

Key Points
IKZF1 associations with high-risk B-ALL may differ by age and sex.
A novel variant on chromosome 14, rs189434316, is associated with over a 3.5-fold risk of normal cytogenetic B-ALL.
Abstract
The incidence and mortality rates of B-cell acute lymphoblastic leukemia (B-ALL) differ by age and sex. To determine if inherited genetic susceptibility contributes to these differences we performed 2 genome-wide association studies (GWAS) by age, sex, and subtype and subsequent meta-analyses. The GWAS included 446 B-ALL cases, and 3027 healthy unrelated blood and marrow transplant (BMT) donors as controls from the Determining the Influence of Susceptibility Conveying Variants Related to One-Year Mortality after BMT (DISCOVeRY-BMT) study. We identified 1 novel variant, rs189434316, significantly associated with odds of normal cytogenetic B-ALL (odds ratio from meta-analysis [ORmeta] = 3.7; 95% confidence interval [CI], 2.5, 6.2; P value from meta-analysis [Pmeta] = 6.0 × 10−9). The previously reported pediatric B-ALL GWAS variant, rs11980379 (IKZF1), replicated in B-ALL pediatric patients (ORmeta = 2.3; 95% CI, 1.5, 3.7; Pmeta = 1.0 × 10−9), with evidence of heterogeneity (P = .02) between males and females. Sex differences in single-nucleotide polymorphism effect were seen in those >15 years (OR = 1.7; 95% CI, 1.4, 2.2, PMales = 6.38 × 10−6/OR = 1.1; 95% CI, 0.8, 1.5; PFemales = .6) but not ≤15 years (OR = 2.3; 95% CI, 1.4, 3.8; PMales = .0007/OR = 1.9; 95% CI, 1.2, 3.2; PFemales = .007). The latter association replicated in independent pediatric B-ALL cohorts. A previously identified adolescent and young-adult onset ALL-associated variant in GATA3 is associated with B-ALL risk in those >40 years. Our findings provide more evidence of the influence of genetics on B-ALL age of onset and we have shown the first evidence that IKZF1 associations with B-ALL may be sex and age specific.
Visual Abstract
Introduction
Acute lymphoblastic leukemia (ALL) is a disease primarily impacting children, however, one-third of cases are in adults (>20 years of age).1 The 5-year relative survival for children 1 to 5 years of age is >90%, but for those over 60 years of age, the 5-year relative survival is <20%.2,3 Causation appears to be multifactorial, including exogenous or endogenous exposures and genetic susceptibility.4 Studies of environmental risk contributing to ALL are inconclusive. Suggested risk factors include obesity and smoking in individuals >55 years of age, occupational exposures, chemotherapy/radiation as therapy for other diseases, and prenatal/early exposures.5-15 These external exposures only account for a small proportion of disease risk, and a strong case for genetic contribution can be made for pediatric and adolescent/young-adult (AYA) ALL.16-19
Genome-wide association studies (GWASs) have identified common genetic variants for pediatric and AYA ALL in 6 regions either within or near these genes: ARID5B, IKZF1, CDNK2A/B, CEBPE, GATA3, BMI-PIP4K2A.20-31 Recently, the largest meta-analysis of 2 pediatric GWASs of B-cell ALL (B-ALL) identified 2 new susceptibility loci within genes LHPP and ELK3.32 Variants in ARID5B, IKZF1, and GATA3 vary in effect by age and cytogenetic subgroups. For example, ARID5B variants are associated with hyperdiploid B-ALL in pediatric patients,20,30,33,34 which accounts for ∼30% of pediatric ALL and is a marker of favorable prognosis.1 In contrast, rs3824662, a variant in GATA3, was consistently associated with AYA ALL regardless of cytogenetic subgroup, but is not associated with pediatric ALL.29 Despite the evidence of potential age effects20,21 (evidenced by the high mortality rate in adult ALL cases3) and sex effects, there is both higher incidence and worse prognosis of ALL in males than females; GWASs have not been conducted in adult-onset ALL nor have they been conducted by sex.3
To this end, we performed the first GWAS in a high-risk B-ALL population treated with unrelated donor (URD) allogeneic blood and marrow transplant (BMT), then further stratified by age, sex, and cytogenetic subgroup.
Methods
This study was conducted in accordance with the Declaration of Helsinki and was reviewed and approved by the Roswell Park Cancer Institute Institutional Review Board. All patient data were deidentified. Summary data are provided in this manuscript.
Study design and population
The cases and controls were selected from an ongoing parent study: Determining the Influence of Susceptibility Conveying Variants Related to One-Year Mortality after BMT (DISCOVeRY-BMT).35-38 Briefly, the parent study was designed to find common germ line genetic variation associated with survival after an URD-BMT. DISCOVeRY-BMT consists of 2 cohorts of ALL, acute myeloid leukemia, and myelodysplastic syndrome patients and their HLA-matched unrelated healthy donors35 (supplemental Methods). For this study, cases were diagnosed with B-ALL and controls were unrelated healthy donors aged 18 to 61 years who passed a comprehensive medical examination and were disease-free at the time of donation. T-cell ALL cases (N = 77) were removed from both cohorts due to the low number of cases and inherent differences between B- and T-cell ALL. All patients and donors provided written informed consent for their clinical data to be used for research purposes and were not compensated for their participation.
Genotyping and quality control
Genotyping was performed at the University of Southern California (USC) Genomics Facility using the Illumina Omni-Express BeadChip containing ∼733 000 single-nucleotide polymorphisms (SNPs). SNPs were removed if the missing rate was >2%, minor allele frequency (MAF) <1%, or for violation of Hardy-Weinberg equilibrium (P < 1.0 × 10−6).
