

Key Points
The antiplatelet effects of 12-HETrE in humans and mice are partly dependent on IP in vitro.
The antithrombotic effects of 12-HETrE are partially dependent on IP in vivo in mice.
Abstract
The dihomo-γ-linolenic acid (DGLA)–derived metabolite of 12-lipoxygenase, 12-hydroxy-eicosatrienoic acid (12-HETrE), was recently shown to potently inhibit thrombus formation without prolonging bleeding in murine models. Although 12-HETrE was found to inhibit platelet activation via the Gαs signaling pathway, the Gαs-coupled receptor by which 12-HETrE mediates its antiplatelet effects has yet to be identified. Defining the receptor by which 12-HETrE exerts its effects is key to determining its therapeutic potential as an antiplatelet drug. Therefore, the goal of this study was to determine the Gαs-coupled platelet receptor through which 12-HETrE exerts its antiplatelet effects. In this study, we showed that pharmacological inhibition of the prostacyclin (IP) receptor in human platelets or genetic ablation of IP in murine platelets prevented 12-HETrE from blocking aggregation in vitro. Furthermore, the antithrombotic effects of 12-HETrE were significantly diminished in IP knockout mice in vivo. Together these data demonstrate that the antiplatelet effects of 12-HETrE are at least partially dependent on IP signaling. Importantly, this work identified 12-HETrE as a novel regulator of IP signaling that may aid in the rationale for design of novel therapeutics to inhibit platelet function. Additionally, this study provides further insight into the mechanism by which DGLA supplementation inhibits platelets function.
Visual Abstract
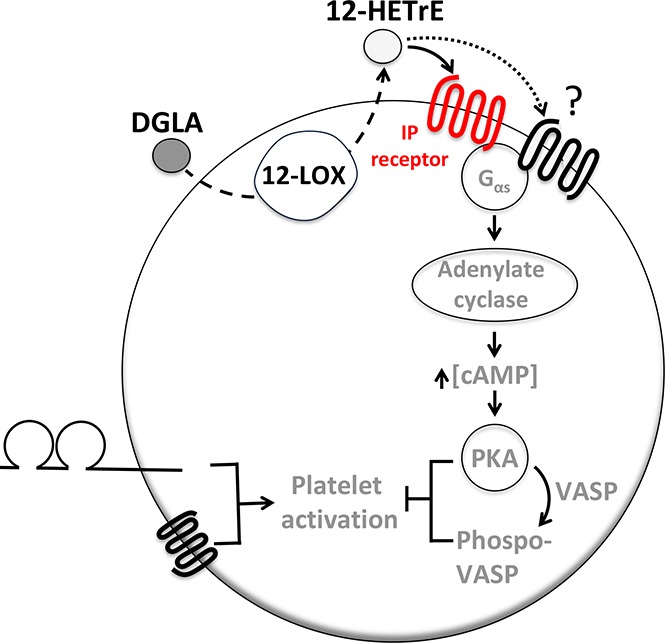
Introduction
The antiplatelet and antithrombotic effects of dihomo-γ-linolenic acid (DGLA), an ω-6 polyunsaturated fatty acid, were recently shown to be dependent on platelet-type 12-lipoxygenase (12-LOX) and its DGLA-derived oxylipin, 12(S)-hydroxyeicosatrienoic acid (12-HETrE).1 12-HETrE was shown to attenuate thrombus formation without disrupting hemostasis in murine models.1 As a potential therapeutic intervention, it is important to understand the most proximal step in the 12-HETrE signaling pathway. 12-HETrE was found to activate the Gαs subunit and exert its antiplatelet effects through adenylyl cyclase, a Gαs pathway component.1 Together these data suggest that 12-HETrE activates a Gαs-coupled platelet receptor; however, the receptor by which 12-HETrE elicits its antiplatelet effect has yet to be identified.
