

Key Points
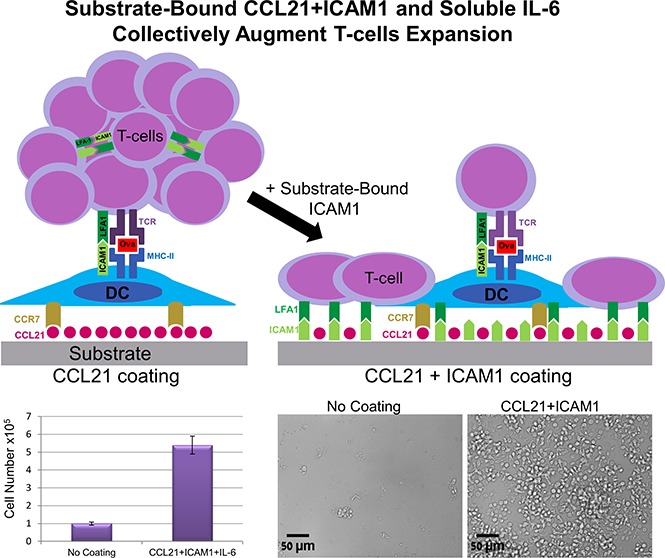
A synthetic immune niche consisting of CCL21, ICAM1 and IL-6 promoted morphological changes of T cell clusters and increased cell expansion.
This synthetic immune niche can serve as an improved approach for efficiently generating antigen-specific immunotherapeutic CD4 T cells.
Abstract
Immune processes within the complex microenvironment of the lymph node involve multiple intercellular, cell-matrix, and paracrine interactions, resulting in the expansion of antigen-specific T cells. Inspired by the lymph node microenvironment, we aimed to develop an ex vivo “synthetic immune niche” (SIN), which could effectively stimulate the proliferation of antigen-activated CD4+ T cells. This engineered SIN consisted of surfaces coated with the chemokine C-C motif ligand 21 (CCL21) and with the intercellular adhesion molecule 1 (ICAM1), coupled with the soluble cytokine interleukin 6 (IL-6) added to the culture medium. When activated by ovalbumin-loaded dendritic cells, OT-II T cells growing on regular uncoated culture plates form nonadherent, dynamic clusters around the dendritic cells. We found that functionalization of the plate surface with CCL21 and ICAM1 and the addition of IL-6 to the medium dramatically increases T-cell proliferation and transforms the culture topology from that of suspended 3-dimensional cell clusters into a firm, substrate-attached monolayer of cells. Our findings demonstrate that the components of this SIN collectively modulate T-cell interactions and augment both the proliferation and survival of T cells in an antigen-specific manner, potentially serving as a powerful approach for expanding immunotherapeutic T cells.
Visual Abstract
Introduction
Adaptive immunity is based on specific responses against pathogenic targets, involving complex cellular processes and intercellular interactions that occur in specific “niches” within the lymphatic system.1-4 Mimicry of such niches by engineering artificial lymphoid tissues or synthetic immune niches (SINs) is an emerging field, with important implications for cell-based immune therapies.5
A major challenge for T-cell-based immunotherapies is the necessity to expand antigen-specific T cells in large quantities while maintaining their functionality. Synthetic ex vivo activation and expansion of antigen-specific T cells can serve for adoptive therapies of malignancies and infections, whereas expansion of specific regulatory T cells (Tregs) can be harnessed for suppression of autoimmune processes.6,7 Moreover, SINs could provide novel tools for basic research into the mechanisms underlying immunological processes by enabling the controlled regulation and perturbation of specific factors potentially involved in cell–cell or cell–matrix interactions.
In recent years, a number of SIN engineering approaches have been described, based on various geometries, physical structures, and chemical and metabolic compositions.8-21 The development of SINs for the selective stimulation of specific T cells is a particularly challenging mission, as it must encompass the broad diversity of natural immune niches and the complex interplay between the stromal and immune cell types that reside within them. These studies have provided valuable insights into the molecular complexity and specific functionalities of the various factors residing in natural immune niches, but have yielded limited information on the synergy between them, nor have they addressed the role of topology in their effective integration.
These considerations motivated us to design novel SINs, which combine an antigen-mediated activation of T cells with 3 categories of molecular elements, namely, chemoattractants, adhesion molecules, and soluble cytokines, aiming at an effective expansion of functional T-cell populations. The choice of specific molecules of each category for the design of the SIN was largely based on the current knowledge concerning the main cellular interactions that take place within lymph nodes in vivo.
Ample recent data22 indicate that well-orchestrated interactions with the microenvironment enable T cells and antigen-loaded dendritic cells (DCs) to meet and bind to each other, via matching epitopes and adhesion molecules, thereby supporting the survival and expansion of antigen-specific T cells.
The lymphatic stroma, a network of fibroblastic reticular cells (FRCs) and associated reticular fibers, provides suitable spaces for cells to interact.23,24 Extracellular matrix proteins secreted by these FRCs facilitate the adhesion and apparent “crawling” of immune cells on the FRC surface.25 In addition to mediating adhesive processes, FRCs produce diverse chemokines, cytokines, and growth factors that recruit and promote the survival and proliferation of immune cells.26-29
In an attempt to increase T-cell proliferation, we chose to integrate CC-chemokine ligand 21 (CCL21), secreted by lymphatic stroma and endothelium,30 with the intercellular adhesion molecule 1 (ICAM1) and the cytokine interleukin 6 (IL-6). CCL21 displays therapeutic potential,31-33 as it induces several processes of great importance to the immune response: colocalization and recruitment of T cells and DCs,26,27 improved cell migration,34,35 priming of T cells for synapse formation,36 and costimulation of naive T-cell expansion and Th1 polarization.37
CCL21 also plays an important role in T-cell adhesion and clustering. Following activation, T cells begin to proliferate and aggregate to form cell clusters around antigen-presenting DCs.38-40 Within these clusters, both T–DC and T–T bonds are forged and mediated through the immobilization of ICAM1 and lymphocyte function-associated 1 (LFA1).38,39 By binding to its CC-chemokine receptor 7 (CCR7) on T cells, CCL21 increases LFA1 responsiveness to ICAM1 and mediates the arrest of motile lymphocytes on ICAM1-expressing DCs, endothelial cells, and other T cells.30,41
On the basis of the central role of CCL21 and ICAM1 in mediating efficient T–DC and T–T interactions, we hypothesized they could also be exploited in a “minimal,” yet effective, synthetic environment to modulate intercellular interactions and improve expansion of antigen-specific T cells. In addition, we explored the effect of diverse cytokines on the expansion of T cells in the engineered SIN and found that IL-6 increases cell survival. Taken together, T-cell stimulation with antigen-loaded DC, coupled with a unique combination of a chemokine, adhesion molecule, and cytokine, enables the efficient stimulation and augmented proliferation of antigen-specific T cells.
