

Key Points
MLF1 suppresses C/EBPα degradation and AML development induced by the Trib1-COP1 complex.
MLF1 binds to COP1 to prevent the interaction between Trib1 and COP1, thereby inhibiting COP1’s ubiquitin ligase activity for C/EBPα.
Abstract
C/EBPα is a key transcription factor regulating myeloid differentiation and leukemogenesis. The Trib1-COP1 complex is an E3 ubiquitin ligase that targets C/EBPα for degradation, and its overexpression specifically induces acute myeloid leukemia (AML). Here we show that myeloid leukemia factor 1 (MLF1) stabilizes C/EBPα protein levels by inhibiting the ligase activity of the Trib1-COP1 complex. MLF1 directly interacts with COP1 in the nucleus and interferes with the formation of the Trib1-COP1 complex, thereby blocking its ability to polyubiquitinate C/EBPα for degradation. MLF1 overexpression suppressed the Trib1-induced growth advantage in a murine bone marrow (BM) culture and Trib1-induced AML development in BM-transplanted mouse models. MLF1 was expressed in hematopoietic stem cells and myeloid progenitors (common myeloid progenitors and granulocyte-macrophage progenitors) in normal hematopoiesis, which is consistent with the distribution of C/EBPα. An MLF1 deficiency conferred a more immature phenotype on Trib1-induced AML development. A higher expression ratio of Trib1 to MLF1 was a key determinant for AML development in mouse models, which was also confirmed in human patient samples with acute leukemia. These results indicate that MLF1 is a positive regulator that is critical for C/EBPα stability in the early phases of hematopoiesis and leukemogenesis.
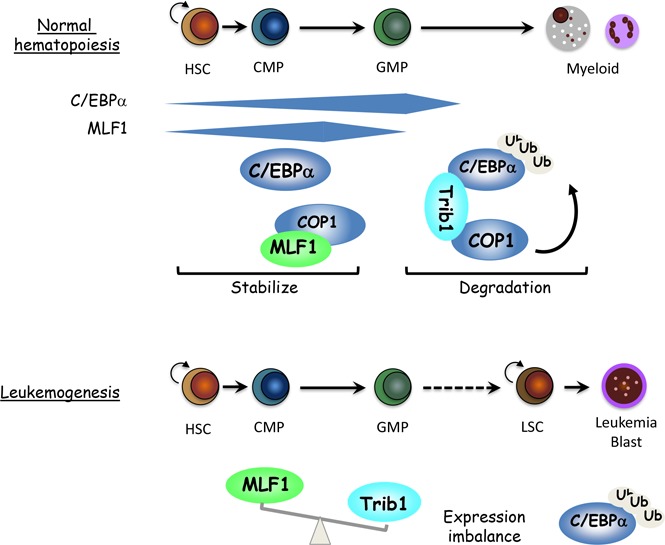
Visual Abstract
Introduction
The sequential expression of lineage-specific transcription factors strictly controls tissue-specific differentiation and proliferation. The deregulation of these transcription factors has been directly linked to the blockade of differentiation and consequential accumulation of undifferentiated cells, leading to tumorigenesis. CCAAT/enhancer-binding protein α (C/EBPα) is a critical transcription factor regulating myeloid differentiation in normal hematopoiesis and is frequently deregulated in acute myeloid leukemia (AML).1 C/EBPα is mutated in ∼10% to 15% of AML with a normal karyotype and is functionally suppressed in various manners in other AMLs, such as transcriptional repression by the leukemic fusion protein AML1-ETO,2 posttranslational modifications by the oncogenic kinase Flt3-ITD,3 and protein degradation by the tribbles homologs, Trib1 and Trib2.4,5
A link between tribbles and the C/EBP family was initially discovered in flies. Tribbles promotes the proteasome-dependent degradation of slbo, the Drosophila homolog of the C/EBP family, which is critical for cell migration during oogenesis.6 Mammals have 3 homologs of tribbles, Trib1, Trib2, and Trib3, which are characterized by a pseudokinase domain and C-terminal COP1-binding motif.7,8 Trib1 and Trib2, but not Trib3, downregulate C/EBPα and induce AML in mouse models.4,9,10 Constitutive photomorphogenic 1 (COP1) is an E3 ubiquitin ligase that is conserved from plants to mammals. In plants, COP1 targets transcription factors (HY5, HYH, HFR1, and LAF1) in order to promote morphogenesis in response to light signals.11 In mammals, putative substrates of COP1 include transcription factors (c-Jun, ETV1, p53, C/EBPα, TORC2, and FOXO1) and enzymes (acetyl-CoA carboxylase) that are involved in tumorigenesis and metabolism.7,12-18 Because COP1 functions in many biological responses in mammalian cells, the tribbles family appears to be an adaptor protein that recruits COP1 to a specific substrate for proteasome-mediated degradation. The Trib1-COP1 ligase complex specifically targets C/EBPα for degradation in hematopoiesis, and its overexpression causes AML in mouse models.15 The COP1-binding motif of Trib1 and ligase activity of COP1 are both required for C/EBPα degradation and AML development,9,15 which indicates that the formation of the Trib1-COP1 complex is critical for these activities.
