

Key Points
An in vitro model shows that hemolysis could be due to the presence of a subclinical PNH clone causing a negative C3b/d DAT.
Changes to decay-accelerating factor and membrane inhibitor of reactive lysis may lead to overt hemolysis after minor mismatched transfusions.
Abstract
Minor ABO-mismatched transfusions are a common occurrence, although infrequent transfusion reactions occur. We sought to investigate the regulation of complement C3 activation induced by anti-A. In vitro complement C3 activation was observed with 10 of 30 group O samples and correlated with immunoglobulin M (IgM) anti-A titers. We developed an in vitro paroxysmal nocturnal hemoglobinuria (PNH) model of hemolysis in which group A1 red blood cells (RBCs) were chemically treated with 2-aminoethylisothiouronium (AET) to alter regulators of complement C3 activation. Intravascular hemolysis was simulated by incubating an IgG nonhemolytic group O plasma (titer = 32) with A1 RBCs. IgG was detected on the RBCs, but hemolysis was observed with AET-treated RBCs only. When treated and untreated RBCs were tested together (1:4), we determined that the failure to observe C3b/d deposition on RBCs was due to the complete hemolysis of the AET-treated minor RBC population. A group O patient with a 9% CD59-deficient PNH clone was sensitized with an IgM anti-I. Hemolysis, with a weak positive direct antiglobulin test (DAT) resulting from C3b/d, was observed after incubation with fresh AB serum. Flow cytometry showed an 86% reduction of the PNH clone. Our work indicates that the transfusion of minor ABO-mismatched plasma could cause hemolysis with a negative DAT C3b/d. We propose that the presence of a PNH clone is 1 possible cause of unexplained anemia for recipients of ABO-mismatched product. This work suggests that other acquired or inherited defects of decay-accelerating factor and membrane inhibitor of reactive lysis could be responsible for infrequent but clinically important hemolysis after ABO-mismatched transfusions.
Visual Abstract
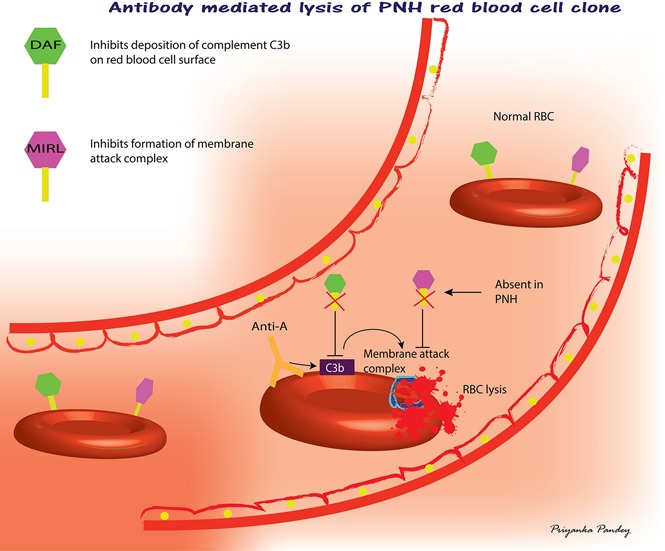
Introduction
Minor ABO-mismatched transfusions in United States are an accepted clinical practice.1 Minor ABO mismatches occur when the donor has an ABO isoagglutinin against ABO antigens expressed on the recipient’s red blood cells (RBCs). A summary of recent studies indicate that ∼13% of platelet transfusions are minor ABO-mismatched.2 This practice leads to the transfusion of 270 mL of plasma containing ABO isoagglutinins in apheresis platelet products transfused to ABO-incompatible recipients. Changes to hemoglobin levels are associated with minor ABO-mismatched platelet transfusions,3 and clinically significant acute intravascular hemolysis secondary to the transfusion of donor isoagglutinins have been reported despite the use of low titered products.4-6 Hemolysis related to minor ABO-mismatched platelet transfusions has been found to be associated with unfavorable outcomes.7 Such incompatible transfusions are transfused to some surgical patients.8,9 Moreover, alterations to the molecules that regulate complement deposition on RBCs, decay-accelerating factor (DAF, CD55) and membrane inhibitor of reactive lysis (MIRL, CD59), are known to be risk factors for hemolysis. Paroxysmal nocturnal hemoglobinuria (PNH) cells are devoid of surface molecules, DAF, and MIRL, and minor ABO-mismatched transfusions cause hemolytic reactions.10 Several myelopathies are known to be associated with the presence of subclinical PNH.11 Subclinical PNH is a condition in which a subject has a minor clone of PNH cells and has no clinical or laboratory evidence of hemolysis.11 DAF and MIRL express antigens of the Cromer and CD59 blood group systems, respectively, with Cromer Inab representing a null phenotype.12,13 DAF regulates the activity of C3 convertase and MIRL inhibits the formation of membrane attack complex.14 Other disease-related deficiencies in the expression of these molecules could cause increased susceptibility of RBCs to complement-mediated intravascular hemolysis.15