Materials and methods
Mice
C57BL/6 mice were obtained from Harlan Laboratories (Rehovot, Israel), and OT-II mice from Jackson Laboratories (Bar Harbor, ME). All mice were 5 to 12 weeks old, maintained at the Weizmann Institute’s Lorry Lokey Pre-Clinical Research Facility, and were cared for in accordance with national and institutional guidelines. Experiments were approved by the Institutional Animal Care and Use Committee.
Cell isolation and culture handling
Naive CD4+ T cells were purified (>95%) from a cell suspension harvested from spleens of OT-II mice, using a MACS CD4+CD62L+ T Cell Isolation Kit II (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer's instructions. Similarly, DCs were purified (>85%) from spleens of C57BL/6 mice, using MACS CD11c microbeads (Miltenyi Biotec). T cells and DCs were cultured in a 3:1 ratio, respectively, with 1 μg/mL Ovalbumin peptide (OVA323-339, InvivoGen, San Diego, CA) in RPMI 1640 medium without phenol red, supplemented with 10% serum, 100 U/mL penicillin, 100 mg/mL streptomycin, 2 mM glutamine, 10 mM HEPES, 1 mM sodium pyruvate, and 50 mM β-mercaptoethanol (Biological Industries, Beit Haemek, Israel).
Surface functionalization and soluble factors
Surface functionalization was performed by overnight incubation in phosphate-buffered saline of 5 μg/mL CCL21, 50 μg/mL ICAM1 (R&D Systems, Minneapolis, MN), or 20 μg/mL anti-LFA1 (leaf-aCD11a; BioLegend, San Diego, CA).
For blocking LFA1 in culture media, 5 μg/mL blocking antibody was added 24 hours after cell seeding, so as not to disturb DC–T-cell interactions.
IL-6 was added immediately after seeding of the cells as a soluble cytokine, 100 ng/mL culture media (BioLegend).
Labeling with membrane-permeable dyes
Before seeding, naive CD4+ T cells were labeled with 10 μM CPDE-450 (Cell Proliferation Dye eFluor-450), 5 µM CPDE-670 (eBioscience, San Diego, CA), or 2 µM carboxyfluorescein diacetate succinimidyl ester (CFSE; Invitrogen, Carlsbad, CA) for 10 minutes at 37°C. Excess dye was removed by 3 cold washes with 7 volumes RPMI before cell seeding.
Cell proliferation assay
Before seeding, T cells were stained with 5 μM CFSE (Biolegend) for 20 minutes at 37°C, according to the manufacturer’s instructions. Excess dye was removed by washing with 5 volumes RPMI. T cells (30-90 × 103) were seeded with DCs (10-30 × 103) and 1 μg/mL ovalbumin peptide (OVA323-339; InvivoGen, San Diego, CA) in a 96-well plate with 250 µL complete RPMI medium. Five days later, cells were detached from the surface after 10-minute incubation with phosphate-buffered saline without calcium and magnesium. Propidium iodide (1 μg/mL) was added to each well for staining of dead cells. Single-cell suspensions were taken for flow cytometry analysis (Becton Dickinson and FlowJo Software). Live single cells (negative PI staining) were gated, and the mean fluorescence intensity of their CFSE was used to evaluate the degree of their proliferation.
Live staining of T cells for culture height measurements
For measuring culture height using Z-stack imaging, 5 μg/mL anti-CD44 (stains activated T cell; BioLegend) and 10 mM CyTRACK Orange (eBioscience, San Diego, CA) were gently added by pipetting into the culture medium of each well, so not to disturb cell contacts and structures. Cells were incubated for 1 hour before Z-stack imaging and height calculation.
Microscopy and image analysis
Wide-range phase-contrast images were acquired and stitched, using the 30× objective of a Ti-eclipse microscope (Nikon Instruments, Konan, Minato-ku, Tokyo, Japan) equipped with an automated stage, and an incubator with a 5% CO2 atmosphere.
Time-lapse movies were acquired using a DeltaVision Elite microscope (Applied Precision, GE Healthcare, Issaquah, WA) mounted on an inverted IX71 microscope (Olympus, Center Valley, PA) connected to a Photometrics CoolSNAP HQ2 camera (Roper Scientific, Martinsried, Germany). The primary image processing software used was SoftWorX 6.0. This microscope was also used for the production of deconvolution-based 3-dimensional (3D) image reconstructions, rendering supported by BITPlan software (Willich-Schiefbahn, Germany).
High-content/high-throughput microscopy was conducted on 96-well plates using a Hermes microscope (IDEA Bio-Medical Ltd., Rehovot, Israel) equipped with automated scanning optics, high-precision autofocus, and a closed environmental chamber. For cell counting and cell death analysis, we used WiSoft software (IDEA Bio-Medical Ltd.).
Cluster analysis was carried out using Fiji software: background was subtracted by means of a rolling ball method, and textured cluster areas were enhanced by a standard deviation filter before thresholding using a fixed value. Morphological filtering was applied to fill holes and connect broken clusters; finally, single cells were discarded by size and circularity. Areas of individual clusters and the total area of all clusters in a given image were measured for each condition.
Cell viability assay and enumeration
A microscopic cell-counting assay was performed using automated analysis of fluorescent microscope images. Live cells were identified by staining the cell nuclei with 1 μg/mL Hoechst (33342; ImmunoChemistry Technologies, Bloomington, MN) and with 250 ng/mL propidium iodide (Sigma Aldrich, St. Louis, MO), for elimination of dead cells from the analysis. At the experimental end-point, cells were pipetted to breakdown clusters and spun down. Fluorescent images were taken using a Hermes microscope (IDEA Bio-Medical Ltd.), and image analysis software (WiSoft; IDEA Bio-Medical Ltd.) was used to quantify viable cell numbers.
A metabolic viability assay was performed by adding 20 μL CellTiter-Blue (Promega Corporation, Madison, WI) per 100 μL culture medium for 3 hours. Results were quantified using a fluorescent plate reader (excitation, 560 nm; emission, 590 nm) and a linear equation of a calibration column.
Scanning electron microscopy
Cells were seeded on glass coverslips placed inside wells of a 24-well plate. After 72 hours, wells were gently washed in 0.1 M cacodylate buffer, fixed with Karnovsky fixative (2% glutaraldehyde, 3% paraformaldehyde, in 0.1 M cacodylate buffer), and incubated overnight at 4°C. Cover glasses were dehydrated in increasing concentrations of ethanol (30%, 50%, 70%, 96%, and 100%), followed by critical-point drying in BAL-TEC CPD030 and sputtering in a gold palladium sputter coater (Edwards, Crawley, United Kingdom). Images were taken using a secondary electron detector in a high-resolution Ultra 55 scanning electron microscope (Zeiss, Oberkochen, Germany).