This study sought to determine the Gαs-coupled platelet receptor by which 12-HETrE inhibited platelet activation. Human platelets express a number of Gαs-coupled receptors, including 4 that are known to be regulated by oxylipins: prostacyclin (prostaglandin I2 [PGI2]) receptor (IP), prostaglandin E2 (PGE2) receptors (EP2 and EP4), and prostaglandin D2 (PGD2) receptor (DP1).2 We demonstrated that pharmacological inhibition of IP, but not EP2, EP4, or DP1, in human platelets reduced the antiaggregatory effects of 12-HETrE. Furthermore, the genetic ablation of IP in mice completely abolished the ability of 12-HETrE to inhibit platelet aggregation in vitro and severely reduced the antithrombotic effects of 12-HETrE in vivo. These data suggest that 12-HETrE exerts its antiplatelet effects through IP. This study provides further mechanistic support for a cardioprotective role of DGLA and identifies a potential ligand of IP.
Methods
Reagents
The IP antagonist RO3244794 was kindly provided by Adam Lauver (Michigan State University). The other prostanoid receptor antagonists, MK 0524, TG4-155, and CJ-42794, were purchased from Cayman Chemicals (Ann Arbor, MI). 12-HETrE was produced as previously reported.1 All other chemicals were purchased from Sigma Aldrich (St Louis, MO) unless otherwise specified.
Isolation of human platelets
The University of Michigan Institutional Review Board approved all experiments involving human participants, and written informed consent was obtained from self-reported healthy volunteers before blood collection. Whole blood was collected via venipuncture into vacutainers containing sodium citrate (3.2%; Becton, Dickinson and Company, Franklin Lakes, NJ). Platelets were isolated from whole blood via serial centrifugation, as previously reported.3 Briefly, platelet-rich plasma acquired via centrifugation of whole blood at 200 g for 10 minutes was treated with acid citrate dextrose (2.5% sodium citrate tribasic, 1.5% citric acid, 2.0% D-glucose) and apyrase (0.02 U/mL) and then spun for 10 minutes at 2000g. Platelets were resuspended to a final concentration of 3.0 × 108 platelets/mL in Tyrode’s buffer (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid 10 mM, sodium bicarbonate 12 mM, sodium chloride 127 mM, potassium chloride 5 mM, monosodium phosphate 0.5 mM, magnesium chloride 1 mM, and glucose 5 mM) as determined by a complete blood cell counter (Hemavet 950FS; Drew Scientific, Miami Lakes, FL).
Isolation of mouse platelets
Approval for all studies involving animals was obtained from the University of Michigan Institutional Animal Care and Use Committee. The wild-type (WT) C57BL/6 mice used for these studies were purchased from Jackson Laboratory (Bar Harbor, ME), and the IP-deficient (IP−/−) mice were kindly provided by Garret Fitzgerald (University of Pennsylvania). Blood was drawn from the inferior vena cava of 8- to 12-week-old anesthetized mice into a syringe containing sodium citrate (3.8%). An equal volume of Tyrode’s buffer was added to mouse whole blood, and platelets were isolated by serial centrifugation, as described in previous paragraph. Pelleted mouse platelets were resuspended in Tyrode’s buffer to 3.0 × 108 platelets/mL.
Platelet aggregation
Platelet aggregation was performed under stirring conditions (1100 rpm) at 37°C for 6 minutes using a Chrono-log Model 700D lumi-aggregometer (Havertown, PA). Human platelets were incubated with a prostanoid receptor antagonist for 2 minutes, then treated with 12-HETrE for an additional 2.5 minutes before agonist stimulation with PAR4-activating peptide (PAR4-AP; AYPGKF; GL Biochem, Shanghai, China), collagen (Chrono-log), or U46619 (Cayman Chemicals). Platelets isolated from WT or IP−/− mice were treated with 12-HETrE for 2.5 minutes before PAR4-AP stimulation. The concentrations of antagonist, 12-HETrE, and PAR4-AP used for each experiment are reported where appropriate.
VASP phosphorylation
Platelets were incubated with 12-LOX oxylipins, forskolin (5 µM), or dimethyl sulfoxide (DMSO) for 2.5 minutes at 37°C, and reactions were stopped by the addition of 5X Laemmli sample buffer (Tris-Cl 300 mM; pH, 6.8; 10% sodium dodecyl sulfate, 50% glycerol, 25% β-mercaptoethanol, 0.05% bromophenol blue). Platelet lysates were run on a 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and western blots were performed with antibodies to phosphorylated serine 157 and total vasodilator-stimulated phosphoprotein (VASP) (Enzolife sciences, Farmingdale, NY).
