchwala M, Wu L, Celen C, Li L, Liang H, Zhang S, Maples T, Nguyen LH, Wang SC, Signer RAJ, Sorouri M, Nassour I, Liu X, Xu J, Wu M, Zhao Y, Kuo Y-C, Wang Z, Xing C, Zhu H. Suppression of the SWI/SNF Component Arid1a Promotes Mammalian Regeneration. Cell Stem Cell. 2016;18:456–466. doi: 10.1016/j.stem.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, Jiang L, Fan B, Terracciano L, Armeanu-Ebinger S, Ribback S, Dombrowski F, Evert M, Chen X, Monga SPS. Activation of β-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology. 2014;147:690–701. doi: 10.1053/j.gastro.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Xu E, Zhao Y, Singh S, Li X, Couchy G, Chen X, Zucman-Rossi J, Chikina M, Monga SPS. Modeling a human hepatocellular carcinoma subset in mice through coexpression of met and point-mutant β-catenin. Hepatology. 2016;64:1587–1605. doi: 10.1002/hep.28601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirona RG, Lee W, Leake BF, Lan L-B, Cline CB, Lamba V, Parviz F, Duncan SA, Inoue Y, Gonzalez FJ, Schuetz EG, Kim RB. The orphan nuclear receptor HNF4alpha determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nat Med. 2003;9:220–224. doi: 10.1038/nm815. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, Feilitzen von K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist P-H, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- Wang Y-P, Yu G-R, Lee M-J, Lee S-Y, Chu I-S, Leem S-H, Kim D-G. Lipocalin-2 negatively modulates the epithelial-to-mesenchymal transition in hepatocellular carcinoma through the epidermal growth factor (TGF-beta1)/Lcn2/Twist1 pathway. Hepatology. 2013;58:1349–1361. doi: 10.1002/hep.26467. [DOI] [PubMed] [Google Scholar]
- Wiwi CA, Waxman DJ. Role of hepatocyte nuclear factors in transcriptional regulation of male-specific CYP2A2. J. Biol. Chem. 2005;280:3259–3268. doi: 10.1074/jbc.M409294200. [DOI] [PubMed] [Google Scholar]
- Wu JN, Roberts CWM. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discovery. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H-B, Wang X-F, Zhang Q, Tang Z-Q, Jiang Y-H, Fan H-Z, Sun Y-H, Yang P-Y, Liu F. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis. 2014;35:867–876. doi: 10.1093/carcin/bgt398. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Kuick R, Tipton C, Wu R, Sessine M, Wang Z, Baker SJ, Fearon ER, Cho KR. Arid1a inactivation in an Apc- and Pten-defective mouse ovarian cancer model enhances epithelial differentiation and prolongs survival. J. Pathol. 2015;238:21–30. doi: 10.1002/path.4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Gong C, Bing Y, Li T, Liu Z, Liu Q. Hypermethylation-repressed methionine adenosyltransferase 1A as a potential biomarker for hepatocellular carcinoma. Hepatol. Res. 2013;43:374–383. doi: 10.1111/j.1872-034X.2012.01099.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biology. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen J, Lin H, Jin R, Liu J, Liu X, Meng N, Cai X. The Clinicopathologic Significance of BAF250a (ARID1A) Expression in Hepatocellular Carcinoma. Pathol. Oncol. Res. 2015 doi: 10.1007/s12253-015-0022-9. [DOI] [PubMed] [Google Scholar]
- Zhu H, Shah S, Shyh-Chang N, Shinoda G, Einhorn WS, Viswanathan SR, Takeuchi A, Grasemann C, Rinn JL, Lopez MF, Hirschhorn JN, Palmert MR, Daley GQ. Lin28a transgenic mice manifest size and puberty phenotypes identified in human genetic association studies. Nature Genetics. 2010;42:626–630. doi: 10.1038/ng.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ. Integrative analysis of ChIP-chip and ChIP-seq dataset. Methods Mol. Biol. 2013;1067:105–124. doi: 10.1007/978-1-62703-607-8_8. [DOI] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pagès H, Lin SM, Lapointe DS, Green MR. ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. doi: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.