Transmission electron microscopy
Cells were seeded on a coverslip placed inside a single well of a 24-well plate. After 72 hours, cells were fixed in Karnovsky fixative for 2.5 hours at room temperature and then washed 4 times with 0.1 M cacodylate buffer. Samples were then postfixed for 1 hour in 1% OsO4, 0.5% K2Cr2O7, and 0.5% K4[Fe(CN)6]3H2O in 0.1 M cacodylate buffer and washed 3 times in double-distilled water, followed by 1 hour incubation with uranyl acetate (2% in double-distilled water). Samples were then washed in double-distilled water and dehydrated in increasing concentrations of ethanol (30%, 50%, 70%, 96%, and 100%). Epon (EMS, Hatfield, PA) was gradually infiltrated into the cultures in increasing concentrations (30%, 50%, and 70% in propylene oxide) for 2 hours each: 100% for 16 hours, and again 100% 3 times for 2 hours each. Samples were then transferred in Epon to backing molds and baked overnight in an oven. The Epon blocks were disconnected from the cover glass by cooling using liquid nitrogen, and then by heating with a hot plate. Finally, 70- to 100-nm ultrathin slices were cut and stained with uranyl acetate 2% in ethanol and lead citrate. Grids were analyzed in a CM-12 Spirit FEI electron microscope; images were taken with an Eagle 2kx2k FEI (Eindhoven, Netherlands).
T-cell activation with beads
Freshly isolated CD4 T cells were incubated 1:1 with activation beads freshly prepared according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany) and with IL-2 (30 U/mL; BioLegend, San Diego, CA).
Statistical analysis
Data are shown in each graph as an average of 3 to 10 replicates that were cultured and measured separately in a single independent experiment, representative of more than 3 separate repeated experiments. Error bars indicate standard error of the mean, and statistical significance was calculated using a standard t-test.
Results
Surface-bound CCL21 increases the number and size of T-cell clusters over time
As a model for antigen-specific T-cell stimulation, we used CD4+ T cells expressing ovalbumin-specific T-cell receptors, isolated from TCR transgenic OT-II mice. The cells were cocultured with DCs, loaded with the corresponding cognate ovalbumin peptide. DCs and T cells were prelabeled with different cell-permeable dyes to distinguish between them. Cell clusters that formed after DC stimulation of T cells consisted almost entirely of proliferating CD4+ T cells, with only 1 to 2 DCs per cluster, most of which were dead after 24 hours (Figure 1A-D).
Figure 1.

Substrate-immobilized CCL21 increases the number and size of dynamic T-cell clusters. (A-D) Representative images of OT-II T cells (blue) and ovalbumin-loaded DCs (green) labeled before seeding with membrane-permeable dyes and cocultured on an uncoated substrate (A-B) or on a CCL21-coated substrate (C-D). After 72 hours of incubation, the majority of DCs were dead (red, propidium iodide in B,D), and large T-cell clusters were visible, especially in the CCL21-coated cultures (E-F; clusters marked with red arrows). Scale bars, 50 μm (A-D) and 200 μm (E-F). (G-H) Bar graphs illustrating the number (G) and total projected areas (H) of clusters formed on uncoated and CCL21-coated surfaces at different incubation times (data are representative of at least 3 independent experiments with 5 replicates each; error bars represent standard error of the mean; calculated P values [using t test] are as indicated in the figure). Note that substrate-bound CCL21 increased the number and projected areas of T-cell clusters. (I-J) Two examples of projected images of Z-stack series of single clusters formed on CCL21-coated surfaces (cluster borders are denoted by a yellow line). OT-II CD4+ cells, labeled before seeding with membrane-permeable dyes, green, blue, or red, were mixed in equal numbers and cocultured with antigen-loaded DCs on CCL21-coated surfaces for 72 hours. Note that the clusters are polyclonal, as evidenced by the fact that they contain cells tagged by all 3 colors. Scale bars, 20 μm. (K-P) Time-lapse frames depicting the dynamic properties of clusters (supplemental Video 1). As shown, clusters can merge; also, single cells can leave existing clusters and join new ones. Cluster merger events are color-coded as follows: red + pink (K, L) = dark green (M); dark green + light green (M) = light blue (N); light blue + dark blue (O) = purple (P). Scale bars, 50 μm.
Plating the DC-activated T cells on CCL21-functionalized surfaces induced a major increase in cluster size (compare Fig. 1B-C,E-F) Dynamic morphometry indicated that the T-cell clusters continuously expanded in size for ∼48 to 120 hours (Figure 1G-H), especially on CCL21. From that point onward, they disassembled dramatically and scattered into single cells (supplemental Figure 1). Notably, a similar phenomenon is also observed in the natural niche, when T cells detach from the clusters, leave the lymph node, and translocate into the circulation.42
Comparing T-cell clusters formed on CCL21-coated and CCL21-uncoated surfaces indicates that CCL21 induced a greater than twofold increase in the number of clusters formed (Figure 1G), and more than a fivefold increase in cluster-projected area (Figure 1H). To examine the cellular clonality of these large CCL21-induced clusters, we mixed equal numbers of OT-II T cells, prelabeled with 3 different dyes, and subjected individual clusters to 3D, deconvolution-based fluorescence imaging. As seen in Figure 1I-J, all clusters contained cells labeled with all 3 dyes, confirming they are formed by a merger of multiple T-cell clones. Live cell imaging demonstrated that the clusters are highly dynamic, and change in size as a result of cell proliferation, as well as by merger or splitting of clusters (Figure 1K-P; supplemental Video 1).
Substrate-attached ICAM1, in the presence or absence of CCL21, transforms T-cell clusters into substrate-attached monolayers
Because large clusters may limit the access of cells in the cluster’s core to environmental signals or nutrients, we thought to suppress cluster formation by further functionalizing the CCL21-coated culture substrate with the adhesion molecule ICAM1. Light and electron microscopy revealed a major morphological transformation of CD4+ T cells plated on the double-functionalized surface (Figure 2). In contrast to the clusters formed on the uncoated surface (Figure 2A1-A2) or the enlarged clusters on the CCL21-coated surface (Figure 2B1-B2), T cells associated with the CCL21-ICAM1-coated surface formed 2D monolayers of flat cells (Figure 2C1-C2).
Figure 2.

Substrate-immobilized ICAM1, with or without CCL21, reduces T-cell clustering and induces their spreading on surfaces. (A1-C2) Representative scanning electron micrographs, showing T-cell clusters formed in cultures without surface coating (A1, A2) and in cultures with CCL21 coating (B1, B2) or CCL21+ICAM1 coating (C1, C2). Note that ICAM1 coating (C1, C2) induces cell spreading, resulting in significantly smaller and flatter clusters. Scale bars in A1-C1, 100 μm; in A2-C2, 10 μm. (D-F) Transmitted light microscopy images, depicting cell spreading and a decrease in cluster size after their culturing for 72 hours on a surface coated with ICAM1+CCL21 (E), compared with CCL21 alone (D). Addition of anti-LFA1 blocking antibodies to the culture medium of cells on CCL21-coated surfaces inhibits cluster formation (F). Scale bars, 200 μm; white squares show enlarged area in which the scale bar is 100 μm. (G-J) Representative phase contrast images, showing cells 83 hours after their seeding on coated substrates. Note that the ICAM1 surface coating modifies single-cell morphology. Nonclustered cells on an uncoated surface are mostly round, with hardly any cell–cell contacts (G), whereas on CCL21-coated surfaces, cells commonly display an elongated morphology (H). On ICAM1-coated surfaces, the majority of the cells are elongated, with multiple cell–cell contacts (I, J). Scale bars, 50 μm (supplemental Video 2).