Myeloid leukemia factor 1 (MLF1) was originally identified as a carboxyl-terminal component of the leukemic fusion protein NPM-MLF1 generated by the t(3;5) chromosomal translocation in patients with AML.19 The role of NPM in leukemogenesis has been established by the finding of cytoplasmic mutants of NPM1 specific to AML.20,21 In contrast, the contribution of MLF1 to leukemogenesis remains unclear; however, previous findings have suggested the involvement of MLF1 in normal hematopoiesis and a tumor suppressor pathway. The expression of human MLF1 is preferentially detected in CD34+ primitive cells and declines during the lineage commitment in normal hematopoiesis.22 The ectopic expression of murine MLF1 disturbs the differentiation program toward the erythroid lineage, thereby promoting a lineage switch to myelomonocytes in an in vitro culture system.23 In clinical studies, MLF1 is overexpressed in >25% of myelodysplastic syndrome–associated AML and the malignant transformation phase of myelodysplastic syndrome.22 These findings imply that MLF1 normally regulates the development of primitive hematopoietic cells, and its deregulation leads to hematopoietic dysplasia and leukemogenesis. We previously reported that MLF1 induces cellular growth arrest in a manner that is dependent on the tumor suppressor p53.24 NPM and MLF1 are both shuttling proteins that are independently involved in the stabilization of p53 through the Arf-Mdm2-p53 pathway and the COP9 signalosome complex (CSN)–COP1-p53 pathway.25-27 However, previous findings including those from genetically engineered mouse models suggest that COP1 behaves differently from Mdm2, an E3 ligase specific to p53.28 The absence of a COP1-binding motif in p53 implies that p53 may not be a direct substrate of COP1, rather COP1 simply functions upstream of the p53 pathway. The leukemic fusion of MLF1 with NPM relocalizes the majority of MLF1 from the cytoplasm to the nucleolus and abolishes growth-suppressing activity,19,27 indicating that a shuttling imbalance in MLF1 results in the instability of its downstream factors and increases susceptibility to malignant transformation. In the present study, we investigated a critical issue regarding why the deregulation of MLF1 specifically causes AML, and also the possibility that MLF1 stabilizes other substrates of COP1 in myeloid leukemogenesis. We herein show for the first time that MLF1 inhibits the formation of the Trib1-COP1 complex by directly binding to COP1 in the nucleus, thereby stabilizing the C/EBPα protein.
Methods
Cell culture, transfection, and retroviral production
The 293T human embryonic kidney cells and NIH 3T3 (Arf-null, p53 wild-type [WT]) mouse fibroblasts (provided by C. J. Sherr and M. F. Roussel) were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL of penicillin, and 100 μg/mL of streptomycin (GIBCO/BRL) and transfected with expression vectors via the calcium phosphate-DNA precipitation method.29 Regarding viral production, complementary DNAs encoding Trib1, COP1, and MLF1 in the retroviral vectors, pMSCV-puro-GFP (for the expression of GFP-fused proteins), pMSCV-IRES-GFP (a gift from Owen Witte, for the expression of GFP-coexpressed proteins), and pMSCV-neo (for the expression of untagged proteins), were cotransfected into 293T cells together with a plasmid encoding an ecotropic helper virus containing a defective virion-packaging sequence. Culture supernatants containing retroviruses were harvested 48 to ∼72 hours after transfection and used for infection.