Laser-induced cremaster arteriole thrombosis model
The laser-induced cremaster arteriole thrombosis model was performed as previously described.1,4 Briefly, the cremaster muscle of anesthetized WT or IP−/− mice (8-12 weeks of age) was prepared under a dissecting microscope with constant superfusion of 37°C bicarbonate-buffered saline. Antiplatelet (DyLight 488 anti-GPIb 1 µg/g; Emfret, Eibelstadt, Germany) and antifibrin (Alexa Fluor 647 0.3 µg/g; a gift from Rodney Camire at Children’s Hospital of Philadelphia) antibodies were administered simultaneously with either 12-HETrE (6 mg/kg) emulsified in a formulation of 5% DMSO and 45% PEG300 in phosphate-buffered saline (PBS) or vehicle (5% DMSO and 45% PEG300) via jugular vein catheter into mice 10 minutes before induction of laser-induced injury of the cremaster arterioles (30-50 µm diameter) by a laser ablation system (Ablate! photo-ablation system; Intelligent Imaging Innovations, Denver, CO). Multiple laser injuries were performed in each mouse, with each new injury induced upstream of all prior injuries. Images of thrombus formation were taken at .2-s intervals using a Zeiss Axio Examiner Z1 fluorescent microscope with a ×63 objective and a high-speed sCMOS camera. Images were analyzed using Slidebook 6.0 (Intelligent Imaging Innovations).
FeCl3-induced carotid artery thrombosis model
Gel-filtered platelets were isolated from genotype-matched donor mice using a Sepharose 2B chromatography column and fluorescently labeled with calcein acetoxymethyl (1 μg/mL). Fluorescence-labeled platelets (3 × 106 platelets per gram) were injected into the tail vein of recipient mice (8-10 weeks old). Recipient mice were anesthetized, and the right common carotid artery was exposed. After surgery, 12-HETrE (6 mg/kg) emulsified in a formulation of 5% DMSO and 45% PEG300 in PBS or vehicle (5% DMSO and 45% PEG300 in PBS) was injected via jugular vein catheter. Ferric chloride (FeCl3)–induced thrombus formation was initiated by the topical application of Whatman filter paper saturated with a 10% FeCl3 solution to the carotid artery for 2 minutes. The carotid artery was continuously recorded under 5× objective with a Zeiss Axio Examiner Z1 fluorescent microscope beginning 1 minute before induction of FeCl3 injury until vessel occlusion was reached or the recording was terminated at 30 minutes postinjury. All the images were recorded and analyzed using Slidebook 6.0 program.
Statistical analysis
Data analysis was performed with GraphPad Prism 7 software (GraphPad Software, La Jolla, CA). Data are reported as means ± standard deviations, and statistical tests used for individual experiments are listed in figure legends where appropriate.
Results
Antiplatelet effects of 12-HETrE are partially dependent on IP in human platelets
Our laboratory recently showed that 12-HETrE inhibits platelet activation via an unidentified Gαs-coupled receptor.1 Human platelets express 4 known Gαs-coupled receptors that are activated by oxylipins, including IP, EP2, EP4, and DP1.2 To determine if 12-HETrE signals via a known oxylipin-activated Gαs-coupled platelet receptor, washed human platelets were incubated with various receptor antagonists in the presence or absence of 12-HETrE. Platelets were then stimulated with EC100 levels of PAR4-AP, resulting in full aggregation in DMSO-treated (vehicle) treated platelets. Because platelet aggregation varies among individuals,5-7 the concentration of PAR4-AP required to elicit full platelet aggregation ranged from 25 to 50 μM. Treatment with the IP antagonist RO3244794 (250 nM) fully blocked the antiaggregatory effects of 12-HETrE at all concentrations of 12-HETrE tested (5-20 μM; Figure 1A). In contrast, 12-HETrE inhibition of platelet activation persisted in the presence of both EP2 (TG4-155; 2 μM) and EP4 (CJ-42794; 80 nM) antagonists as well as the DP1 antagonist (MK 0524; .4 nM; Figure 1B-C). As expected, the pharmacological inhibitors of IP, EP2/EP4, and DP1 completely reversed the antiplatelet effects of their cognate ligands, PGI2 (4 nM), PGE2 (20 μM), and PGD2 (30 nM), respectively.
Figure 1.