Further experiments demonstrated that cell–cell interactions induced by immobilized CCL21 are LFA1-dependent, as the formation of large clusters on CCL21 (Figure 2D) was dramatically reduced by adding either ICAM1 coating (Figure 2E) or LFA1 blocking antibody to the culture (Figure 2F). More specifically, when closely examining images of clusters formed on CCL21 coating alone (Figure 2D), we distinguished between 2 distinct types of cell clusters; namely, large spherical 3D clusters (average projected area, 4.3 × 104 ± 1.6 × 104 μm2) and small, relatively flat clusters (average projected area, 3.6 × 103 ± 2.2 × 103 μm2). The number of large clusters per 1 mm2 on CCL21 alone was ∼3.7, and the number of small and flat clusters was about 20.2. Notably, the large 3D clusters were present only in cultures stimulated with CCL21 alone (Figure 2D) and were absent from cultures functionalized with CCL21 combined with either ICAM1 (Figure 2E) or anti-LFA1 antibodies (Figure 2F).
As indicated, when cultured over CCL21+ICAM1 (Figure 2E), the large clusters disappeared and the density of small clusters increased to ∼45/mm2 (more than double the number of clusters on CCL21). The average projected area of the small and flat clusters also increased to 8 × 103 ± 5.7 × 103 μm2. The addition of anti-LFA1 to CCL21 (Figure 2F) abolished the formation of large, 3D clusters, yet the number of small and flat clusters per 1 mm2 was ∼12, and their average projected area was 2.2 × 103 ± 0.9 × 103 μm2.
ICAM1 and CCL21 also exerted a strong effect on the morphology of single T cells (Figure 2G-J; supplemental Video 2). Cells interacting with uncoated surfaces were mostly round, with hardly any stable cell contacts (Figure 2G), whereas on the CCL21-coated surface, they developed an elongated morphology (Figure 2H). ICAM1 coating, with or without CCL21, induced massive cell spreading on the surface and multiple cell–cell contacts, some by means of long, thin tethers (Figure 2I-J). Notably, cultures on ICAM1 alone, without CCL21 (Figure 2I), contained lower cell densities then that of CCL21+ICAM1 (Figure 2J), especially as incubation time increased (supplemental Video 2).
Transmission electron microscopy images revealed that T cells within the large clusters are loosely packed, despite multiple filopodia-mediated contacts between them (Figure 3A-C). When cultured on a CCL21+ICAM1-coated surface, cells spread and form extended and tight membrane contacts with the surface, along with peripheral interactions with neighboring cells (Figure 3D-H).
Figure 3.

ICAM1-coated surfaces, with or without CCL21, decrease culture height. (A-C) Transmission electron microscopy images of T-cell clusters formed on surfaces coated with CCL21. The experiment demonstrates that clusters are loosely packed, despite multiple membrane contacts between cells within the cluster. Scale bars, 5 μm (A), 1 μm (B), and 0.5 μm (C). (D-H) Transmission electron microscopy images of transverse sections of T cells, cultured on a CCL21+ICAM1-coated surface (surface marked with red arrows in panel D). Note that the T cells formed multiple, noncontinuous membrane contacts, both with the coated surface (D-E,H) and with neighboring cells (D,F-G). Scale bars, 5 μm (D), 200 nm (E), 500 nm (F), 200 nm (G), and 500 nm (H). (I1-L2) Representative fluorescent Z-stack projected images of single T-cell clusters, formed on different coatings, are shown in top views (I1,J1,K1,L1) and in matching side views (I2,J2,K2,L2). T-cell membranes are live-stained green, and cell nuclei are live-stained red (supplemental Video 3). (M) Culture heights were calculated on the basis of Z-stack deconvolution imaging (data are representative of at least 2 independent experiments with 10 replicates each; error bars indicate standard error of the mean; calculated P values [using t test] are as indicated in the figure). Note that clusters formed on surfaces coated with ICAM1, either with (L1,L2,M) or without (K1,K2,M) CCL21, are thinner compared with those formed on uncoated surfaces (I1,I2,M), whereas very large and thick clusters are induced by CCL21 coating (J1,J2,M). Scale bars, 20 μm.
Comparative 3D imaging of T cells (Figure 3I1-M; supplemental Video 3) showed that clusters forming on the CCL21-coated surface were significantly thicker (h = 26 µm, on average) compared with those forming on the uncoated surface (h = 16 µm). However, the clusters forming on the dual (CCL21+ICAM1) coating became thinner (h = 11 µm), and even thinner still (h = 7 µm) when ICAM1 alone was used (Figure 3M).
By culturing cells on a patterned surface characterized by separate areas coated with ICAM1, CCL21, or both, we further verified that the observed differences in T-cell clustering, cell spreading, and cell density are indeed local, confined to the coated area, and unaffected by both the relative order of the coatings on the surface and the number of seeded cells (Figure 4; supplemental Figure 2; supplemental Video 4). Taken together, these findings confirm that ICAM1 drives T-cell adhesion to the surface and can effectively compete with their ability to cluster with each other.
Figure 4.

The effects of CCL21 and ICAM1 on cell morphology are confined to functionalized areas along the substrate. (A) Experimental layout: silicon molds containing 4 separate, bottomless chambers were placed on a 35-mm glass-bottomed dish (A1). Different coating solutions were placed in each chamber to form separate strips containing no coating, ICAM1 coating, CCL21 coating, and CCL21+ICAM1 coating on the glass surface (A2). After overnight incubation, the coating solutions and silicon molds were removed (A3), and the surfaces were thoroughly rinsed. OT-II T cells were then cocultured with ovalbumin-loaded DCs (A4) and imaged by phase-contrast microscopy (supplemental Figure 2; supplemental Video 4). (B) A stitched image of the surface with cultured T cells, taken at 36 hours, in which each area is marked underneath the horizontal lines: no coating is marked in yellow, ICAM1 in blue, CCL21 in green, and CCL21+ICAM1 in purple. Magnified single fields are shown for each area (C-F). T cells are found mainly within the coated areas, which display a higher cell density (D-F) compared with that of the uncoated area (C). The induction of large clusters by CCL21 coating (B,E) and the cell spreading and culture flattening induced by ICAM1 (B,D,F) are confined to the coated area and do not affect the morphology of T cells in other areas that share the same original cell pool and culture medium. Scale bars, 500 μm.