Targeted disruption of the mouse Mlf1 gene
The Mlf1 targeting vector was constructed by subcloning a 5.5-kb genomic DNA fragment containing the sequence upstream of the initial methionine and a 2.2-kb genomic DNA fragment downstream of exon 1 into the pKSloxPNT vector at the ClaI and BamHI sites, respectively. The targeting vector was linearized with SalI and electroporated into mouse RF8 ES cells. ES clones selected in 200 μg/mL G418 and 0.2 μM FIAU were subjected to a Southern blot analysis by using a probe external to the 3′ end of the targeting construct. Mlf1+/− ES cells were microinjected into blastocyst stage C57BL/6 mouse embryos. Chimeric males were crossed with C57BL/6 females (CLEA Japan Inc.), and offspring were genotyped by genomic polymerase chain reaction (PCR) using the following Mlf1-specific primers: a (5′-AAG AAG AGG CTA TAA GGT GTA ATA GGG ATG-3′), b (5′-CAC TAA TAT GGC AGT TAT GAT CAC CAG TTA-3′), and neomycin-resistant gene-specific primer c (5′-CCT GCG TGC AAT CCA TCT TGT TCA CA-3′).
BM transplantation and analyses
Bone marrow (BM) cells were aseptically isolated from the femurs and tibias of 8-week-old C57BL/6 mice (CLEA Japan Inc.) that had been IV injected with 5-fluorouracil (5-FU) (150 mg/kg of body weight; Kyowa Hakko Kogyo) 5 days before, incubated in BM medium (Dulbecco’s modified Eagle medium, 15% heat-inactivated fetal bovine serum, 2 mM glutamine, 100 units/mL of penicillin, 100 μg/mL of streptomycin, 5% WEHI-3B conditioned medium, 6 ng/mL mouse interleukin-3 [mIL-3], 10 ng/mL human IL-6 (hIL-6), and 50 ng/mL mouse stem cell factor [mSCF]) (recombinant cytokines were from R&D Systems Inc.), and infected with a retroviral supernatant according to the spin infection procedure described previously in the presence of polybrene (6 μg/mL).27
Infected, unsorted BM cells (0.5–1 × 106 cells) were injected into the veins of 8- to 10-week-old C57BL/6 mice that had been sublethally irradiated (10 Gy; M-150WE; SOFTEX) several hours before the injection. All mice were maintained in the Nara Institute of Science and Technology Animal Facility in accordance with the Nara Institute of Science and Technology guidelines. Blood samples were routinely analyzed with an automated cell counter (F-820 analyzer; Sysmex) and also by inspecting blood smears after staining with May-Grünwald Giemsa solution (Merck).
In the liquid culture, BM cells were cultured in BM medium for 5 days after retroviral infection, and GFP-positive populations of BM cells were isolated by cell sorting with FACSAria (BD Biosciences) and then split and maintained in BM medium. For cell line experiments, infected GFP-positive BM cells were cultured and maintained in medium containing 10% WEHI-3B conditioned medium as a source of IL-3. In the colony formation assay, sorted GFP-positive BM cells were plated at 2 × 103 cells per 35-mm dish onto methylcellulose-based medium containing mIL-3 (10 ng/mL), hIL-6 (10 ng/mL), mSCF (50 ng/mL), and human erythropoietin (3 U/mL) (MethoCult GF M3434; StemCell Technologies Inc.), and colonies were enumerated after 9 days. Cells for second and third plating were collected from methylcellulose-based medium, counted, and replated at 1 × 104 cells per 35-mm dish into fresh methylcellulose-based medium containing mIL-3 (10 ng/mL), hIL-6 (10 ng/mL), and mSCF (50 ng/mL) (MethoCult GF M3534).
Flow cytometric analysis
PharM Lyse (BD Bioscience) was used to remove red blood cells. BM and spleen cells were analyzed and sorted with FACSAria (BD Biosciences) after staining with antibodies to CD3 (145-2C11), B220 (RA3-6B2), TER-119 (TER-119), Mac-1 (CD11b, M1/70), Gr-1 (RB6-8C5), c-Kit (ACK2), Sca-1 (D7), and CD34 (RAM34) (all from eBioscience), as well as anti-FcγRII/III (2.4G2; BD Bioscience), which were conjugated with phycoerythrin (PE), PE-Cy5, allophycocyanin, Pacific Blue, or biotin, and together with streptavidin-PE-Cy7.