Problematic samples were removed based on the sample missing rate, duplicates, reported-genotyped sex mismatch, abnormal heterozygosity, cryptic relatedness, and population outliers (supplemental Methods). All quality-control (QC) measures were implemented in R and Plink statistical software.39,40
Imputation
Genotype data were imputed using Impute2 v2.041-43 with a reference panel of haplotypes from the 1000 Genomes phase 1v3.44,45 QCTOOL46 was used to remove imputed genotypes with a MAF <0.01 and info and certainty score <0.7 and <0.9, respectively45,46
Statistical analyses
Descriptive statistics, including χ2 and Student t tests, were performed on demographic variables (age, sex, and cytogenetic subgroup) by case-control status. Logistic regression assuming an additive model implemented in SNPTESTv2.5 was used to perform genome-wide association analyses adjusted for age in cases and controls for overall B-ALL and subtype-specific B-ALL analyses.40,43 The following subtype-specific analyses were performed: hyperdiploid negative (<51 chromosomes), Philadelphia chromosome negative (Ph−), abnormal and normal cytogenetic subgroups. Some high-risk subtypes have been omitted due to data availability and insufficient sample sizes. GWASs by age group were done for individuals <20 years (pediatric), 20 to 40 years (young adult), and >40 years (older adult) compared with all controls. These age categories represent an average of age-specific categories from clinical literature and prior pediatric and AYA GWASs. Sex-specific analyses were also performed in males and females; sex status of individuals was self-reported and further confirmed by genotyping. Cohorts 1 and 2 were combined with METAL software (odds ratio from meta-analysis [ORmeta] and P value from meta-analysis [Pmeta])47,48 using standard error weighted meta-analysis; genome-wide significance was defined as Pmeta < 5 × 10−8 with Pmeta < 5.0 × 10−6 considered suggestive of association. The Cochran Q method (P values) and I2 were used to test for heterogeneity across subgroups.49 To determine whether age or sex could be considered a mediator50,51 (thus significantly impacting the effect of the association between SNP and B-ALL risk), the Sobel mediation test was used.52,53 The proportion of the total effect mediated by age or sex was determined by comparing the difference between the β-coefficients for B-ALL before and after adjustment for age: (βunadj − βadj)/βunadj, where βunadj and βadj are the total effect and the direct effect, respectively.52,53
We defined significant evidence of age mediating the SNP-disease relationship when P was <.05 and if all of the Sobel test criteria for a mediator were met.52,53 To interrogate the role of genetics in ALL age of onset further, we also performed a case-only analysis, using age as outcome (continuous and categorical) in regression models for each genome-wide significant association initially identified. Interaction analyses of SNP and sex, as well as OR, and comparisons of stratified estimates were used to determine whether sex was acting as an effect modifier.
Replication of sex-specific effects
DISCOVeRY-BMT ALL cases are predominantly young adults and older adults, thus sex- specific findings for our pediatric associations were tested within a meta-analysis of 2 previously performed pediatric B-ALL GWASs. These 2 data sets were previously used in a meta-analyses with 2 other published GWASs.54-60 Briefly, the cases consisted of 437 children of European ancestry with B-ALL treated on The Children’s Oncology Group P9904 protocol58,61 and 427 pediatric B-ALL cases from the German GWAS.55,62 Controls included 475 from the German GWAS and European ancestry controls (N = 958) from the Genetic Association Information Network (supplemental Methods).54 Stratified analyses by sex were performed within the cohort and we report ORs and P values for the logistic additive model. These data are only pediatric and thus replication for adult GWAS associations could not be performed.
Heritability and polygenic risk scores
Two approaches were used to better understand heritability and the aggregate contribution of genetic variation to B-ALL risk. We estimated the proportion of phenotypic variance explained by common SNPs genome-wide in males and females separately using genome-wide complex trait analysis (GCTA).63-66 Second, polygenic risk scores (PRSs) were calculated by combining significant loci weighted by effect sizes estimated from the logistic regression GWAS using PRSice software67 (supplemental Methods).
Results
Demographics
DISCOVeRY-BMT consists predominantly (>95%) of individuals self-reported as European American (EA) with 3073 BMT recipients and 3144 BMT donors initially genotyped. Sample QC on self-reported EA recipients and donors was performed on each cohort separately (supplemental Methods; supplemental Figure 1), which yielded 2111 recipients and 2219 donors in cohort 1 and 777 recipients and 808 donors in cohort 2.
Analyses described herein include either the 364 individuals with B-ALL who received a BMT (cases) and 2219 donors (controls) in cohort 1 and 82 B-ALL cases and 808 controls in cohort 2, or a subset of these (supplemental Table 1). Ages of cases ranged from 1 to 68 years, whereas controls ranged from 18 to 61 years due to minimum and maximum ages for donation. Controls are predominantly male, which reflects clinical selection bias against parous females who may increase risk of graft-versus-host disease. However, the proportion of males in the control group mirrors that of cases. In both cohorts, cases were also skewed to a hyperdiploid-negative karyotype, which is a more aggressive subtype of B-ALL frequently treated with BMT.
Genome-wide associations
The final genotyping data in recipients consisted of 637 655 typed SNPs in cohort 1 and 632 823 typed SNPs in cohort 2 (supplemental Methods; supplemental Figure 1) from which ∼8.5 million imputed variants were available for genome-wide analyses. Quantile-quantile (Q-Q) plots of SNP association with B-ALL (supplemental Figure 2) show no evidence of genomic inflation due to cryptic population structure (λ = 1.001) and none of the principal components (PCs) were associated with risk of B-ALL, therefore, PCs were not included in the regression analyses. We report on a novel genome-wide association with normal cytogenetic B-ALL, as well as genome-wide significant associations in genes previously identified by GWASs, considering age and sex in exploratory analyses (supplemental Table 2; supplemental Figure 3).
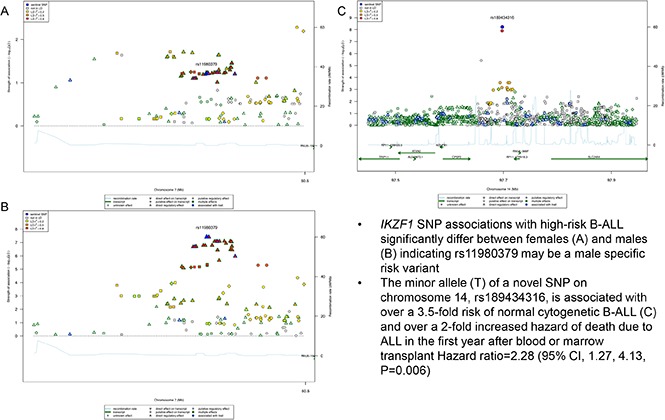
A novel association, rs189434316 (92 697 912 bp), with normal cytogenetic B-ALL was identified on chromosome 14 (Table 1; Figure 1) between SLC24A4 and CPSF2. The T allele (MAF = 0.07) increases odds of normal cytogenetic B-ALL by over 3.5-fold compared with controls (ORmeta = 3.7; 95% confidence interval [CI], 2.5-6.2; Pmeta = 6.0 × 10−9). This genome-wide association was seen only with normal cytogenetic B-ALL and was not observed overall or in other subtypes (all B-ALL cases, Pmeta = 2.6 × 10−5; hyperdiploid negative, Pmeta = 1.6 × 10−6; Ph−, Pmeta = 3.4 × 10−6; abnormal cytogenetic B-ALL, Pmeta = .3). To further explore the biological relevance of this novel variant, we analyzed the association of the SNP with both death due to disease and progression free survival, defined as the time to disease progression or death following transplant. The T allele is associated with death due to disease (hazard ratio [HR]meta = 2.28; 95% CImeta = 1.27, 4.13; Pmeta = .006) and worse progression-free survival (HRmeta = 1.45; 95% CImeta = 1.03, 2.03; Pmeta = .03) in normal cytogenetic B-ALL cases.