CCL21, ICAM1, and IL-6 collectively augment the expansion of antigen-specific T cells
The effect of the surface coating on CD4+ T cells was not limited to morphological rearrangement, but also exerted a major effect on T-cell yield. To quantify that effect, we used nuclear staining and automated image analysis to count the number of viable cells at different time points during culture (Figure 5A-I). Cell numbers in the cultures with either CCL21 alone or ICAM1 alone were up to twofold greater than those cultured on uncoated surfaces (Figure 5I). When CCL21 and ICAM1 were combined, the increase in cell number rose to 4.5-fold at 7 days (Figure 5I; supplemental Figure 3A) and was restricted to antigen-specific activated T cells. This was demonstrated by image quantification of OT-II-GFP-expressing cells that were mixed with unstained non-ova T cells at a ratio of 1:99 (Figure 5J-L). OT-II cells preferentially expanded more than eightfold on uncoated surfaces, and more than 16-fold on surfaces coated with CCL21+ICAM1, after 7 days in culture (Figure 5L), whereas the vast majority of non-ova T cells died (supplemental Figure 4).
Figure 5.

Immobilized CCL21 and ICAM1 increase viable cell numbers of antigen-specific T cells. (A-H) Representative fluorescent images of T cells grown on different coated surfaces after the breaking down of clusters and their spin-down. Images were taken after 72 hours (A-D) and 7 days (E-H) of culture. Cell nuclei are stained blue in all cells, and red only in dead cells. Scale bars, 50 μm. (I) Bar graph illustrating viable cell numbers, as quantified using automated image analysis after cluster breakdown, and their spin-down (data are representative of at least 3 independent experiments with 3 to 5 replicates [wells] and 20 fields of view in each well; see supplemental Figure 3 for summary of all independent tests). Error bars represent standard error of the mean. Immobilized CCL21 or ICAM1 alone doubled live cell numbers at all time points. Combining the 2 further increased the yield of live cells from 48 hours onward, with a more than fourfold increase at 7 days. (J-K) Representative fluorescent images of OT-II T cells (ova specific) expressing ubiquitin-green fluorescent protein (green), and non–ova-specific T cells, mixed at a 1:99 ratio. Cells were cultured with ova-loaded DCs for 7 days on an uncoated surface (J) or on a surface coated with CCL21+ICAM1 (K). Images were taken after the breaking down of clusters and their spin-down, facilitating automated cell counting. Cell nuclei are stained blue, dead cell nuclei are in red (propidium iodide). Scale bars, 50 μm. (L) Bar graph illustrating the ratio of viable OT-II T cells to the originally seeded cell number, as quantified by automated image analysis (Data are representative of at least 3 independent experiments with 6 replicates (wells) and 20 fields of view in each well. Error bars represent standard error of the mean). Coating with CCL21+ICAM1 enriches specifically activated T cells (see supplemental Figure 4 for data of non-ova T cells). Calculated P values in panels I and L (using t test) are as indicated in the figure.
To determine whether the increase in viable cell number on the double-coated surface was merely the result of geometrical changes and physical tethering of the cells to the surface, we examined T-cell adhesion and expansion on a surface coated with anti-LFA1 blocking antibodies. Similar to findings on the ICAM1 coating, the immobilized anti-LFA1 mediated T-cell binding to the surface via the LFA1 receptor (Figure 6A-C). Nevertheless, there was no apparent increase in cell expansion in these cultures, indicating that stimulation of cell proliferation is not driven merely by adhesion to the substrate and reduction of the clusters per se but, rather, depends on genuine ICAM1-LFA1 interactions (Figure 6D). It should be noted that the effects of surface coatings on cells are direct and not mediated by the DCs, as when the cells were transferred to the coated surface (at the 24-hour point) the vast majority of the DCs were already dead (data not shown).
Figure 6.

Increased cell expansion on CCL21+ICAM1-coated surfaces depends on ICAM1-LFA1 signaling. (A-C) Representative images of OT-II T cells activated for 24 hours with antigen-loaded DCs and reseeded for an additional 48 hours on different surface coatings, with or without an immobilized blocking antibody against LFA1. The degree of cell spreading was similar when CCL21 (A) was combined with either ICAM1 (B) or blocking antibody against LFA1 (C). Scale bars, 100 μm. (D) Bar graph illustrating the number of live cells, measured using a metabolic cell viability assay (data are representative of at least 3 independent experiments with 3 to 4 replicates; error bars represent standard error of the mean). Number of T cells seeded per well: 150 × 103. Despite similar degrees of cell spreading and attachment to the surface through the LFA1 T-cell receptor, there were 4.5 times more live cells when CCL21 coating was combined with ICAM1 than when combined with the anti-LFA1 blocking antibody, indicating that the effect on expansion required actual ICAM1-LFA1 signaling and could not be replaced by the immobilized anti-LFA antibody. Calculated P values (using t test) are as indicated in the figure.
Finally, we examined the possibility of further augmenting cell number by adding selected cytokines to the culture medium at a wide range of concentrations (5-200 ng/mL). Tested cytokines included IL-2, IL-12, IL-6, IL-7, tumor necrosis factor α, interferon γ, tumor necrosis factor β, IL-4, and granulocyte-macrophage colony-stimulating factor. Most of the cytokines tested had no significant effect on T-cell expansion, either alone or in combination with CCL21 and/or ICAM1 coatings (data not shown). However, we found that IL-6 decreased the percentage of dead cells/late apoptosis/necrosis by up to 20% (Figure 7E; supplemental Figure 5), and increased T-cell expansion without coating by more than threefold (Figure 7F). More important, this IL-6-induced increase was greatly enhanced when cells were seeded on CCL21+ICAM1-coated surfaces, yielding an up to eightfold increase in the number of viable T cells at 7 days compared with the uncoated surface, and up to a 1.6-fold increase, compared with the CCL21+ICAM1-coated surface (Figure 7F; supplemental Figure 3).
Figure 7.