Inhibition of IP in human platelets diminishes 12-HETrE antiplatelet effects PAR4-mediated platelet aggregation. The minimal concentration of PAR4-AP (25-50 µM) inducing full platelet aggregation (>70%) in DMSO-treated human platelets was used to stimulate platelets incubated with increasing concentrations of 12-HETrE (5-25 µM) in the presence of a vehicle control or an antagonist to IP (R03244794; 250 nM; n = 3-6) (A), EP2 (TG4-155; 2 μM) and EP4 (CJ-42794; 80 nM) (n = 5) (B), or DP1 (MK 0524; .4 nM; n = 5) (C). Two-way statistical analysis of variance was performed. Known antiplatelet prostanoid receptor agonists PGI2 (4 nM), PGE2 (20 μM), and PGD2 (30 nM) were used to confirm the efficacy of R03244794, TG4-155/CJ-42794, or MK 052480, respectively. Two-tailed, paired t test was performed; *P < .05, **P < .01, ***P < .001. Data represent mean ± standard deviation.
12-HETrE has been shown to inhibit platelet aggregation in response to all agonists tested, including collagen and U46619, the thromboxane mimetic.1,8 To assess if 12-HETrE inhibited platelet aggregation induced by agonists other than PAR4-AP in an IP-dependent manner, RO3244794- (250 nM) or vehicle control–treated platelets were incubated with 12-HETrE and then stimulated with collagen or U46619. Treatment of platelets with RO3244794 reversed the inhibitor effects of 10 or 20 μM of 12-HETrE on U46619-mediated platelet aggregation (Figure 2A). In response to collagen stimulation, the antiaggregatory properties of 12-HETrE were fully reversed at 10 μM of 12-HETrE but only partially reversed at 20 μM of 12-HETrE (Figure 2B). To evaluate whether IP was required for 12-HETrE to induce Gαs signaling, VASP (S157) phosphorylation, the major substrate of PKA, was measured in platelets treated with 12-HETrE (10 or 20 μM) in the presence or absence of RO3244794. Relative to DMSO, 12-HETrE induced VASP phosphorylation in control-treated platelets but not in RO3244794-treated platelets (Figure 2C). To determine if RO3244794 selectively inhibited Gαs signaling in response to IP stimulation, platelets were incubated with RO3244794 and then treated with PGI2, a known IP agonist, or ligands to either EP2 or DP1, PGE2 or PGD2, respectively. As expected, PGE2 (20 μM) or PGD2 (30 nM) caused robust VASP phosphorylation in either vehicle control– or RO3244794-treated platelets (Figure 2D). PGI2 was capable of eliciting VASP phosphorylation in control-treated platelets but not in platelets treated with RO3244794. To determine whether 12-HETrE functions independently of endogenous IP agonists such as the COX metabolites, PGI2 and PGE1, the ability of 12-HETrE to inhibit PAR4-mediated aggregation in platelets treated with aspirin, a COX inhibitor, was assessed. In platelets pretreated with aspirin, 12-HETrE inhibited PAR4-medated aggregation compared with DMSO (Figure 2E).
Figure 2.

12-HETrE is partially dependent on IP to inhibit human platelet activation. RO3244794- (250 nM) or vehicle control–treated human platelets were incubated with 12-HETrE (10 or 20 μM) or DMSO and then stimulated with U46619, the thromboxane mimetic (A), or collagen (B). One-way statistical analysis of variance with Dunn’s multiple comparison post-test was performed. Asterisks denote statistical differences between control- and RO3244794-treated groups: *P < .05, **P < .01, ***P < .001. Circles denote statistical differences between 12-HETrE–treated platelets and controls: •P < .05, ••P < .01, •••P < .001. (C) Human platelets were treated with DMSO, forskolin (5 µM), or 12-HETrE (10 or 25 µM) for 2.5 minutes and lysed, separated on a sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and immunoblotted with antibodies specific for phosphorylated (pS157; green) and total (red) VASP. The amount of phosphorylated VASP was normalized to the amount of total VASP in each sample, and data are reported as fold change in VASP phosphorylation relative to the DMSO treated sample. (D) VASP phosphorylation was measured by western blot using the lysates of human platelets incubated with RO3244794 or vehicle control and then treated with PGI2 (4 nM), PGE2 (20 μM), or PGD2 (30 nM). (E) Human platelets pretreated with aspirin were stimulated with PAR4-AP (75 µM) in the presence of 12-HETrE (10 μM) or vehicle control. Data represent mean ± standard deviation. One-way statistical analysis of variance with Dunn’s multiple comparison post-test was performed. *P < .05, **P < .01, ***P < .001. ETOH, ethanol.