Mitigation strategies to avoid hemolytic reactions are being adopted, including the use of low-titer ABO platelet products and volume reduction to prevent significant hemolysis in the transfusion of minor ABO-mismatched platelet products.16-18 Avoiding high ABO isoagglutinin titers (eg, ≥100) most directly addresses the risk of RBC hemolysis. However, underlying patient-inherited immune factors could lead to transfusion-associated hemolysis, including complement receptor–dependent monocyte/macrophage-mediated phagocytosis or unregulated complement-mediated intravascular hemolysis.19
We sought to explore the effect on the direct antiglobulin test (DAT) and hemolysis associated with deficiencies in the complement regulating surface molecules DAF and MIRL. We developed a PNH-like clonality model to evaluate complement-mediated ABO isoagglutinin-dependent hemolysis.
Methods
Preparation of MIRL-deficient RBCs
Group A1 RBCs and anti-A obtained from group O blood donors were used in the study. For removal of MIRL, 200 µL 6% solution 2-aminoethylisothiouronium (AET; Sigma Aldrich, MO), brought to pH 8.0 using 5N sodium hydroxide was incubated with 200 µL of saline washed packed RBCs for 20 minutes at 37°C water bath. After incubation, cells were saline washed 3 times at 700g for 3 minutes.
Removal of MIRL from the surface of treated RBCs was evaluated by flow cytometry (FACSCalibur, BD Biosciences, CA) using fluorescein isothiocyanate anti-human CD55 (Clone IA10, BD Biosciences) and phycoerythrin (PE) anti-human CD59 (Clone p282 [H19], BD Biosciences) along with appropriate isotype controls. Untreated and unstained group A1 RBCs were used to gate the cells.
Anti-A evaluation of plasma samples
Initially, 30 healthy group O donor plasma samples were evaluated for direct (immunoglobulin M [IgM]) and indirect (IgG) anti-A and were titered by doubling dilution in phosphate-buffered saline (PBS)/6% bovine serum albumin (BSA) using buffered or IgG gel agglutination cards (Ortho Clinical Diagnostics, Pompano Beach, FL), respectively. A standardized grading system was used to score hemagglutination.20
Complement activation of anti–A-sensitized RBCs
To evaluate the ability of anti-A to activate complement, 400 µL of 1:100 diluted group O plasma in PBS/0.2% BSA was combined with 100 µL of 5% group A1 RBCs in PBS/0.2% BSA for 30 minutes at 37°C. A 100-µL aliquot was washed and evaluated for IgG sensitization using a murine monoclonal anti-IgG (γ-clone, Immucor, Norcross, GA). The remaining anti–A-sensitized RBCs were washed and resuspended in 200 µL of fresh AB serum for 60 minutes at 37°C. After incubation, the reaction mix was centrifuged and visually inspected for hemolysis and a red cell pellet. RBCs were washed 4 times with 0.85% saline and evaluated for C3 activation using a murine monoclonal anti-human C3b/d blend (γ-clone, Immucor). Complete hemolysis was assumed when there was no red cell pellet after incubation at 37°C.
Correlation analysis
A correlation analysis was performed between IgM or IgG anti-A titers and complement activation. Pearson’s correlation coefficient (r) was calculated with a 2-tailed P < .05 considered as the cutoff score for significance.
Anti–A-dependent complement C3 activation studies
A single sample with an IgM titer of 16 and an IgG titer of 32 that did not activate complement was selected to evaluate complement activation using untreated RBCs and AET-treated RBCs.