Table 1.
Genome-wide significant associations with B-ALL by age (<20, 20-40, >40 y) in EAs
| Variant information | Overall (age-adjusted) by subtype | <20 y* | 20-40 y* | >40 y* | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP†/nearest gene | Chr:base pair position | Alleles (EAF) | Cases | C1/C2 | ORmeta (95% CI)/ Pmeta | C1/C2 | ORmeta (95% CI)/ Pmeta | C1/C2 | ORmeta (95% CI)/ Pmeta | C1/C2 | ORmeta (95% CI)/ Pmeta | ||||
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | ||||||||
| rs3824662/GATA3 | 10:8104207 | A/C (0.20) | B-ALL | 1.9 (1.5, 2.3)/ 1.9 (1.2, 2.6) | 6.7 × 10−11/ .001 | 1.9 (1.3, 2.7)/ 3.3 × 10−13 | 1.9 (1.3, 2.6)/ 1.4 (0.4, 2.5) | .0002/.33 | 1.9 (1.0, 3.1)/ .0002 | 1.8 (1.2, 2.4)/ 2.4 (0.9, 3.8) | .0003/ .007 | 1.9 (1.1, 3.3)/ 6.4 × 10−6 | 2.2 (1.5, 3.0)/ 1.9 (0.7, 3.2) | 1.8 × 10−6/ .03 | 2.2 (1.1, 4.0)/ 2.4 × 10−7 |
| rs11980379/IKZF1 | 7:50469980 | C/T (0.30) | B-ALL | 1.6 (1.3, 1.9)/ 1.4 (1.0, 1.9) | 4.4 × 10−7/ .04 | 1.5 (1.2, 2.0)/ 6.9 × 10−8 | 2.4 (1.6, 3.1)/ 2.3 (1.7, 2.9) | 3.6 × 10−8/ .007 | 2.3 (1.5, 3.7)/ 1.0 × 10−9 | 1.5 (1.1, 1.9)/ 1.3 (0.6, 2.0) | .006/.4 | 1.4 (1.0, 2.1)/ .005 | 1.3 (0.9, 1.6)/ 1.1 (0.5, 1.7) | .1/.7 | 1.2 (0.9, 1.7)/ .1 |
| rs189434316/CPSF2‡ | 14:92697912 | A/T (0.07) | Normal Cytogenetic B-ALL | 3.6 (3.1, 4.1)/ 5.4 (4.3, 6.4) | 1.0 × 10−6/ .003 | 3.7 (2.5, 6.2)/ 6.0 × 10−9 | — | — | — | — | — | — | — | — | — |
P genome-wide significance <5.0 × 10−8 meta. Number of cases: B-ALL overall: overall (C1 = 364/C2 = 82) <20 y (C1 = 113/C2 = 21); 20-40 y (C1 = 130/C2 = 30); >40 y (C1 = 121/C2 = 31).
C1, cohort 1; C2, cohort 2; Chr, chromosome; EAF, effect allele frequency (the effect allele is bolded); OR, odds ratio of logistic additive model; ORmeta, odds ratio for fixed-effects model meta-analysis of DISCOVeRY-BMT cohorts 1 and 2; Pmeta, P value for fixed-effects model meta-analysis of DISCOVeRY-BMT cohorts 1 and 2.
Age-specific ORs and CIs are not reported due to low EAF of rs189434316.
Both variants in GATA3 and IKZF1 are typed, rs189434316 is imputed (info score = 0.87 and certainty = .95).
Nearest gene.
Figure 1.

Regional plot of SNP associations with normal cytogenetic B-ALL. Regional plot showing association with normal cytogenetic B-ALL in a region of chromosome 14 (rs189434316). The x-axis is the position on the chromosome (Mb) and y-axis is the −log10 P value of the SNP association with normal cytogenetics B-ALL. Filled colors indicate linkage disequilibrium (LD), as measured by r2, with the most significant SNP (shown in purple); red shows a high degree of LD whereas blue indicates lower r2. Meta-analyses show that the T allele (MAF = 0.07) increases odds of normal cytogenetic B-ALL by over 3.5-fold compared with controls (Pmeta = 5.6 × 10−9). This association was seen only with normal cytogenetic B-ALL and not observed in other subtypes. CPSF2 is the nearest gene to the association signal.
We found evidence of genome-wide associations in GATA3 and IKZF1, both identified in published AYA and pediatric ALL GWASs, respectively.20-23,29 With the exception of GATA3 and IKZF1, other previously published pediatric and AYA GWAS associations (CDKN2A/B, ARID5B, BMI-PIP4K2A, CEBPE, LHPP, ELK3) did not replicate in DISCOVeRY-BMT (supplemental Table 2). The variant in GATA3, rs3824662, previously shown to be associated with AYA B-ALL,29 increased odds of B-ALL overall (Pmeta = 3.29 × 10−13), within hyperdiploid-negative (Pmeta = 2.95 × 10−13), Ph− (Pmeta = 1.16 × 10−12), and normal cytogenetic B-ALL (1.09 × 10−8) (Table 1; supplemental Table 2). Another GATA3 variant, rs569421, showed a 60% increased risk of B-ALL in cases compared with controls (Pmeta = 4.4 × 10−8). This variant has not been reported to be associated with B-ALL overall or by subtype, however, joint analyses with rs3824662 indicated rs569421 is not an independent risk variant.