IP is required for 12-HETrE to inhibit the activation of mouse platelets
Because of potential off-target effects of pharmacological inhibitors, we chose to further evaluate the requirement of IP in the antiplatelet functions of 12-HETrE using platelets from IP−/− mice. Platelets from WT mice treated with 12-HETrE before stimulation with either 25 or 50 μM of PAR4-AP had a significant decrease in platelet aggregation compared with platelets treated with vehicle (Figure 3A). Consistent with pharmacological inhibition of IP in human platelets, 12-HETrE was unable to inhibit PAR4-mediated aggregation in platelets from IP−/− mice (Figure 3A).
Figure 3.

IP is required for 12-HETrE to inhibit mouse platelet activation. (A) Platelets isolated from WT (black bars) or IP−/− (red bars) mice (n = 4-5) were stimulated with 25 or 50 µM of PAR4-AP in the presence of DMSO or 12-HETrE (10 or 25 μM). Two-tailed paired t test; ***P < .001. (B) The lysate of platelets from WT or IP−/− mice that had been treated with DMSO, forskolin (Forsk; 5 µM), 12-HETE (HETE; 25 µM), or 12-HETrE for 2.5 minutes were separated on sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with antibodies specific for phosphorylated (pS157; green) and total (red) VASP. The amount of phosphorylated VASP was normalized to the amount of total VASP in each sample, and data are reported as fold change in VASP phosphorylation relative to DMSO-treated sample. One-way statistical analysis of variance with Dunn’s multiple comparison post-test was performed comparing DMSO with each condition. *P < .05, **P < .01, ***P < .001. Data represent mean ± standard deviation.
To determine if IP was required for 12-HETrE–induced Gαs signaling, platelets from WT or IP−/− mice were treated with 12-HETrE, and VASP (S157) phosphorylation was assessed by western blot as a surrogate for PKA activation. As expected, 12-HETrE was able to elicit robust VASP (S157) phosphorylation in platelets from WT but not IP−/− mice compared with vehicle (Figure 3B). Forskolin (5 μM), a direct adenylyl cyclase activator, elicited VASP phosphorylation in platelets from WT and IP−/− mice, suggesting that cyclic adenosine monophosphate formation was not disrupted downstream of adenylyl cyclase in IP−/− mice. As expected, there was no increase in VASP phosphorylation compared with vehicle control in platelets treated with 12-HETE, a 12-LOX–derived proaggregatory oxylipin (Figure 3B). Using a genetic approach, these data further support that IP is required for 12-HETrE to induce Gαs signaling in platelets.
Antithrombotic effects of 12-HETrE are at least in part dependent on IP expression in platelets
To determine if the previously reported in vivo antithrombotic effects of 12-HETrE were dependent on IP, WT and IP−/− mice were IV injected with 12-HETrE before a laser-induced injury at the inner face of the cremaster muscle arteriole wall. Consistent with previous work,1 WT mice treated with 12-HETrE had a significant reduction in thrombus formation as measured by both platelet accumulation and fibrin deposition compared with control mice (Figure 4; supplemental Videos 1 and 2). Similar to previous studies,9 IP−/− mice exhibited an increase in thrombus size and delayed resolution compared with WT mice (Figure 4). 12-HETrE–treated IP−/− mice had a reduction in platelet accumulation, but not fibrin deposition, compared with vehicle-treated IP−/− mice after laser injury; however, the ability of 12-HETrE to reduce platelet accumulation was significantly reduced in IP−/− mice (∼50% reduction) compared with WT mice (∼90% reduction) (Figure 4; supplemental Videos 3 and 4). The ability of 12-HETrE to partially inhibit platelet accumulation in vivo supports a requisite role for IP in 12-HETrE–mediated signaling in platelets.
Figure 4.