Protein analyses
Cell lysis, cell fractionation, immunoprecipitation, gel electrophoresis, and immunoblotting were performed as described.24 A mouse monoclonal antibody to MLF1 (3E9) and rabbit polyclonal antibody to COP1 were generated using bacterially produced polypeptides in our laboratory.24 Mouse monoclonal antibodies to hemagglutinin (HA) peptide epitopes (12CA5) and γ-tubulin (GTU-88) were obtained from Roche and Sigma, respectively. A rabbit polyclonal antibody to an HA epitope (HA.11) and a mouse monoclonal antibody to a FLAG epitope (M5) were purchased from BAbCO and Eastman Kodak Company, respectively. A mouse monoclonal antibody to polyubiquitin chains (FK2) was obtained from Nippon Bio-Test Laboratories.
In the in vitro binding assay, complementary DNAs corresponding to the region of MLF1 were amplified by PCR, confirmed by sequencing, and inserted into the vector pGEX (GE Healthcare), and a series of MLF1 deletion mutant proteins were expressed in bacteria and purified as described.24 A crude cell extract containing the HA-tagged Trib1 protein was prepared from 293T cells transfected with the HA-Trib1 expression vector. A binding assay was performed as described.24 Glutathione S-transferase (GST) proteins were quantitated by staining with Coomassie brilliant blue after separation by sodium dodecyl sulfate (SDS) PAGE.
In the in vivo ubiquitination assay, 293T cells were transfected with a combination of vectors encoding FLAG-C/EBPα, GFP-COP1, HA-Trib1, HA-Ub, and HA- or GFP-tagged MLF1 and harvested 4 days after transfection. Cells were lysed in SDS-sample buffer (40 mM tris(hydroxymethyl)aminomethane–HCl, pH 6.8, 0.1 M dithiothreitol, 1% SDS, 10% glycerol, and 0.05% Bromophenol Blue). After boiling, cell lysates were diluted with EBC buffer (50 mM tris(hydroxymethyl)aminomethane–HCl, pH 7.5, 120 mM NaCl, 0.5% NP-40, and 1 mM EDTA containing 5 mg/mL of aprotinin, 1 mM phenylmethylsulfonyl fluoride, 0.1 mM NaF, 0.1 mM NaVO4, and 1 mM dithiothreitol) and immunoprecipitated with an antibody to a FLAG epitope followed by immunoblotting with an antibody to polyubiquitin chains.
Quantitative RT-PCR
Total RNA was isolated using the ISOGEN reagent (Nippon Gene) and reverse transcribed using RNase-free Superscript reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Semiquantitative reverse transcription PCR (semi-qRT-PCR) was performed within a linear range as described previously22 and data were normalized to the expression levels of β-actin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for each sample. We confirmed that reactions were quantitatively performed within a linear range by conducting several control experiments. The following oligonucleotide primers specific to mouse Trib1, MLF1, β-actin, and human GAPDH were used: mouse Trib1, 5′-GGA CTT TGG AGA CAT GCA CTC CT-3′ (sense) and 5′-GAC CAA AAG CGT ATA GAG CAT CAC C-3′ (antisense); mouse MLF1, 5′-CCC ATA ATC GTC GAG AAC GTG ATG AT-3′ (sense) and 5′-CTT TTA CTC CTC CTG GAG CCC TTC-3′ (antisense); mouse β-actin, 5′-CTT CTA CAATGA GCT GCG TGT-3′ (sense) and 5′-CAA CGT CAC ACT TCA TGA TGG-3′ (antisense); human GAPDH, 5′- CCA TCA CCA TCT TCC AGG AG-3′ (sense) and 5′- CCT GCT TCA CCA CCT TCT TG-3′ (antisense).