SNP rs11980379 in IKZF1 is a known pediatric B-ALL risk variant and perfectly correlated (r2 = 1.0) with rs413260, which has also previously been associated with pediatric B-ALL.20,21,23-28,30-32 SNP rs11980379 in IKZF1 was significantly associated at the genome-wide level with Ph− (Pmeta = 3.6 × 10−9) and normal cytogenetics B-ALL (age-adjusted) (Pmeta = 4.6 × 10−8) (supplemental Table 2). However, rs11980379 demonstrated smaller effect sizes and higher P values with increasing age (Table 1). In pediatric patients, the C allele in this SNP conferred 2.3-fold increased odds of B-ALL, an ∼1.4-fold increased risk of B- ALL in young adults (Pmeta = .005), and a 1.2-fold increased risk of B-ALL in older adults (Pmeta = .13) (Table 1; Figures 2-3). Stronger associations with smaller P values were also observed within the pediatric age group across all cytogenetic subgroups (hyperdiploid negative, Ph−, abnormal and normal) (data not shown) and thus this age-specific association is not attributable to differences in underlying subtype distributions. Sobel tests of mediation estimate that the proportion of the total effect (percentage of mediation) of the SNP that is mediated by age is 4% (P = .055), indicating some evidence for age as a pathway mediator.
Figure 2.

Manhattan plots for SNP associations with B-ALL by age. (A) Pediatric (<20 years), (B) young adults (20-40 years), and (C) older adults (>40 years). The x-axis indicates the chromosome and the y-axis is the −log10 P value from meta-analyses of SNP associations with B-ALL in cohorts 1 and 2. The dashed red line indicates genome-wide significance of P > 5.0 × 10−8. The SNPs highlighted in red on chromosome 7 (pediatric) are known variants in IKZF1.
Figure 3.

Regional plot of age-specific (<20, 20-40, >40 years) associations with B-ALL in IKZF1. Regional plot showing age-stratified association with B-ALL within IKZF1. The x-axis is the position on the chromosome (Mb), and y-axis is the −log10 P value. The circles (older adults), diamonds (young adults) and squares (pediatric) show the P value for the SNP association with risk of B-ALL, rs11980379. Filled colors indicated the LD with the most significant SNP shown in purple. Red indicates a high degree of LD whereas blue indicates lower r2. This plot shows a significant association in pediatrics, but no evidence of association with B-ALL in young adults and older adults.
In addition to age-specific effects, there are strong sex-specific associations for IKZF1, however, unlike age, sex does not mediate the effect of the variant on the risk of B-ALL (Sobel mediation, P = .5) but rather there is evidence of heterogeneity of effect between males and females. The C allele in rs11980379 showed an 80% increased risk of B-ALL in males (OR = 1.8; 95% CI, 1.24-2.50; Pmeta = 3.8 × 10−8), whereas there is no significant association in females (OR = 1.26; 95% CI, 0.92-1.70; Pmeta = .06) (Figure 4; Table 2; supplemental Figure 4), with evidence of significant heterogeneity between males and females (I2 = 80; Q = 0.02). When comparing crude and sex-adjusted OR, the crude OR for the IKZF1 SNP is 1.58, whereas the sex-adjusted OR is 1.56, yielding about a 1% difference; the ORs between the crude and adjusted should be different if sex is a confounder. The stratified ORs for males (1.9 [95% CI, 1.4, 2.3]) and females (1.2 [95% CI, 0.9, 1.6]) differ and in turn differ from the crude OR. Allele frequencies of controls by sex show no difference between males and females thus the SNP is not just associated because it has a higher frequency in men in the general population. This evidence indicates that sex could be modifying the effect of this IKZF1 variant on ALL risk. This sex-specific genetic effect shows some evidence of an age association as well. The C allele is not a risk factor in females >15 years (OR = 1.1; 95% CI, 0.8-1.5; P = .6) but appears to be more strongly associated in females <15 years (OR = 1.9; 95% CI, 1.2-3.2; P = .007). In contrast males show little change in OR for those >15 years at diagnosis (OR = 1.74; 95% CI, 1.4- 2.2; P = 6.38 × 10−6) vs <15 years (OR = 2.34; 95% CI, 1.4-3.8; P = .0007). In analyses of cases >15 years of age, the evidence of heterogeneity based on the Cochran Q statistic remains between males and females (I2 = 60), whereas in cases <15 years of age (replication cohort) there is no significant evidence in effect heterogeneity between males and females (supplemental Table 3). This finding for >15 years is also supported by evidence of statistical interaction between the SNP and sex variable (P = .02) in models of ALL susceptibility.
Figure 4.

Regional plot of sex-specific associations with B-ALL in IKZF1. Regional plot of sex-specific associations with B-ALL in the IKZF1 region. The x-axis is the position on the chromosome (Mb), and y-axis is the −log10 P value. The squares (male) and circles (female) show the P value for the SNP association with risk of B-ALL. Filled colors indicate the LD with the most significant SNP shown in purple. Red indicates a high degree of LD whereas blue indicates lower r2.
Table 2.
Genome-wide significant associations with the C allele in rs11980379 (IKZF1) and B-ALL by sex in EAs
| Sex | N cases (Cohort 1/Cohort 2) | Cohort 1 | Cohort 2 | ORmeta (95% CI) | Pmeta | I2 (P) | ||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |||||
| Male | 219/53 | 1.9 (1.4, 2.3) | 1.5 × 10−7 | 1.5 (0.9, 2.2) | .05 | 1.8 (1.2, 2.5) | 3.8 × 10−8 | 80.2 (.02) |
| Female | 145/29 | 1.2 (0.9, 1.6) | .13 | 1.4 (0.6, 2.1) | .3 | 1.3 (0.9, 1.7) | .06 | |
I2 and P reflect heterogeneity in the SNP associations with B-ALL between males and females.
OR, odds ratio of logistic additive model risk is presented in terms of the C (minor) allele.
Additional genome-wide significant variants previously identified in pediatric and AYA B-ALL GWASs, in genes ARID5B, BMI1-PIP4K2A, CEBPE, CDKN2A/B, ELK3, and LHPP, did not reach genome-wide significance in our B-ALL GWAS either overall, or by subtype, age, or sex. The most significant association in these genes was seen in CDKN2B, rs1333035, Pmeta = 2.09 × 10−5, in LD (r2 = 0.8) with rs3218005, previously identified in pediatric genome-wide analyses of ALL (supplemental Table 2).
Replication analysis of sex-specific findings
Replication analysis of sex-specific findings for those ages 1 to 15 years was performed in meta-analyses of 2 B-ALL pediatric GWASs. The rs11980379 association replicated in both the male and female B-ALL pediatric population conferring a 40% increased risk (ORmeta = 1.4; 95% CI, 1.1-1.9; P = 3.9 × 10−5) in males and a 60% increased risk in females (ORmeta = 1.6; 95% CI, 1.2, 2.3; P = 1.4 × 10−8) (supplemental Table 3). There was no evidence of heterogeneity in effect sizes between males and females <15 years, similar to our findings. The novel significant variant, rs189434316, could not be replicated in this pediatric population as there were no cytogenetically normal cases; rs189434316 was not associated with abnormal cytogenetic B-ALL in either our data or the replication (P = .3).