Mixed red cell population studies
An in vitro study was designed using a mixed red cell format meant to mimic the in vivo clinical setting of ABO-mismatched apheresis platelets transfusion to a recipient with a minor population of DAF- and/or MIRL-deficient RBCs. The ratio of anti-A plasma to RBCs was based roughly on the transfusion of 300 mL of plasma from a platelet apheresis product to a recipient with a blood volume of 5000 mL. Thus, IgG sensitization with 400 μL of a 1:100 dilution of plasma mixed with 100 μL of 5% RBCs would approximate the in vivo situation for a recipient with a 45% hematocrit. Two red cell populations were tested: (1) 100% untreated A1 RBCs and (2) 80% untreated A1 RBCs combined with 20% AET-treated RBCs.
Results
Characterization of DAF- and MIRL-deficient RBCs
Flow cytometry showed that MIRL-deficient RBCs were produced. The treated RBCs had overlapping profiles that corresponded to their isotype-specific antibody controls (Figure 1).
Figure 1.

Flow cytometric evaluation of MIRL. The graphs represent fluorescence counts on the y-axis vs channel PE (MIRL) on the x-axis. (A) Untreated group A1 RBCs expressing MIRL, (B) AET-treated group A1 RBCs lost MIRL and have an overlapping profile with the isotype control and unstained RBCs. Green peak, isotope control; pink peak, unstained control; purple peak, MIRL-positive cells.
Evaluation of anti-A plasma samples
Thirty blood donor samples were evaluated for their direct (IgM) and indirect (IgG) anti-A titer and the ability to activate complement C3b with group A1 (untreated) RBCs. All plasma samples had direct agglutinating IgM anti-A titers ≤64 and 4 of 30 had IgG titers <100. Of the 30 samples, 10 activated complement as determined by C3b/d deposition using an indirect antiglobulin test with fresh AB serum as a source of complement. Two of the 4 samples with IgG titers <100 failed to activate complement (Table 1). One of the 2 samples with a titer <100 that did not activate complement was chosen for the in vitro experiments (described in “Methods”).
Table 1.
Anti-A characteristics of 30 group O blood donors
IgM titer evaluated by direct hemagglutination using ID-Microtyping System buffered cards.
IgG titer evaluated using ID-Microtyping System anti-IgG cards.
RBC-bound complement C3b evaluated with anti-C3b/d after anti-A sensitization followed by washing and incubation with AB serum.
Correlation analysis
A positive correlation was observed between IgM titers and complement activation (r = 0.727, P < .0001). There was no correlation observed between IgG titers and complement activation (r = 0.001, P > .05).
Anti–A-dependent complement C3 activation studies
Untreated and AET-treated RBCs were evaluated for anti–A-associated complement C3 activation. No hemolysis was observed with untreated cells, whereas hemolysis was observed with AET-treated RBCs (Table 2). Further, no C3b/d deposition was observed with the untreated cells, which was consistent with the initial evaluation of the lack of complement activation (Table 1). In contrast, microscopic C3b/d deposition was observed for the AET-treated RBCs because few RBCs remained intact to evaluate C3b/d deposition (Table 2).
Table 2.
Anti-A sensitization and complement activation
| A1 RBCs | Sensitization* | 6% albumin IgM | Anti-human IgG | Complement-mediated lysis† | Anti-human C3b/d |
|---|---|---|---|---|---|
| Untreated | Group O plasma | M+ | 2+ | No hemolysis | 0 |
| Pooled AB plasma | 0 | 0 | No hemolysis | 0 | |
| AET-treated | Group O plasma | W | 2+ | Hemolysis | M+ |
| Pooled AB plasma | 0 | 0 | No hemolysis | 0 |
*Hemagglutination and hemolysis were scored according to Judd et al20: 0, negative reaction, no agglutination; M+, agglutinates visible under microscope only; W, weak reaction, many tiny agglutinates; 2+, many small visible agglutinates.
Presence of hemolysis visualized after incubation with AB serum at 37°C for 60 min.