Coating with CCL21+ICAM1 increases T-cell proliferation following antigen-specific and nonspecific activation, whereas IL-6 increases cell survival. (A-D) Representative images of OT-II T cells expressing ubiquitin-green fluorescent protein (green) grown in the different indicated conditions for 7 days, after the breaking down of the clusters, and their spin-down. Dead cell nuclei are stained in red. Scale bars, 50 μm. (E-F) Bar graphs illustrating the percentage of dead cells (E) and the number of viable cells (F), as quantified using automated image analysis (number of T cells seeded per well: 12 × 103; data are representative of at least 3 independent experiments with 5 replicates (wells) and 20 fields of view in each well (see supplemental Figure 3 for summary of all independent expansion tests). Error bars represent standard error of the mean. IL-6 decreased the percentage of dead cells by approximately twofold on both coated and uncoated surfaces (E). IL-6 increased live cell numbers by 7.7-fold when combined with the CCL21+ICAM1 coating. CCL21+ICAM1 coating with no IL-6 increased cell numbers by 4.9-fold compared with the uncoated surface (F). (G-H) Histogram and bar graph illustrating the increase in cell proliferation induced by CCL21+ICAM1 with or without IL-6 following antigen-specific activation, as measured by CFSE dilution (data are representative of at least 3 independent experiments with 3 replicates each; error bars represent standard error of the mean). Color code in panel G: green, nonactivated T cells; red, T cells activated without coating; purple, T cells activated on CCL21+ICAM1 coating; orange, T cells activated on CCL21+ICAM1 coating with IL-6. (I) Bar graph illustrating live cell number, measured using a metabolic cell viability assay (data are representative of at least 3 independent experiments with 4 replicates each; error bars represent standard error of the mean) of T cells activated with either antigen-loaded DCs or activation beads, with or without CCL21+ICAM1 coating. Number of T cells seeded per well was 60 × 103. CCL21+ICAM1 increases T-cell yield in both activation methods. (J-M) Representative images demonstrating higher cell densities in cultures with CCL21+ICAM1 compared with no coating, activated with either activation beads (J-K) or antigen-loaded DCs (L-M). Scale bars, 50 μm. (N-O) Histogram and bar graph illustrating the increase in cell proliferation induced by CCL21+ICAM1 with and without IL-6, following activation with beads, as measured by CFSE dilution (data are representative of at least 3 independent experiments with 3 replicates each; error bars represent standard error of the mean). Color code in panel N is the same as that described in panel G. Calculated P values in E-F, H-I, and O (using t test) are as indicated in the figures.
Notably, the CCL21+ICAM1 coating did not significantly promote cell survival (Figure 7E). However, CFSE staining of cells before seeding and flow cytometry analysis demonstrated that the contribution of CCL21+ICAM1 to cell yield was mediated primarily through increased proliferation, as demonstrated by a nearly fourfold decrease in CFSE fluorescence intensity compared with the uncoated cultures (Figure 7G-H).
Furthermore, when testing the effect of CCL21+ICAM1 on T cells activated with commercially available anti-CD3 anti-CD28 coated beads, we found that activation with beads yields significantly fewer T cells than with DCs (Figure 7I-M). Nonetheless, CCL21+ICAM1 coating significantly augments T-cell proliferation and yield compared with T cells without coating (Figure 7I-K,N-O).
Discussion
In this study, we aimed to create an ex vivo environment for the efficient expansion of functional, antigen-specific CD4+ T cells, using cell-bound and secreted components of the lymph node niche. We show that the combined presentation of the chemokine CCL21 and the adhesion molecule ICAM1 on the substrate on which the cells were cultured induced a dramatic shift in the organization and morphology of the cells, as well as greatly improved the expansion of antigen-specific T cells. This combined effect was further enhanced by the addition of soluble IL-6, leading to a nearly eightfold increase in overall cell yield.
A hallmark of T-cell activation, both in vitro and in vivo39 is the formation of T-cell clusters. Our findings demonstrate that the size and number of such clusters are greatly elevated when the cells are plated onto a CCL21-coated substrate (Figure 1). This effect can be attributed to the known activation of LFA1,30,41 and stimulation of cell migration by CCL21.34,35 These large clusters were shown here to originate from multiple clones of specifically activated T cells that are loosely packed around 1 to 2 DCs, rather than from a single, proliferating T cell. Furthermore, these clusters are highly dynamic, frequently merging, splitting, and exchanging cells with other clusters (Figure 1).
The formation of large T-cell clusters driven by a CCL21 coating could potentially exert either beneficial or detrimental effects on T-cell activation. On the one hand, clustering may support the activation of many T cells by a small number of DCs, as well as the interplay between T cells residing within the same cluster. Clustered T cells secrete various regulatory factors with autocrine and paracrine activity, and the close proximity of cells within the cluster facilitates intercellular communication in a regulated microenvironment.3,40,43,44
On the other hand, large T-cell clusters may limit or even downregulate T-cell proliferation by restricting the cells’ access to essential signals or nutrients, or even by creating inhibitory environmental conditions. For example, clustering was shown to limit cytotoxic T-cell effector function and differentiation by limiting the T cells’ exposure to antigen during activation, upregulating inhibitory receptors (CTLA-4) and downregulating the expression of effector molecules.45 In vivo work further supports this duality, and some studies hint at the importance of clusters in T-cell activation and differentiation processes.3,40 However, ICAM1-mediated interactions, per se, are not obligatory for cell expansion to occur, as ICAM1-deficient T cells, which are unable to aggregate, can still proliferate.45
As CCL21 coating induced the formation of very large, yet short-lived, 3D clusters, we assumed that the conditions within these clusters might be less favorable. Hence, we examined the possibility that coating of the culture substrate with the adhesion molecule ICAM1 might attract LFA1-expressing cells, thereby reducing cell–cell interactions and, as a consequence, massive clustering. This, in turn, may help avoid some of the limiting factors inherent in clusters, enabling better control over the environmental conditions the cells experience.
Indeed, substrates double-coated with CCL21 and ICAM1 prevented large cluster formation and induced cell spreading and flattening on these substrates (Figures 2-4) in a direct, local manner limited to the coated area, but not affecting cells in adjacent, noncoated areas (Figure 4). Notably, T-cell clusters on uncoated or CCL21-coated surfaces were loosely attached to each other and to the culture substrate and could be easily displaced by gentle mechanical perturbation, whereas detaching the cells from the ICAM1-coated surface was difficult, even with vigorous pipetting. This demonstrates that surface-bound ICAM1 forms tighter, more stable contacts with cellular LFA1 compared with intercellular ICAM1-LFA1 interactions, and might thus reinforce and sustain the cells’ interactions with the stimulatory, substrate-bound CCL21. T cells on CCL21+ICAM1-coated surfaces also formed contacts with neighboring cells, using thin membranal extensions (Figure 2I-J), similar to those previously described.46
Most important, we found that the combination of CCL21 and ICAM1 increases T-cell yield and enriches antigen-specific activated CD4+ T cells (Figure 5; supplemental Figure 3A). We attribute the increase in cell numbers induced by the CCL21+ICAM1 surface coating primarily to enhanced cell proliferation, rather than to a reduction in cell death (Figure 7G-H). The strength of this combination might arise from preserving the potentially beneficial effects of intercellular interactions through LFA1-ICAM1 while avoiding the down-regulatory activity observed in the large 3D clusters. Another possible explanation for the mechanism underlying the collective effect of CCL21-ICAM1 involves the activation of LFA1, the ICAM1 receptor, by CCL21, thereby reinforcing the cells’ interactions with the surface. Our findings indicate that the effect of surface-bound ICAM1 is not merely attributable to the physical tethering of LFA1 to the surface, but rather to a genuine signaling event, induced by the exogenous ICAM1 (Figure 6).