qRT-PCR was performed on a LightCycler 96 Real-time PCR system (Roche) by using FastStart Universal Probe Master (Roche) with Universal ProbeLibrary (Roche). All data are presented as relative expression levels normalized to β-actin expression. The following oligonucleotide primers and Universal ProbeLibrary probes specific to mouse Trib1, MLF1, β-actin, and human MLF1 were used: mouse Trib1, 5′-TCAGGAAGTTCGTCTTCTCCA-3′ (sense) and 5′-GATCTCGGGGCTCACGTA-3′ (antisense) and #46 probe; mouse MLF1, 5′-TGAGAAGTGTTGGCCATGAG-3′ (sense) and 5′-TCTCATTGCTTTTCTGGTTTCTT-3′ (antisense) and #1 probe; mouse β-actin 5′-GGATGCAGAAGGAGATTACTGC-3′ (sense) and 5′-CCACCGATCCACACAGAGTA-3′ (antisense) and #63 probe; human MLF1 5′-TGAGAAGTGTTGGCCATGAG-3′ (sense) and 5′-GGCTGGACTTTGTTGAGGTT-3′ (antisense) and #36 probe.
Bioinformatics analysis of human gene expression
All human gene expression profiling data were obtained from the National Center for Biotechnology Information Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/). For the relative expression analysis of TRIB1 and MLF1 in human leukemic progression, the GEO data sets, accession numbers GSE33075 (normal BMs), GSE1159 (AML), and GSE5900 (acute lymphoblastic leukemia [ALL]), were retrieved and analyzed statistically using R version 3.2.2 (R Foundation for Statistical Computing, Vienna, Austria). Patients with AML were classified into 2 groups based on negative and positive mutational status and cytogenetic abnormalities and analyzed in a similar fashion.
Results
MLF1 specifically interacts with COP1 in the nucleus
We previously showed that MLF1 suppressed the activity of COP1 toward the tumor suppressor p53 through the third component of the COP9 signalosome complex (CSN3).24 Because the CSN complex acts as a modifier in signaling pathways,30 MLF1 may be directly involved in the regulation of COP1. We investigated the physical interaction between endogenous MLF1 and COP1 proteins in mouse fibroblast NIH3T3 cells and detected a large amount of the COP1 protein in the anti-MLF1 immunoprecipitate (Figure 1A, left panel). In a reciprocal experiment, the MLF1 protein was not detected in the anti-COP1 immunoprecipitate because the anti-COP1 antibody raised in our laboratory recognized the same region of the COP1 amino acid sequence that MLF1 requires for the physical interaction with COP1 (data not shown). To avoid the problem of the anti-COP1 antibody, we used tagged proteins for the complex formation between MLF1 and COP1. In 293T cells cotransfected with FLAG-tagged MLF1 and HA-tagged COP1 proteins, we detected a large amount of the MLF1 protein in the anti-HA immunocoprecipitate (Figure 1A, right panel). MLF1 is a shuttling protein and exerts growth-suppressing activity in the nucleus.27 When we separated the nuclear and cytoplasmic fractions from cells expressing endogenous MLF1 and COP1, we detected the interaction of MLF1 with COP1 in the nucleus, but not in the cytoplasm (Figure 1B). A pull-down assay with a series of GST-tagged MLF1 deletion mutants produced in Escherichia coli and ectopically expressed COP1 in 293T cells revealed that COP1 bound to an MLF1 mutant lacking 39 C-terminal amino acids (MLF1/1-229), but not to that lacking 102 amino acids (MLF1/1-166) (Figure 1C-D), suggesting that residues 167 to 229 are required for the interaction with COP1. Although Tribbles (Trib1, Trib2, and Trib3) have a consensus COP1-binding motif at the carboxyl terminus, we were unable to detect it in the residues of MLF1.
Figure 1.

Myeloid leukemia-associated MLF1 specifically interacts with COP1 in the nucleus. (A) Specific interaction between endogenous MLF1 and COP1 proteins (left panels). Endogenous MLF1 and COP1 proteins were immuno-coprecipitated from the NIH3T3 cell lysate shown at the top and analyzed by immunoblotting with antibodies to COP1 (upper panel) and MLF1 (lower panel). Specific interaction between ectopically expressed MLF1 and COP1 proteins (right panels). The 293T cells were cotransfected with FLAG-tagged MLF1 and HA-tagged COP1. Ectopic MLF1 and COP1 proteins were immuno-coprecipitated from cell lysates shown at the top. The immune complex was analyzed by immunoblotting with antibodies to an HA epitope (upper panel) and MLF1 (lower panel). NRS, normal rabbit serum. (B) Nuclear and cytoplasmic fractions from NIH3T3 cells were separated by sequential elutions with digitonin-containing buffer and analyzed by immunoblotting using antibodies to COP1 and MLF1. (C) Schematic representation of MLF1 deletion mutants. The results of COP1 binding are summarized on the right. (D) The region of MLF1 involved in binding to COP1 in vitro. GST-control, GST-MLF1 WT, and GST-MLF1 mutant fusion proteins shown at the top of the panel were incubated with 293T cell lysates containing HA-tagged COP1 proteins. Bound proteins were detected by immunoblotting with an antibody to COP1. The amounts of GST proteins absorbed on the beads were evaluated by Coomassie brilliant blue (CBB) staining.