GCTAs and PRS
GCTA estimates of male and female heritability (h2) were 0.83 (standard error [SE] = 0.34; P = .001) and 0.52 (SE = 0.18; P = .0002), respectively (supplemental Table 4). The correlation between male and female heritability estimates was weak with a large standard error (ρ = 0.11; SE = 0.29) (supplemental Table 4).
The PRS distributions and medians differ between cases (overall and by age group) and controls, with cases having a significantly higher median risk score than controls (P < .001) (supplemental Figure 5). In cohort 1, the high-risk group (carriers of the most high-risk alleles from SNPs) had a threefold increased risk of having B-ALL (P = 2.4 × 10−14) and the medium-risk group conferred an almost twofold increased risk of disease (P = .0006), when compared with the low-risk group (reference) (supplemental Table 5).
When stratified by age, pediatric individuals with high PRS had an almost fourfold increased risk of B-ALL (P = 1.5 × 10−7); the medium PRS group had 40% increased odds of having B-ALL (P = .3), although nonsignificant, compared with those with low PRS score. In the young adults and older adults, high-risk PRS score individuals had threefold increased odds of disease, P = 4.3 × 10−7 and P = 3.7 × 10−6, respectively. The medium-risk groups showed an ∼1.8 and twofold difference from the low-risk group in young adults (P = .02) and older adults (P = .01), respectively (supplemental Table 5). Males had a 3.5-fold increased risk of B-ALL for the high- risk PRS group compared with the low-risk group (P = 4.1 × 10−11). Females also showed an increased risk of B-ALL (OR = 2.5) in the high-risk group compared with the low-risk group (P = .0001). The high-risk median PRS was not significantly different between males and females (P = .34).
Discussion
In these GWASs of B-ALL susceptibility across age and between sexes in a high-risk BMT population, we found evidence for novel associations within subtypes, as well as sex- and age- specific associations in previously identified variants. Besides these variants, we did not replicate genome-wide or suggestive associations with other prior known pediatric GWAS regions. Most likely this is due to these loci being associated with favorable-risk ALL (pediatric) and the cases that comprise DISCOVeRY-BMT are high-risk ALL.
The established GATA3 B-ALL risk variant showed association regardless of cytogenetic subgroup. GATA3 encodes for a transcription factor that is critical for lymphoid cell lineage commitment and early T-cell differentiation, and loss-of-function somatic mutations have been discovered in early T-cell precursor ALL.68,69 Alterations in GATA3 have been linked to other blood cancers, including Hodgkin lymphoma.70 Our GATA3 findings in conjunction with the evidence for association of the GATA3 risk variant with pediatric Ph-like ALL,22 a more adverse type of ALL, and AYA associations suggest GATA3 is linked to high-risk ALL.
The strength of the IKZF1 variant in our high-risk pediatric B-ALL group indicates that this variant most likely contributes to risk of both adverse ALL (hyperdiploid negative) and favorable ALL as it has previously been strongly associated in pediatric ALL cases (hyperdiploid) with favorable outcomes. IKZF1 is a transcription factor that is needed for development of hematopoietic stem cells to lymphoid precursors.71,72 It is frequently targeted by copy-number alterations in ALL blast cells (particularly in high-risk ALL); deletions and mutations result in loss of function or dominant-negative isoforms73 and are associated with a poor prognosis.72,73 Two SNPs (rs6964969 and rs11978267) strongly correlated with the IKZF1 variant (r2 > .95) are cis-expression quantitative trait loci in monocytes and whole blood.74-76 In addition, rs6964969 is predicted to affect NFKB1 transcription factor binding, which can lead to inappropriate immune cell development or delayed cell growth when there are problems with normal binding/expression.77
The age-specific findings reinforce the idea that genetic variation may contribute differently to risk of pediatric vs adult high-risk B-ALL. This is reasonable as other genetic features, for example, chromosome aberrations, also differ between pediatric and adult ALL.
Unlike age, sex is not a mediator variable in the relationship between the IKZF1 genetic variant and B-ALL risk, but rather is modifying the effect of the SNP on risk of B-ALL. Interestingly, it is possible that this sex effect is age-specific. Our analyses of genetic association at an approximate pre- and postpuberty age may indicate that there is some relationship between female sex hormones (activated during puberty) and this SNP, manifesting in similar risk attributable to the IKZF1 variant before puberty in males and females, but not for females following puberty. Analyses of the (prepuberty) pediatric patients, and thus those with favorable B-ALL subtypes, who comprise the replication data set, clearly demonstrate that pediatric ALL germ line susceptibility IKZF1 associations do not differ by sex. Our data demonstrate that as age increases (>15 years), sex-specific associations are observable. Although we analyzed <15 vs >15 years, further investigation into the role of age and sex in large sample sets with greater variance in B-ALL subtype is warranted.
The PRS models show that the high-risk group and the medium-risk group have a significantly different risk than the low-risk category, with nonoverlapping CIs. Given the significantly increased risk of B-ALL in those in the high-risk group, it is valuable to consider these variants together and how they are contributing to disease risk.
Our novel finding on chromosome 14, rs189434316, was associated with normal cytogenetic B-ALL but not abnormal cytogenetic B-ALL. This variant is significantly associated with increased hazard of death due to disease and worse progression-free survival, adding to the biological plausibility of this novel finding. It is not immediately clear how this SNP could be correlated with outcome, as functional annotation does not demonstrate evidence for impacting gene expression or transcription factor binding. Replication in another high-risk population is an important next step.78
Although this is the first B-ALL susceptibility study of a high-risk population across the age spectrum, our study has some limitations. We had 80% power to detect ORs in line with previous reports, 2 to 1.5, for MAF ranging from 40% to 10%, respectively; this was reduced for the age and sex subgroup analyses. Also, specific translocations (eg, BCR-ABL, ETV- RUNX, and MLL) were not considered and analyses were limited to EAs, thus, these variants may not be valid for other continental ancestry groups. We used a standard genome-wide association significance level of P < 5.0 × 10−8 for each of our GWASs; a more stringent threshold might need to be considered given that we are testing 3 age groups and sex as well.
Our age-specific GWAS identified inherited variants that strongly influence B-ALL susceptibility in adults and validate AYA findings, shedding new light on age-related differences in ALL biology. To date, GWASs of ALL either adjusted for sex or did not report sex-specific results, hence our study provides the first evidence that sex is an effect modifier and different genetic variants are contributing to ALL in males and females.