In vitro mixed RBC population studies
The mixed cell populations were evaluated for anti-A sensitization and complement C3 activation. There was no hemolysis after incubation with AB serum when 20% AET-treated RBCs sensitized with AB plasma were tested; complement C3b/d was not detected on the RBCs. Hemolysis was observed when 20% AET-treated RBCs were incubated with AB serum after sensitization with group O plasma. The remaining RBCs evaluated for complement activation had no evidence of C3b/d deposition, consistent with the remaining untreated RBCs (Table 3).
Table 3.
Mixed-cell complement lysis: simulation study
| Study RBC population | Sensitization* | 6% albumin IgM | Anti-human IgG | Complement-mediated lysis† | Anti-human C3b/d |
|---|---|---|---|---|---|
| 20% AET-treated | Group O plasma | W | 2+s | Weak hemolysis | 0 |
| Pooled AB plasma | 0 | 0 | No hemolysis | 0 | |
| PNH sample (9%) | Anti-I | 1+ | 2+ | Hemolysis | 1+ |
| Pooled AB plasma | 0 | 0 | No hemolysis | 0 |
Hemagglutination and hemolysis were scored according to Judd et al20: 0, negative reaction, no agglutination; W, weak reaction, many tiny agglutinates; 1+, very small just visible agglutinates; 2+, many small visible agglutinates; 2+s, medium sized visible agglutinates.
†Presence of hemolysis visualized after incubation with AB serum at 37°C for 60 min.
Evaluation of a patient with PNH
A patient with PNH showing normal CD55 expression and a 9% CD59 deficiency was evaluated using our model. This sample was group O therefore, a low titer (32) IgM anti-I was used at a dilution of 1:4 to sensitize the patient’s RBCs. Afterward, the sensitized red cells were incubated in fresh AB serum. The sample showed an 86% reduction of the CD59-deficient clone (Figure 2), and weak complement C3 activation was observed (Table 3).
Figure 2.

Antibody-mediated lysis of PNH clone. The graphs represent MIRL-positive cells on the y-axis in a PE channel. The x-axis represents a blank fluorescein isothiocyanate channel. A group O RBC sample with a clone of patient PNH RBCs was used for the study. (A) Negative control: RBCs sensitized with pooled AB plasma. (B) An 86% reduction in the size of PNH clone was observed upon sensitization of RBCs with anti-I because of the lysis of PNH clone.
Discussion
We confirmed that DAF and MIRL are important regulators of complement activation on RBCs; removing DAF and MIRL increases RBC susceptibility to complement dysregulation and hemolysis. The effects of chemical treatment on the red cell membranes related to susceptibility to hemolysis are unknown. Treatment with these chemicals did not cause RBCs to lyse on storage nor when incubated with fresh AB serum at a normal physiologic pH, contrary to other treatments.21 Moreover, published studies have shown that AET treatment mimics PNH biochemically, serologically, and by electron microscopy.22-25 We expanded on the current knowledge about the regulation of complement activation by demonstrating low or absent MIRL antigen expression with or without DAF predispose RBCs to hemolysis. Our study showed that a low titer anti-A isoagglutinin, which normally failed to activate complement in vitro with healthy RBCs, can potentially cause significant hemolysis resulting from RBC-related factors. We found a significant positive correlation between the IgM class of anti-A titers and in vitro complement activation and hemolysis. Specifically, we showed that DAF- and/or MIRL-deficient cells result in significant complement activation and hemolysis with a noncomplement-activating anti-A. The in vitro model was supported by using a confirmed PNH sample under similar experimental conditions.