MLF1 inhibits the polyubiquitination of C/EBPα induced by the Trib1-COP1 complex
We examined whether MLF1 affects the level of the C/EBPα protein and its polyubiquitination induced by the Trib1-COP1 complex. Consistent with previous studies, the coexpression of Trib1 and COP1 induced a decrease in ectopic C/EBPα protein levels in 293T cells, whereas the additional coexpression of MLF1 induced a marked increase in protein levels (Figure 2A). This result indicates that MLF1 stabilizes the C/EBPα protein by counteracting Trib1-COP1 ligase activity. Because we previously reported optimum conditions for the ubiquitination assay of C/EBPα in order to evaluate COP1 ligase activity with a minimum amount of Trib1,15 we investigated whether MLF1 inhibits the polyubiquitination of C/EBPα induced by the Trib1-COP1 complex. When we coexpressed either HA-tagged or GFP-tagged MLF1 with Trib1 and COP1, the polyubiquitination of C/EBPα was markedly suppressed with a subsequent increase in C/EBPα (Figure 2B). We hypothesized that MLF1 prevents the direct binding of Trib1 to COP1. In order to test this, we transiently expressed HA-tagged Trib1 with COP1 and MLF1, respectively, and together with COP1 and MLF1 in 293T cells. COP1 efficiently coprecipitated with HA-tagged Trib1, whereas complex formation between COP1 and Trib1 was markedly weaker when MLF1 was coexpressed (Figure 2C). Taken together, these results suggest that MLF1 interferes with complex formation, triggering COP1 ligase activity toward C/EBPα.
Figure 2.

MLF1 inhibits the polyubiquitination of C/EBPα by interfering with Trib1-COP1 complex formation. (A) 293T cells were transfected with a combination of vectors shown at the top. Cell lysates were analyzed by immunoblotting with antibodies to a FLAG epitope, MLF1, COP1, an HA epitope, and γ-tubulin. (B) 293T cells were transfected with a combination of vectors shown at the top. Cell lysates were analyzed by immunoprecipitation with an antibody to a FLAG epitope followed by immunoblotting with an antibody to polyubiquitin chains (poly-Ub), and by immunoblotting with antibodies to COP1, MLF1, and a FLAG epitope. Total RNA was analyzed by semi-qRT-PCR using a pair of primers specific to Trib1 and human GAPDH. (C) 293T cells were transfected with a combination of vectors shown at the top. Cell lysates were analyzed by immunoprecipitation with an antibody to an HA epitope followed by immunoblotting with antibodies to an HA epitope, MLF1, and COP1.
MLF1 suppresses the Trib1-induced growth advantage and is expressed in normal hematopoiesis
MLF1 induces cell cycle arrest in murine primary fibroblasts including murine embryonic fibroblasts and NIH3T3 cells.24,27 If MLF1 is a direct inhibitor of COP1 activity, it is expected to suppress Trib1-promoted cellular proliferation. Therefore, we investigated the effects of MLF1 on the Trib1-induced growth advantage in mouse primary hematopoietic progenitor cells in culture. We transduced primary BM cells with either MLF1-IRES-GFP or control MigR1 retroviruses in the presence or absence of pMSCV-neo-Trib1 and sorted GFP-positive cells for further experiments. Growth curves showed that the cells expressing MLF1 hardly grew and ceased to proliferate in a liquid culture (BM medium containing IL-3, IL-6, and SCF), whereas those expressing Trib1 grew rapidly. The cells coexpressing Trib1 with MLF1 grew markedly slower than those expressing Trib1 alone (P < .0005; Figure 3A). We established IL-3–dependent cell lines expressing Trib1 alone and Trib1/MLF1 by transferring cells from BM medium to IL-3–containing medium (medium supplemented with WEHI-conditioned medium). The cells expressing Trib1 alone grew faster than those coexpressing Trib1/MLF1 in an IL-3–dependent culture (P < .01; Figure 3B) under the condition that Trib1 was equally overexpressed in both cell lines (Figure 3B, right panel). The growth-suppressing activity of MLF1 was also observed in the replating colony formation assay. An equal number of GFP-positive cells from each retrovirally transduced cell was sorted and subjected to initial plating. We detected a smaller number of colonies in MLF1 and MLF1/Trib1-transduced cells than in MigR1 control and Trib1-transduced cells. Secondary replating showed few colonies in MLF1-transduced cells and a smaller number of colonies in MLF1/Trib1-cotransduced cells than the markedly higher number in Trib1-transduced cells (*P < .05; Figure 3C). Consistent with the results obtained for primary fibroblasts, MLF1 induced growth arrest in primary hematopoietic progenitors and suppressed Trib1-promoted cellular proliferation.