Antithrombotic effects of 12-HETrE are partly dependent on IP. (A) Representative images of platelet (anti-GPIb; green) and fibrin (antifibrin; red) accumulation at the indicated times after laser-induced injury of cremaster muscle arteriole in WT mice treated with vehicle (n = 3; 23 thrombi) or 12-HETrE (6 mg/kg; n = 3; 24 thrombi) or IP−/− mice treated IV with vehicle control (n = 3; 21 thrombi) or 12-HETrE (6 mg/kg; n = 3; 24 thrombi). Thrombus growth was quantified in real time after laser-induced injury of the cremaster muscle arterioles of WT or IP−/− mice by measuring the mean fluorescence intensity (MFI) of platelet and fibrin accumulation. Laser-induced injury and subsequent imaging were acquired using a 3I intravital microscopy system on a Zeiss Examiner at ×63 magnification. Two-way analysis of variance. Data represent mean ± standard deviation (SD). (B) The time to vessel occlusion after FeCl3-induced injury of the carotid artery was measured in WT and IP−/− mice treated with either control (DMSO and PEG300) or 12-HETrE. Two-tailed paired t test was performed; **P < .01. Data represent mean ± SD.
To determine if 12-HETrE impedes occlusive thrombus formation in large vessels in an IP-dependent manner, an FeCl3-induced carotid artery injury model of thrombosis was used. WT mice treated with 12-HETrE had an increased time to vessel occlusion in response to FeCl3-induced injury of the carotid artery compared with WT mice treated with control (DMSO and PEG300; Figure 4B). The time to vessel occlusion in IP−/− mice treated with either 12-HETrE or vehicle control was similar in response to FeCl3-induced injury of the carotid artery (Figure 4B). These data suggest that 12-HETrE prevents FeCl3-induced vessel occlusion of the carotid artery in an IP-dependent manner.
Discussion
12-HETrE, the 12-LOX–derived metabolite of DGLA, was recently demonstrated to inhibit platelet activation through a yet-to-be identified Gαs-coupled platelet receptor.1 The goal of this study was to determine if 12-HETrE inhibited platelet activation through a known lipid-activated Gαs-coupled platelet receptor. We demonstrate that inhibition of IP, a known lipid-activated Gαs-coupled platelet receptor, either pharmacologically or genetically reduced the antiaggregatory effects of 12-HETrE in vitro and that 12-HETrE was less potent at inhibiting thrombus formation in IP−/− mice compared with WT mice in vivo. Together these data establish a role for IP in mediating the antiplatelet and antithrombotic effects of 12-HETrE.
This study is the first to identify a platelet G protein–coupled receptor (GPCR) that is regulated by a 12-LOX–derived oxylipin. This finding is consistent with previous reports demonstrating that the 12-LOX–derived oxylipin of AA, 12-HETE, binds the orphan GPCR GPR31 in cancer cells.10-12 GPR31 has not yet been shown to be expressed on platelets, and on the basis of the data presented here (Figure 1), it is unlikely that 12-HETE and 12-HETrE signal through the same receptor, because 12-HETE was unable to elicit VASP signaling in platelets.
The inhibitory effects of 12-HETrE at 10 µM were fully reversed by RO3244794, an IP antagonist, in response to platelet stimulation with the minimal concentration of PAR4-AP, collagen, or U46619 required to cause full platelet aggregation. The antiplatelet functions of higher concentrations of 12-HETrE (20 µM) were fully reversed by RO3244794 in PAR4-stimulated platelets (Figure 1), but only partially reversed by RO3244794 in response to U46619 or collagen stimulation (Figure 2A-B). One explanation for the observed agonist-dependent effects may be the enhanced potency by which 12-HETrE has been shown to inhibit collagen- and U46619-mediated platelet activation compared with PAR4-mediated platelet activation. Interestingly, the IP antagonist fully blocked the phosphorylation of VASP on serine 157 in response to 12-HETrE (20 µM), suggesting the inhibitory effect of 12-HETrE observed at this concentration was not a result of stimulation of another Gαs-coupled platelet receptor. However, we have not excluded that the inhibition of IP may indirectly antagonize another receptor or signaling pathway. The mechanism by which 12-HETrE exerts its residual non–IP-dependent inhibition of platelet function at higher concentrations remains an active area of research in our laboratory.