The implication for the clinical setting is that intravascular hemolysis of a minor population of RBCs could go undetected in a DAT. We propose 1 hypothesis to explain DAT-negative hemolysis: an unrecognized (subclinical) PNH clonal disorder. And although PNH is generally uncommon, the frequency of a PNH clone among the various recipient populations is not known. Undiagnosed PNH clones have been observed with a frequency of 15% in DAT-negative hemolytic anemia and 53% in bone marrow failure.26,27 In a previous incidental report, a patient was found to have PNH after a minor ABO-mismatched whole blood transfusion.10 Moreover, DAF expression can vary widely, and in the absence of DAF, MIRL prevents hemolysis to some degree but does not prevent complement C3 activation.12 Complement C3 activation has clinical implications for extravascular hemolysis because it causes increased macrophage-mediated phagocytosis.28
Hemolysis following ABO-mismatched platelet transfusions is uncommon, with a clinical incidence of ∼0.01%.16,29 However, it is becoming increasingly clear that persistent hemolysis may be clinically relevant in terms of organ failure, thrombosis, and mortality. Free hemoglobin is deleterious because it can cause multiple organ damage.30 This observation is particularly important because studies have reported association of ABO-mismatched red cell and platelet transfusions with increased morbidity and mortality.31-33 An international survey of red cell transfusions reported that 54% of group O blood is transfused to non–group O recipients.34 Furthermore, patients given multiple ABO nonidentical transfusions of platelets routinely develop IgG-positive direct antiglobulin tests and require increased red cell transfusion needs suggesting low-grade hemolysis.7,33
As for the reported incidence of hemolysis in platelet transfusion, transfusing out-of-group platelets is common because transfusing ABO-matched platelets is not practical for every patient. The incidence and factors predisposing patients to hemolysis from ABO-incompatible platelet transfusions are unknown. Thus, institutions are adopting mitigation strategies to reduce ABO-associated hemolysis for minor ABO-mismatched transfusions of platelet components. Screening donors for a predefined high titered anti-A/anti-B likely reduces the incidence, but to date, there is no consensus on immunoglobulin class or what level constitutes a critical titer.6,35-37 Volume reduction or replacing the plasma with saline may mitigate such minor ABO-mismatched hemolysis but compromise platelet integrity and function.38
Our work suggests that it is prudent to investigate a clinically significant drop in hemoglobin for minor ABO-mismatched recipients of minor ABO-mismatched products who may have a hematologic myelopathy. The transfusion of low antibody titer platelet products is intended to mitigate significant hemolysis. However, our study with a low-titer anti-A indicates that in spite of safe titers of ABO isoagglutinins in platelet product, hemolysis can potentially occur because of patient-related factors. Further, we do not know if small volumes of high(er) titer ABO antibodies in red cell products can cause incidental clinically significant hemolysis. Our study provides 1 potential disease state to evaluate when hemolytic reactions are associated with minor ABO-mismatched transfusions: subclinical PNH. Evaluation of other potential causes of DAF12,13 and MIRL dysregulation15 could be explored because alterations to these molecules may be the root cause of intravascular hemolysis.
Acknowledgments
The authors thank Brian R. Curtis for providing the paroxysmal nocturnal hemoglobinuria sample for analysis.
This work was funded by a research grant to G.A.D. from the Commonwealth Transfusion Foundation (formerly the Virginia Blood Foundation).
Authorship
Contribution: P.P. performed the experiments, analyzed the data, and wrote the manuscript; W.Q.A. edited the manuscript and contributed to the content of the introduction and discussion; J.L.G. contributed to the content of the discussion; G.A.D. conceived the study, developed the model, analyzed the data, and confirmed the final version of the manuscript; and all authors reviewed the final version of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Gregory A. Denomme, BloodCenter of Wisconsin, 638 N 18th St, PO Box 2178, Milwaukee, WI 53201-2178; e-mail: gregory.denomme@bcw.edu.