Understanding genetic contribution can aid our understanding of B-ALL etiology. Also, identification of people at high risk for B-ALL enables the integration of genetic and clinical risk factors to improve patient stratification.79 This study is 1 step closer to achieving more personalized inherited susceptibility to this heterogeneous and devastating disease.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
K.O., E.H., and A.S. performed the replication analyses while K.O. and E.H. were employed at The University of Chicago.
This work was supported by the National Institutes of Health: National Cancer Institute grant R03 CA188733 (L.E.S.-C. and T.H.), National Heart, Lung, and Blood Institute grant R01 HL102278 (which funded DISCOVeRY-BMT) (L.E.S.-C. and T.H.), and National Cancer Institute grant P30 CA016056 (which partially supported the Roswell Park Cancer Institute Biostatistics & Bioinformatics Core).
Replication data sets were supported by grants from the National Institutes of Health (Eunice Kennedy Shriver National Institute of Child Health and Human Development HD0433871; National Cancer Institute CA129045 and CA40046 [K.O.]; National Institute of Mental Health R01 MH101820; National Cancer Institute U01CA176063 and National Institute of General Medical Sciences U01GM92666); the St. Baldrick’s Foundation (K.O.); the American Cancer Society, Illinois Division (K.O.); and the Cancer Research Foundation (K.O.).
The Children’s Oncology Group GWAS was also supported by grants from the National Institutes of Health, National Cancer Institute (the Chair’s grant U10 CA98543 and Human Specimen Banking grant U24 CA114766).
The German GWAS was supported by the German Ministry of Education and Research (Bundesministeriums für Bildung und Forschung) through the National Genome Research Network, the popgen biobank, the Federal Radiation Protection Agency (project no. 3609S30013), the Deutsche Krebshilfe, and the Madeleine Schickedanz Kinderkrebs-Stiftung; received infrastructure support through the Deutsche Forschungsgemeinschaft excellence cluster “Inflammation at Interfaces”; and was conducted within the frame of the International BFM Study Group.
The Center for International Blood and Marrow Transplant Research was supported by US Public Health Service grant/cooperative agreement 5U24-CA076518 from the National Cancer Institute, the National Heart, Lung, and Blood Institute, and the National Institute of Allergy and Infectious Diseases; grant/cooperative agreement 5U10HL069294 from the National Heart, Lung, and Blood Institute and the National Cancer Institute; contract HHSH250201200016C with the Health Resources and Services Administration/US Department of Health and Human Services; grants N00014-15-1-0848 and N00014-16-1-2020 from the Office of Naval Research; and grants from Alexion; Amgen, Inc*; anonymous donation to the Medical College of Wisconsin; Astellas Pharma US; AstraZeneca; Be the Match Foundation; Bluebird Bio, Inc*; Bristol-Myers Squibb Oncology*; Celgene Corporation*; Cellular Dynamics International, Inc; Chimerix, Inc*; Fred Hutchinson Cancer Research Center; Gamida Cell Ltd; Genentech, Inc; Genzyme Corporation; Gilead Sciences, Inc*; Health Research, Inc; Roswell Park Cancer Institute; HistoGenetics, Inc; Incyte Corporation; Janssen Scientific Affairs, LLC; Jazz Pharmaceuticals, Inc*; Jeff Gordon Children’s Foundation; The Leukemia & Lymphoma Society; Medac, GmbH; MedImmune; The Medical College of Wisconsin; Merck & Co, Inc*; Mesoblast; MesoScale Diagnostics, Inc; Miltenyi Biotec, Inc*; National Marrow Donor Program; Neovii Biotech NA, Inc; Novartis Pharmaceuticals Corporation; Onyx Pharmaceuticals; Optum Healthcare Solutions, Inc; Otsuka America Pharmaceutical, Inc; Otsuka Pharmaceutical Co, Ltd Japan; Patient-Centered Outcomes Research Institute; Perkin Elmer, Inc; Pfizer, Inc; Sanofi US*; Seattle Genetics*; Spectrum Pharmaceuticals, Inc*; St. Baldrick’s Foundation; Sunesis Pharmaceuticals, Inc*; Swedish Orphan Biovitrum, Inc; Takeda Oncology; Telomere Diagnostics, Inc; University of Minnesota; and Wellpoint, Inc* (*corporate members).
The views expressed in this article do not reflect the official policy or position of the National Institutes of Health, the Department of the Navy, the Department of Defense, the Health Resources and Services Administration, or any other agency of the US government.
Authorship
Contribution: A.I.C.-G. performed research, analyzed/interpreted data, and wrote the paper; T.H. conceived and designed the study, acquired data, and interpreted data analyses; L.M.P., K.O., A.S., E.H., C.A.H., D.O.S., L.P., X.S., A.W.B., and S.N.J.S. analyzed and interpreted data; E.E., D.E., M.S., J.C., and L.O. contributed data and data analyses and interpreted data; S.S., M.C.P., and D.J.W. acquired data and interpreted data analyses; P.L.M. interpreted data analyses; L.E.S.-C. conceived and designed the study, acquired, analyzed, and interpreted data, and wrote the paper; and all authors participated in the revising of the manuscript, contributed critically important intellectual content, and gave final approval of the version/revised version submitted.
Conflict-of-interest disclosure: T.H. owns stock in Novartis Pharmaceuticals Corporation. D.J.W. provided consulting services to, and served on advisory boards for, Kadmon and Alexion, and received research funding from Alexion. S.S. received compensation for travel, accommodations, and expenses from Astellas Pharma. M.C.P. received honoraria, as well as compensation for travel, accommodations, and expenses, from Baxalta and Atara Biotherapeutics. P.L.M. received honoraria from Celgene, Bristol-Myers Squibb, Janssen Pharmaceutical, Sanofi, and Karyopharm Therapeutics Inc; research funding from Celgene; and compensation for travel, accommodations, and expenses from Celgene and Sanofi. The remaining authors declare no competing financial interests.
Correspondence: Lara E. Sucheston-Campbell, College of Pharmacy, College of Veterinary Medicine, The Ohio State University, 496 W. 12th Ave, 604 Riffe, Columbus, OH 43210; e-mail: sucheston-campbell.1@osu.edu.
References
- 1.National Cancer Institute. Adult acute lymphoblastic leukemia treatment (PDQ)–health professional version. Updated 17 April 2015. http://www.cancer.gov/types/leukemia/hp/adult-all-treatment-pdq. Accessed 28 July 2015.