Similar to the antiplatelet effects of 12-HETrE, the dependence of the antithrombotic effects of 12-HETrE on IP was stimulus dependent. In the FeCl3-induced carotid artery injury model of thrombosis, IP was required for 12-HETrE to prolong the time to vessel occlusion (Figure 4B), whereas the antithrombotic effects of 12-HETrE were only partially dependent on IP in the laser-induced injury model of thrombosis in the cremaster muscle arteriole (Figure 4A). The discrepancy between these 2 models of thrombosis could be as result of the difference in severity of vessel injury or the size of vessel.
This study provides further understanding of the antiplatelet and antithrombotic effects of DGLA. The identification of this stable oxylipin for inhibitory regulation of platelet function through IP and possibly other Gαs-coupled GPCRs will allow for future studies to determine the potential therapeutic implications of 12-HETrE or analogs derived from 12-HETrE to be developed as a new approach for antithrombotic therapeutics.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgment
This work was supported in part by National Institutes of Health grants from the National Institute of General Medical Sciences (R01 GM105671) (M.H. and T.R.H.), National Institute on Minority Health and Health Disparities (R01 MD007880) (M.H.), National Institute on Aging (R01 AG047986) (M.H. and T.R.H.), and National Heart, Lung, and Blood Institute (R01 HL114405 [M.H. and T.R.H.] and F32 HL129491 [B.E.T.]).
Authorship
Contribution: B.E.T. designed and performed the experiments, analyzed and interpreted the data, and wrote the manuscript; Z.R.I., J.C.F., R.A., and M.E. performed experiments, analyzed the data, and edited the manuscript; and T.R.H. and M.H. designed experiments and interpreted data and edited manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Michael Holinstat, University of Michigan Medical School, 1150 W. Medical Center Dr, Room 2220D, Ann Arbor, MI 48109-5632; e-mail: mholinst@umich.edu.
References
- 1.Yeung J, Tourdot BE, Adili R, et al. 12-HETrE, a 12-lipoxygenase oxylipin of dihomo-γ-linolenic acid, inhibits thrombosis via Gαs signaling in platelets. Arterioscler Thromb Vasc Biol. 2016;36(10):2068-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hubertus K, Mischnik M, Timmer J, et al. Reciprocal regulation of human platelet function by endogenous prostanoids and through multiple prostanoid receptors. Eur J Pharmacol. 2014;740:15-27. [DOI] [PubMed] [Google Scholar]
- 3.Tourdot BE, Conaway S, Niisuke K, Edelstein LC, Bray PF, Holinstat M. Mechanism of race-dependent platelet activation through the protease-activated receptor-4 and Gq signaling axis. Arterioscler Thromb Vasc Biol. 2014;34(12):2644-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reheman A, Yang H, Zhu G, et al. Plasma fibronectin depletion enhances platelet aggregation and thrombus formation in mice lacking fibrinogen and von Willebrand factor. Blood. 2009;113(8):1809-1817. [DOI] [PubMed] [Google Scholar]
- 5.Bray PF. Platelet hyperreactivity: predictive and intrinsic properties. Hematol Oncol Clin North Am. 2007;21(4):633-645, v-vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yee DL, Sun CW, Bergeron AL, Dong JF, Bray PF. Aggregometry detects platelet hyperreactivity in healthy individuals. Blood. 2005;106(8):2723-2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edelstein LC, Simon LM, Montoya RT, et al. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nat Med. 2013;19(12):1609-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikei KN, Yeung J, Apopa PL, et al. Investigations of human platelet-type 12-lipoxygenase: role of lipoxygenase products in platelet activation. J Lipid Res. 2012;53(12):2546-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murata T, Ushikubi F, Matsuoka T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388(6643):678-682. [DOI] [PubMed] [Google Scholar]
- 10.Guo Y, Zhang W, Giroux C, et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J Biol Chem. 2011;286(39):33832-33840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen CH, Stadler S, Brenner S, et al. Cancer cell-derived 12(S)-HETE signals via 12-HETE receptor, RHO, ROCK and MLC2 to induce lymph endothelial barrier breaching. Br J Cancer. 2016;115(3):364-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hampson AJ, Grimaldi M. 12-hydroxyeicosatetrenoate (12-HETE) attenuates AMPA receptor-mediated neurotoxicity: evidence for a G-protein-coupled HETE receptor. J Neurosci. 2002;22(1):257-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.