References
- 1.Lozano M, Heddle N, Williamson LM, Wang G, AuBuchon JP, Dumont LJ; Biomedical Excellence for Safer Transfusion Collaborative. Practices associated with ABO-incompatible platelet transfusions: a BEST Collaborative international survey. Transfusion. 2010;50(8):1743-1748. [DOI] [PubMed] [Google Scholar]
- 2.Dunbar NM, Katus MC, Freeman CM, Szczepiorkowski ZM. Easier said than done: ABO compatibility and D matching in apheresis platelet transfusions. Transfusion. 2015;55(8):1882-1888. [DOI] [PubMed] [Google Scholar]
- 3.Mair B, Benson K. Evaluation of changes in hemoglobin levels associated with ABO-incompatible plasma in apheresis platelets. Transfusion. 1998;38(1):51-55. [DOI] [PubMed] [Google Scholar]
- 4.McManigal S, Sims KL. Intravascular hemolysis secondary to ABO incompatible platelet products. An underrecognized transfusion reaction. Am J Clin Pathol. 1999;111(2):202-206. [DOI] [PubMed] [Google Scholar]
- 5.Larsson LG, Welsh VJ, Ladd DJ. Acute intravascular hemolysis secondaryto out-of-group platelet transfusion. Transfusion. 2000;40(8):902-906. [DOI] [PubMed] [Google Scholar]
- 6.Karafin MS, Blagg L, Tobian AA, King KE, Ness PM, Savage WJ. ABO antibody titers are not predictive of hemolytic reactions due to plasma-incompatible platelet transfusions. Transfusion. 2012;52(10):2087-2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sadani DT, Urbaniak SJ, Bruce M, Tighe JE. Repeat ABO-incompatible platelet transfusions leading to haemolytic transfusion reaction. Transfus Med. 2006;16(5):375-379. [DOI] [PubMed] [Google Scholar]
- 8.Blumberg N, Heal JM, Hicks GL Jr, Risher WH. Association of ABO-mismatched platelet transfusions with morbidity and mortality in cardiac surgery. Transfusion. 2001;41(6):790-793. [DOI] [PubMed] [Google Scholar]
- 9.Refaai MA, Fialkow LB, Heal JM, et al. . An association of ABO non-identical platelet and cryoprecipitate transfusions with altered red cell transfusion needs in surgical patients. Vox Sang. 2011;101(1):55-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brecher ME, Taswell HF. Paroxysmal nocturnal hemoglobinuria and the transfusion of washed red cells. A myth revisited. Transfusion. 1989;29(8):681-685. [DOI] [PubMed] [Google Scholar]
- 11.Parker C, Omine M, Richards S, et al. ; International PNH Interest Group. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699-3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Merry AH, Rawlinson VI, Uchikawa M, Daha MR, Sim RB. Studies on the sensitivity to complement-mediated lysis of erythrocytes (Inab phenotype) with a deficiency of DAF (decay accelerating factor). Br J Haematol. 1989;73(2):248-253. [DOI] [PubMed] [Google Scholar]
- 13.Telen MJ, Green AM. The Inab phenotype: characterization of the membrane protein and complement regulatory defect. Blood. 1989;74(1):437-441. [PubMed] [Google Scholar]
- 14.Brodsky RA. Complement in hemolytic anemia. Blood. 2015;126(22):2459-2465. [DOI] [PubMed] [Google Scholar]
- 15.Frazer-Abel A, Sepiashvili L, Mbughuni MM, Willrich MA. Overview of laboratory testing and clinical presentations of complement deficiencies and dysregulation. Adv Clin Chem. 2016;77:1-75. [DOI] [PubMed] [Google Scholar]
- 16.Josephson CD, Castillejo MI, Grima K, Hillyer CD. ABO-mismatched platelet transfusions: strategies to mitigate patient exposure to naturally occurring hemolytic antibodies. Transfus Apheresis Sci. 2010;42(1):83-88. [DOI] [PubMed] [Google Scholar]
- 17.Quillen K, Sheldon SL, Daniel-Johnson JA, Lee-Stroka AH, Flegel WA. A practical strategy to reduce the risk of passive hemolysis by screening plateletpheresis donors for high-titer ABO antibodies. Transfusion. 2011;51(1):92-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fontaine MJ, Mills AM, Weiss S, Hong WJ, Viele M, Goodnough LT. How we treat: risk mitigation for ABO-incompatible plasma in plateletpheresis products. Transfusion. 2012;52(10):2081-2085. [DOI] [PubMed] [Google Scholar]
- 19.Flegel WA. Pathogenesis and mechanisms of antibody-mediated hemolysis. Transfusion. 2015;55(Suppl 2):S47-S58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Judd J, Johnson S, Storry J. Judd’s Methods in Immunohematology. Bethesda, MD: AABB; 2008. [Google Scholar]