- 2.American Cancer Society. What are the key statistics about acute lymphocytic leukemia? http://www.cancer.org/cancer/acute-lymphocytic-leukemia/about/key-statistics.html. Accessed 5 April 2016.
- 3.National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer statistics. Available at: http://seer.cancer.gov. Accessed 5 April 2016.
- 4.Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet. 2013;381(9881):1943-1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasim K, Levallois P, Abdous B, Auger P, Johnson KC. Lifestyle factors and the risk of adult leukemia in Canada. Cancer Causes Control. 2005;16(5):489-500. [DOI] [PubMed] [Google Scholar]
- 6.United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). Sources and Effects of Ionizing Radiation. Vol. I. Vienna, Austria: UNSCEAR, 2000. [Google Scholar]
- 7.Muirhead CR, Bingham D, Haylock RG, et al. Follow up of mortality and incidence of cancer 1952-98 in men from the UK who participated in the UK’s atmospheric nuclear weapon tests and experimental programmes. Occup Environ Med. 2003;60(3):165-172. [DOI] [PMC free article] [PubMed]
- 8.Hayes RB, Yin SN, Dosemeci M, et al. Benzene and the dose-related incidence of hematologic neoplasms in China. J Natl Cancer Inst. 1997;89(14):1065-1071. [DOI] [PubMed] [Google Scholar]
- 9.Rothman N, Smith MT, Hayes RB, et al. Benzene poisoning, a risk factor for hematological malignancy, is associated with the NQO1 609C-->T mutation and rapid fractional excretion of chlorzoxazone. Cancer Res. 1997;57(14):2839-2842. [PubMed] [Google Scholar]
- 10.Thomas A, Mailankody S, Korde N, Kristinsson SY, Turesson I, Landgren O Second malignancies after multiple myeloma: from 1960s to 2010s. Blood. 2012;119(12):2731-2737. [DOI] [PMC free article] [PubMed]
- 11.Ghosh JK, Heck JE, Cockburn M, Su J, Jerrett M, Ritz B Prenatal exposure to traffic- related air pollution and risk of early childhood cancers. Am J Epidemiol. 2013;178(8):1233-1239. [DOI] [PMC free article] [PubMed]
- 12.Vinceti M, Rothman KJ, Crespi CM, et al. Leukemia risk in children exposed to benzene and PM10 from vehicular traffic: a case-control study in an Italian population. Eur J Epidemiol. 2012;27(10):781-790. [DOI] [PMC free article] [PubMed]
- 13.Miligi L, Benvenuti A, Mattioli S, et al. ; SETIL Working Group. Risk of childhood leukaemia and non-Hodgkin’s lymphoma after parental occupational exposure to solvents and other agents: the SETIL Study [published correction appears in Occup Environ Med 2014;71(7):524]. Occup Environ Med. 2013;70(9):648-655. [DOI] [PubMed] [Google Scholar]
- 14.Worrillow LJ, Travis LB, Smith AG, et al. An intron splice acceptor polymorphism in hMSH2 and risk of leukemia after treatment with chemotherapeutic alkylating agents. Clin Cancer Res. 2003;9(8):3012-3020. [PubMed]
- 15.Allan JM, Wild CP, Rollinson S, et al. Polymorphism in glutathione S-transferase P1 is associated with susceptibility to chemotherapy-induced leukemia. Proc Natl Acad Sci USA. 2001;98(20):11592-11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bielorai B, Fisher T, Waldman D, et al. Acute lymphoblastic leukemia in early childhood as the presenting sign of ataxia-telangiectasia variant. Pediatr Hematol Oncol. 2013;30(6):574-582. [DOI] [PubMed] [Google Scholar]
- 17.Xavier AC, Taub JW. Acute leukemia in children with Down syndrome. Haematologica. 2010;95(7):1043-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Westergaard T, Andersen PK, Pedersen JB, et al. Birth characteristics, sibling patterns, and acute leukemia risk in childhood: a population-based cohort study. J Natl Cancer Inst. 1997;89(13):939-947. [DOI] [PubMed] [Google Scholar]
- 19.Kharazmi E, da Silva Filho MI, Pukkala E, Sundquist K, Thomsen H, Hemminki K. Familial risks for childhood acute lymphocytic leukaemia in Sweden and Finland: far exceeding the effects of known germline variants. Br J Haematol. 2012;159(5):585-588. [DOI] [PubMed] [Google Scholar]
- 20.Treviño LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu H, Yang W, Perez-Andreu V, et al. Novel susceptibility variants at 10p12.31-12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105(10):733-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494-1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006-1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42(6):492-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prasad RB, Hosking FJ, Vijayakrishnan J, et al. Verification of the susceptibility loci on 7p12.2, 10q21.2, and 14q11.2 in precursor B-cell acute lymphoblastic leukemia of childhood. Blood. 2010;115(9):1765-1767. [DOI] [PubMed] [Google Scholar]
- 26.Enciso-Mora V, Hosking FJ, Sheridan E, et al. Common genetic variation contributes significantly to the risk of childhood B-cell precursor acute lymphoblastic leukemia. Leukemia. 2012;26(10):2212-2215. [DOI] [PubMed] [Google Scholar]
- 27.Han F, Tan Y, Cui W, Dong L, Li W. Novel insights into etiologies of leukemia: a HuGE review and meta-analysis of CYP1A1 polymorphisms and leukemia risk. Am J Epidemiol. 2013;178(4):493-507. [DOI] [PubMed] [Google Scholar]
- 28.Ross JA, Linabery AM, Blommer CN, et al. Genetic variants modify susceptibility to leukemia in infants: a Children’s Oncology Group report. Pediatr Blood Cancer. 2013;60(1):31-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perez-Andreu V, Roberts KG, Xu H, et al. A genome-wide association study of susceptibility to acute lymphoblastic leukemia in adolescents and young adults. Blood. 2015;125(4):680-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Migliorini G, Fiege B, Hosking FJ, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122(19):3298-3307. [DOI] [PubMed] [Google Scholar]
- 31.Bahari G, Hashemi M, Naderi M, Taheri M. IKZF1 gene polymorphisms increased the risk of childhood acute lymphoblastic leukemia in an Iranian population. Tumour Biol. 2016;37(7):9579-9586. [DOI] [PubMed] [Google Scholar]
- 32.Vijayakrishnan J, Kumar R, Henrion MY, et al. A genome-wide association study identifies risk loci for childhood acute lymphoblastic leukemia at 10q26.13 and 12q23.1. Leukemia. 2017;31(3):573-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Healy J, Richer C, Bourgey M, Kritikou EA, Sinnett D. Replication analysis confirms the association of ARID5B with childhood B-cell acute lymphoblastic leukemia. Haematologica. 2010;95(9):1608-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paulsson K, Forestier E, Lilljebjörn H, et al. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 2010;107(50):21719-21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hahn T, Sucheston-Campbell LE, Preus L, et al. Establishment of definitions and review process for consistent adjudication of cause-specific mortality after allogeneic unrelated-donor hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015;21(9):1679-1686. [DOI] [PMC free article] [PubMed]
- 36.Sucheston-Campbell LE, Preus L, Pasquini M, et al. Combined donor and recipient non-HLA genotypes show evidence of genome wide association with transplant related mortality (TRM) after HLA-matched unrelated donor blood and marrow transplantation (URD-BMT) (DISCOVeRY-BMT study) [abstract]. Blood. 2015;126(23). Abstract 61. [Google Scholar]
- 37.Sucheston-Campbell LE, Clay A, McCarthy PL, et al. Identification and utilization of donor and recipient genetic variants to predict survival after HCT: are we ready for primetime? Curr Hematol Malig Rep. 2015;10(1):45-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hahn T, Preus L, McCarthy PL, et al. Genome-wide association study of overall and progression-free survival after HLA-matched unrelated donor blood and marrow transplantation (DISCOVeRY-BMT study) [abstract]. Blood . 2015;126(23). Abstract 397. [Google Scholar]
- 39.R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015.