Given the potentially improved access of cells on the double-coated surface to signaling molecules supplied in the culture medium, we examined whether addition of various cytokines can further enhance cell yield. Of the many types of cytokines tested (data not shown), only IL-6 induced a pronounced increase in the number of viable T cells beyond that obtained by the combined CCL21-ICAM1 coating, by attenuating cell death (Figure 7; supplemental Figure 3A-B). It should be noted that IL-6 induced the aforementioned changes only when added at the time of cell seeding, and not when added 24 to 48 hours later (data not shown), implying that it affects an early phase, after T-cell activation by DCs. These findings are in line with studies showing that IL-6 supports the survival and expansion of primed T cells in vivo, mainly by reducing apoptosis.28,29 It should be noted that our preliminary data indicate a similar beneficial effect of CCL21-ICAM1 coating on CD8 cytotoxic T cells while maintaining full functionality.
In our model, we chose to use antigen-loaded DCs as T-cell activators, as they display several important advantages over activation with commercially available anti-CD3 anti-CD28 coated beads. In addition to being a more native, antigen-specific, and less artificial form of activation, we found that DC activation results in greater T-cell yield (Figure 7I-M) while requiring a minimal number of T cells, smaller by sevenfold than the amount needed for beads-induced activation. However, similar to DC activation, T-cell activation with beads over the CCL21+ICAM1 coating, with or without IL-6, significantly augments T-cell proliferation and yield (Figure 7I-K,N-O) compared with activation without coating, whereas IL-6 decreased the percentage of cell death/late apoptosis/necrosis (supplemental Figure 5), implying its beneficial effects on other forms of activation.
It should be noted that CCL21-ICAM1 coating does not affect the proportions of the subdifferentiations Th1 and Th2, induced by antigen-loaded DCs, as well as beads (supplemental Figure 6A-B), apart from a decrease in Th2 induction with bead activation (supplemental Figure 6B). However, the conditions used for successful induction of Th1 and Tregs with bead activation were insufficient when using DC activation and failed to provide further subdifferentiation (supplemental Figure 6A-B). Nonetheless, after restimulation with or without induction of Th1, cells grown on CCL21+ICAM1 secreted similar amounts of interferon γ (supplemental Figure 6C). The fact that CCL21 and ICAM1 do not skew the phenotype of cells but, rather, increase their proliferation, enables flexibility in their manipulation and downstream use for therapy.
In conclusion, we demonstrate here the power of a synthetic immune niche to regulate cell behavior and fate. Specifically, we show that functionalization of the culture substrate with a specific combination of the lymph node components CCL21 and ICAM1, and addition of soluble IL-6 to the medium, can exert major effects on T-cell organization, intercellular interactions, and overall yield of antigen-specific T cells. On the basis of these results, we propose that a well-defined synthetic immune niche can regulate efficient expansion of antigen-specific T cells with desired therapeutic properties.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors are grateful to Barbara Morgenstern for her expert help in the style editing of this manuscript and to Yael Paran (IDEA Bio-Medical Ltd.) for assistance with the WiSoft image analysis system.
The authors acknowledge the support of the European Union Seventh Framework Program (FP7/2007-2013), under grant agreement NMP4-LA-1009-229289 NanoII (B.G. and N.F.); a European Research Council Advanced Grant, under grant agreement 294852-SynAd (B.G.); the Israel Science Foundation grant 1254/11 (N.F.); and the I-CORE Program of the Planning and Budgeting Committee and the Israel Science Foundation (N.F.). B.G. is the incumbent of the Erwin Neter Professorial Chair in Cell and Tumor Biology.
Authorship
Contribution: S.A.-L. participated in planning the experiments and their execution, data analysis, and manuscript preparation; I.Z. participated in planning the experiments and their execution and data analysis; H.S. and E.K. assisted in the electron microscopy experiments; O.G. assisted in image analysis and quantification; and B.G. and N.F. participated in planning the experiments, data analysis, and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Benjamin Geiger, Department of Molecular Cell Biology, Weizmann Institute of Science, 234 Herzl St, Rehovot 7610001, Israel; e-mail: benny.geiger@weizmann.ac.il; and Nir Friedman, Department of Immunology, Weizmann Institute of Science, 234 Herzl St, Rehovot 7610001, Israel; e-mail: nir.friedman@weizmann.ac.il.
References
- 1.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411(6835):380-384. [DOI] [PubMed] [Google Scholar]
- 2.Weigelin B, Krause M, Friedl P. Cytotoxic T lymphocyte migration and effector function in the tumor microenvironment. Immunol Lett. 2011;138(1):19-21. [DOI] [PubMed] [Google Scholar]
- 3.Gérard A, Khan O, Beemiller P, et al. . Secondary T cell-T cell synaptic interactions drive the differentiation of protective CD8+ T cells. Nat Immunol. 2013;14(4):356-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie J, Tato CM, Davis MM. How the immune system talks to itself: the varied role of synapses. Immunol Rev. 2013;251(1):65-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adutler-Lieber S, Zaretsky I, Platzman I, et al. . Engineering of synthetic cellular microenvironments: implications for immunity. J Autoimmun. 2014;54:100-111. [DOI] [PubMed] [Google Scholar]
- 6.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3(9):666-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6(5):383-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tibbitt MW, Anseth KS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol Bioeng. 2009;103(4):655-663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmes B, Castro NJ, Zhang LG, Zussman E. Electrospun fibrous scaffolds for bone and cartilage tissue generation: recent progress and future developments. Tissue Eng Part B Rev. 2012;18(6):478-486. [DOI] [PubMed] [Google Scholar]
- 10.Hughes CS, Postovit LM, Lajoie GA. Matrigel: a complex protein mixture required for optimal growth of cell culture. Proteomics. 2010;10(9):1886-1890. [DOI] [PubMed] [Google Scholar]
- 11.Okamoto N, Chihara R, Shimizu C, Nishimoto S, Watanabe T. Artificial lymph nodes induce potent secondary immune responses in naive and immunodeficient mice. J Clin Invest. 2007;117(4):997-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirsadraee S, Wilcox HE, Korossis SA, et al. . Development and characterization of an acellular human pericardial matrix for tissue engineering. Tissue Eng. 2006;12(4):763-773. [DOI] [PubMed] [Google Scholar]