- 21.Ezzell JL, Wilcox LA, Bernshaw NJ, Parker CJ. Induction of the paroxysmal nocturnal hemoglobinuria phenotype in normal human erythrocytes: effects of 2-aminoethylisothiouronium bromide on membrane proteins that regulate complement. Blood. 1991;77(12):2764-2773. [PubMed] [Google Scholar]
- 22.Sirchia G, Dacie JV. Immune lysis of AET-treated normal red cells (PNH-like cells). Nature. 1967;215(5102):747-748. [DOI] [PubMed] [Google Scholar]
- 23.Lewis SM, Lambertenghi G, Ferrone S, Sirchia G. Electron microscope study of PNH red cells and AET-treated normal red cells (PNH-like cells). J Clin Pathol. 1971;24(8):677-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker CJ, Wiedmer T, Sims PJ, Rosse WF. Characterization of the complement sensitivity of paroxysmal nocturnal hemoglobinuria erythrocytes. J Clin Invest. 1985;75(6):2074-2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan FF, Bryant JA, Fletcher A. Protease-modified erythrocytes: CD55 and CD59 deficient PNH-like cells. Immunol Cell Biol. 1995;73(1):66-72. [DOI] [PubMed] [Google Scholar]
- 26.Segel GB, Lichtman MA. Direct antiglobulin (“Coombs”) test-negative autoimmune hemolytic anemia: a review. Blood Cells Mol Dis. 2014;52(4):152-160. [DOI] [PubMed] [Google Scholar]
- 27.Bat T, Abdelhamid ON, Balasubramanian SK, et al. . The evolution of paroxysmal nocturnal haemoglobinuria depends on intensity of immunosuppressive therapy [published online ahead of print 14 August 2017]. Br J Haematol. doi:10.1111/bjh.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig ML, Waitumbi JN, Taylor RP. Processing of C3b-opsonized immune complexes bound to non-complement receptor 1 (CR1) sites on red cells: phagocytosis, transfer, and associations with CR1. J Immunol. 2005;174(5):3059-3066. [DOI] [PubMed] [Google Scholar]
- 29.Menis M, Izurieta HS, Anderson SA, et al. . Outpatient transfusions and occurrence of serious noninfectious transfusion-related complications among US elderly, 2007-2008: utility of large administrative databases in blood safety research. Transfusion. 2012;52(9):1968-1976. [DOI] [PubMed] [Google Scholar]
- 30.Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. 2005;293(13):1653-1662. [DOI] [PubMed] [Google Scholar]
- 31.Lapierre V, Mahé C, Aupérin A, et al. . Platelet transfusion containing ABO-incompatible plasma and hepatic veno-occlusive disease after hematopoietic transplantation in young children. Transplantation. 2005;80(3):314-319. [DOI] [PubMed] [Google Scholar]
- 32.Pai M, Cook R, Barty R, Eikelboom J, Lee KA, Heddle N. Exposure to ABO-nonidentical blood associated with increased in-hospital mortality in patients with group A blood. Transfusion. 2016;56(3):550-557. [DOI] [PubMed] [Google Scholar]
- 33.Zaffuto BJ, Conley GW, Connolly GC, et al. . ABO-immune complex formation and impact on platelet function, red cell structural integrity and haemostasis: an in vitro model of ABO non-identical transfusion. Vox Sang. 2016;110(3):219-226. [DOI] [PubMed] [Google Scholar]
- 34.Zeller MP, Barty R, Aandahl A, et al. ; Biomedical Excellence for Safer Transfusion (BEST) Collaborative. An international investigation into O red blood cell unit administration in hospitals: the GRoup O Utilization Patterns (GROUP) study. Transfusion. 2017;57(10):2329-2337. [DOI] [PubMed] [Google Scholar]
- 35.Cooling LL, Downs TA, Butch SH, Davenport RD. Anti-A and anti-B titers in pooled group O platelets are comparable to apheresis platelets. Transfusion. 2008;48(10):2106-2113. [DOI] [PubMed] [Google Scholar]
- 36.Fung MK, Downes KA, Shulman IA. Transfusion of platelets containing ABO-incompatible plasma: a survey of 3156 North American laboratories. Arch Pathol Lab Med. 2007;131(6):909-916. [DOI] [PubMed] [Google Scholar]
- 37.Josephson CD, Mullis NC, Van Demark C, Hillyer CD. Significant numbers of apheresis-derived group O platelet units have “high-titer” anti-A/A,B: implications for transfusion policy. Transfusion. 2004;44(6):805-808. [DOI] [PubMed] [Google Scholar]
- 38.Veeraputhiran M, Ware J, Dent J, et al. . A comparison of washed and volume-reduced platelets with respect to platelet activation, aggregation, and plasma protein removal. Transfusion. 2011;51(5):1030-1036. [DOI] [PubMed] [Google Scholar]