- 40.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559-575. [DOI] [PMC free article] [PubMed]
- 41.Howie BN, Donnelly P, Marchini J A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529. [DOI] [PMC free article] [PubMed]
- 42.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11(7):499-511. [DOI] [PubMed] [Google Scholar]
- 43.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39(7):906-913. [DOI] [PubMed] [Google Scholar]
- 44.1000 Genomes Project Consortium; Abecasis GR, Auton A, Brooks LD, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda). 2011;1(6):457-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Band G, Marchini J QCTOOL v2: A tool for quality control and analysis of gwas datasets. Oxford, United Kingdom: Wellcome Trust Centre for Human Genetics.
- 47.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26(17):2190-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539-1558. [DOI] [PubMed] [Google Scholar]
- 50.MacKinnon DP, Lockwood CM, Hoffman JM, West SG, Sheets V. A comparison of methods to test mediation and other intervening variable effects. Psychol Methods. 2002;7(1):83-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pierce BL, Tong L, Chen LS, et al. Mediation analysis demonstrates that trans-eQTLs are often explained by cis-mediation: a genome-wide analysis among 1,800 South Asians. PLoS Genet. 2014;10(12):e1004818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sobel M. Direct and indirect effects in linear structural equation models. Sociol Methods Res. 1987;16(1):155-176. [Google Scholar]
- 53.Baron RM, Kenny DA. The moderator-mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J Pers Soc Psychol. 1986;51(6):1173-1182. [DOI] [PubMed] [Google Scholar]
- 54.Hungate EA, Vora SR, Gamazon ER, et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun. 2016;7:10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ellinghaus E, Stanulla M, Richter G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2012;26(5):902-909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orsi L, Rudant J, Bonaventure A, et al. Genetic polymorphisms and childhood acute lymphoblastic leukemia: GWAS of the ESCALE study (SFCE). Leukemia. 2012;26(12):2561-2564. [DOI] [PubMed] [Google Scholar]
- 57.Rudant J, Orsi L, Menegaux F, et al. Childhood acute leukemia, early common infections, and allergy: The ESCALE Study. Am J Epidemiol. 2010;172(9):1015-1027. [DOI] [PubMed] [Google Scholar]
- 58.Borowitz MJ, Devidas M, Hunger SP, et al. ; Children’s Oncology Group. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group Study. Blood. 2008;111(12):5477-5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Evans TJ, Milne E, Anderson D, et al. Confirmation of childhood acute lymphoblastic leukemia variants, ARID5B and IKZF1, and interaction with parental environmental exposures. PLoS One. 2014;9(10):e110255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Metayer C, Milne E, Clavel J, et al. The Childhood Leukemia International Consortium. Cancer Epidemiol. 2013;37(3):336-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Winick N. Delayed intensification (DI) enhances event-free survival (EFS) of children with B-precursor acute lymphoblastic leukemia (ALL) who received intensification therapy with six courses of intravenous methotrexate (MTX): POG 9904/9905: A Children’s Oncology Group Study (COG) [abstract]. Blood. 2007;110(11). Abstract 583. [Google Scholar]
- 62.Krawczak M, Nikolaus S, von Eberstein H, Croucher PJ, El Mokhtari NE, Schreiber S. PopGen: population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Community Genet. 2006;9(1):55-61. [DOI] [PubMed] [Google Scholar]
- 63.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang J, Benyamin B, McEvoy BP, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee SH, Wray NR, Goddard ME, Visscher PM. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88(3):294-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics. 2012;28(19):2540-2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Euesden J, Lewis CM, O’Reilly PF. PRSice: polygenic risk score software. Bioinformatics. 2015;31(9):1466-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wei G, Abraham BJ, Yagi R, et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity. 2011;35(2):299-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yagi R, Zhu J, Paul WE. An updated view on transcription factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. Int Immunol. 2011;23(7):415-420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Enciso-Mora V, Broderick P, Ma Y, et al. A genome-wide association study of Hodgkin’s lymphoma identifies new susceptibility loci at 2p16.1 (REL), 8q24.21 and 10p14 (GATA3). Nat Genet. 2010;42(12):1126-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshida T, Landhuis E, Dose M, et al. Transcriptional regulation of the Ikzf1 locus. Blood. 2013;122(18):3149-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mullighan CG. Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest. 2012;122(10):3407-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Collins-Underwood JR, Mullighan CG. Genomic profiling of high-risk acute lymphoblastic leukemia. Leukemia. 2010;24(10):1676-1685. [DOI] [PubMed] [Google Scholar]
- 74.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464(7289):773-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeller T, Wild P, Szymczak S, et al. Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5(5):e10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boyle AP, Hong EL, Hariharan M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22(9):1790-1797. [DOI] [PMC free article] [PubMed]
- 79.Irving JA, Enshaei A, Parker CA, et al. Integration of genetic and clinical risk factors improves prognostication in relapsed childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2016;128(7):911-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.