- 13.Guillotin B, Guillemot F. Cell patterning technologies for organotypic tissue fabrication. Trends Biotechnol. 2011;29(4):183-190. [DOI] [PubMed] [Google Scholar]
- 14.Cupedo T, Stroock A, Coles M. Application of tissue engineering to the immune system: development of artificial lymph nodes. Front Immunol. 2012;3:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan JK, Watanabe T. Artificial engineering of secondary lymphoid organs. Adv Immunol. 2010;105:131-157. [DOI] [PubMed] [Google Scholar]
- 16.Irvine DJ, Stachowiak AN, Hori Y. Lymphoid tissue engineering: invoking lymphoid tissue neogenesis in immunotherapy and models of immunity. Semin Immunol. 2008;20(2):137-146. [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi Y, Kato K, Watanabe T. Synthesis of functional artificial lymphoid tissues. Discov Med 2011;12(65):351-362. [PubMed]
- 18.Suematsu S, Watanabe T. Generation of a synthetic lymphoid tissue-like organoid in mice. Nat Biotechnol. 2004;22(12):1539-1545. [DOI] [PubMed] [Google Scholar]
- 19.Zheng Y, Chen J, Craven M, et al. . In vitro microvessels for the study of angiogenesis and thrombosis. Proc Natl Acad Sci USA. 2012;109(24):9342-9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giese C, Lubitz A, Demmler CD, Reuschel J, Bergner K, Marx U. Immunological substance testing on human lymphatic micro-organoids in vitro. J Biotechnol. 2010;148(1):38-45. [DOI] [PubMed] [Google Scholar]
- 21.Bénézech C, Mader E, Desanti G, et al. . Lymphotoxin-β receptor signaling through NF-κB2-RelB pathway reprograms adipocyte precursors as lymph node stromal cells. Immunity. 2012;37(4):721-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Andrian UH, Mempel TR. Homing and cellular traffic in lymph nodes. Nat Rev Immunol. 2003;3(11):867-878. [DOI] [PubMed] [Google Scholar]
- 23.Willard-Mack CL. Normal structure, function, and histology of lymph nodes. Toxicol Pathol. 2006;34(5):409-424. [DOI] [PubMed] [Google Scholar]
- 24.Mueller SN, Germain RN. Stromal cell contributions to the homeostasis and functionality of the immune system. Nat Rev Immunol. 2009;9(9):618-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruco LP, Pomponi D, Pigott R, Gearing AJ, Baiocchini A, Baroni CD Expression and cell distribution of the intercellular adhesion molecule, vascular cell adhesion molecule, endothelial leukocyte adhesion molecule, and endothelial cell adhesion molecule (CD31) in reactive human lymph nodes and in Hodgkin's disease. Am J Pathol 1992;140(6):1337-1344. [PMC free article] [PubMed]
- 26.Gretz JE, Kaldjian EP, Anderson AO, Shaw S. Sophisticated strategies for information encounter in the lymph node: the reticular network as a conduit of soluble information and a highway for cell traffic. J Immunol 1996;157(2):495-499. [PubMed]
- 27.Kaldjian EP, Gretz JE, Anderson AO, Shi Y, Shaw S. Spatial and molecular organization of lymph node T cell cortex: a labyrinthine cavity bounded by an epithelium-like monolayer of fibroblastic reticular cells anchored to basement membrane-like extracellular matrix. Int Immunol 2001;13(10):1243-1253. [DOI] [PubMed]
- 28.Dienz O, Rincon M. The effects of IL-6 on CD4 T cell responses. Clin Immunol. 2009;130(1):27-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rochman I, Paul WE, Ben-Sasson SZ IL-6 increases primed cell expansion and survival. J Immunol 2005;174(8):4761-4767. [DOI] [PubMed]
- 30.Stein JV, Rot A, Luo Y, et al. The CC chemokine thymus-derived chemotactic agent 4 (TCA-4, secondary lymphoid tissue chemokine, 6Ckine, exodus-2) triggers lymphocyte function-associated antigen 1-mediated arrest of rolling T lymphocytes in peripheral lymph node high endothelial venules. J Exp Med 2000;191(1):61-76. [DOI] [PMC free article] [PubMed]
- 31.Sharma S, Zhu L, Srivastava MK, et al. . CCL21 chemokine therapy for lung cancer. Int Trends Immun. 2013;1(1):10-15. [PMC free article] [PubMed] [Google Scholar]
- 32.Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328(5979):749-752. [DOI] [PubMed] [Google Scholar]
- 33.Yang SC, Hillinger S, Riedl K, et al. . Intratumoral administration of dendritic cells overexpressing CCL21 generates systemic antitumor responses and confers tumor immunity. Clin Cancer Res 2004;10(8):2891-901. [DOI] [PubMed]
- 34.Gonen-Wadmany M, Oss-Ronen L, Seliktar D. Protein-polymer conjugates for forming photopolymerizable biomimetic hydrogels for tissue engineering. Biomaterials. 2007;28(26):3876-3886. [DOI] [PubMed] [Google Scholar]
- 35.Dikovsky D, Bianco-Peled H, Seliktar D. The effect of structural alterations of PEG-fibrinogen hydrogel scaffolds on 3-D cellular morphology and cellular migration. Biomaterials. 2006;27(8):1496-1506. [DOI] [PubMed] [Google Scholar]
- 36.Friedman RS, Jacobelli J, Krummel MF. Surface-bound chemokines capture and prime T cells for synapse formation. Nat Immunol. 2006;7(10):1101-1108. [DOI] [PubMed] [Google Scholar]
- 37.Flanagan K, Moroziewicz D, Kwak H, Hörig H, Kaufman HL. The lymphoid chemokine CCL21 costimulates naive T cell expansion and Th1 polarization of non-regulatory CD4+ T cells. Cell Immunol. 2004;231(1-2):75-84. [DOI] [PubMed] [Google Scholar]
- 38.Mitchison NA. An exact comparison between the efficiency of two- and three-cell-type clusters in mediating helper activity. Eur J Immunol. 1990;20(3):699-702. [DOI] [PubMed] [Google Scholar]
- 39.Ingulli E, Mondino A, Khoruts A, Jenkins MK. In vivo detection of dendritic cell antigen presentation to CD4(+) T cells. J Exp Med. 1997;185(12):2133-2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuhbauer DM, Mitchison NA, Mueller B. Interaction within clusters of dendritic cells and helper T cells during initial Th1/Th2 commitment. Eur J Immunol. 2000;30(5):1255-1262. [DOI] [PubMed] [Google Scholar]
- 41.Woolf E, Grigorova I, Sagiv A, et al. . Lymph node chemokines promote sustained T lymphocyte motility without triggering stable integrin adhesiveness in the absence of shear forces. Nat Immunol. 2007;8(10):1076-1085. [DOI] [PubMed] [Google Scholar]
- 42.Miller MJ, Safrina O, Parker I, Cahalan MD. Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J Exp Med. 2004;200(7):847-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thurley K, Gerecht D, Friedmann E, Höfer T. Three-dimensional gradients of cytokine signaling between T cells. PLOS Comput Biol. 2015;11(4):e1004206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248(4961):1349-1356. [DOI] [PubMed] [Google Scholar]
- 45.Zumwalde NA, Domae E, Mescher MF, Shimizu Y. ICAM-1-dependent homotypic aggregates regulate CD8 T cell effector function and differentiation during T cell activation. J Immunol. 2013;191(7):3681-3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sowinski S, Jolly C, Berninghausen O, et al. . Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol. 2008;10(2):211-219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.