Figure 3.

MLF1 suppresses the Trib1-induced growth advantage of hematopoietic cells and is expressed in normal hematopoiesis. (A-C) Primary BM cells were infected with control GFP and MLF1-IRES-GFP retroviruses together with or without pMSCV-neo-Trib1. GFP-positive cells in infected BM cells were isolated by cell sorting and maintained in BM medium. Cell numbers were enumerated for growth curves (A). GFP-positive cells in panel A were transferred to and maintained in IL-3–containing medium. Cell numbers were enumerated for growth curves (B). GFP-positive cells in infected BM cells were isolated by cell sorting and plated in a methylcellulose-based medium. Colony numbers were enumerated after 10 days and replated in fresh medium for the replating colony formation assay (C). Data are averages of 3 independent experiments performed in duplicate and shown as the mean value (± standard deviation). Significance was calculated by Student t test (*P < .05; **P < .01). (D) A qRT-PCR analysis of Mlf1 expression in purified hematopoietic stem and progenitor cells. ΔCt values for each sample were standardized by the ΔCt values of β-actin. Data are the average of 4 independent experiments shown as the mean value (± standard deviation). Significance was calculated by Student t test (*P < .05; **P < .01; ***P < .001).
In normal hematopoiesis, human MLF1 is preferentially expressed in CD34+ stem cells, but only slightly in lineage maker–positive cells.22 In order to examine whether MLF1 is involved in the regulation of C/EBPα in mouse models, we investigated the precise distribution of endogenous MLF1 at various stages of hematopoietic differentiation using real-time RT-PCR methods. MLF1 was strongly expressed in LSK (Lin−Sca-1+c-Kit+) stem cells, common myeloid progenitors (CMPs), and granulocyte-macrophage progenitors (GMPs), but at low levels in megakaryocyte-erythroid progenitors (MEPs) and Lin+ mature cells, suggesting that it functions in stem cells and myeloid-committed progenitor cells (Figure 3D).
Ectopic MLF1 expression blocked Trib1-driven leukemogenesis in mouse models
We investigated whether ectopic MLF1 expression affects Trib1-induced AML development in mouse models. We transduced primary BM cells with retroviruses expressing MLF1 (MLF1-IRES-GFP) or control (MigR1) together with Trib1 (pMSCV-neo-Trib1), and IV injected these cells into sublethally irradiated recipient WT mice. Trib1-transduced mice developed AML with complete penetrance 150 to 230 days after BM transplantation. In contrast, nearly half of the mice cotransduced with MLF1 and Trib1 (MLF1/Trib1-transduced mice) developed AML, whereas the other half did not 250 days after BM transplantation when all Trib1 alone–transduced mice died of AML (Figure 4A). In MLF1/Trib1 mice that developed AML, GFP-positive Mac-1+Gr-1lo blast cells replaced normal BM cells and accumulated in the spleen, which was a similar phenotype to that of Trib1 alone mice (Figure 4B-C). However, in MLF1/Trib1 mice that did not develop AML, GFP-positive cells showed normal distribution patterns in BM and the spleen. In order to identify the key factors preventing the onset of AML, we measured the relative expression levels of ectopically transduced Trib1 and MLF1 in the cells of leukemic and nonleukemic MLF1/Trib1 mice. Trib1 was strongly expressed in all MLF1/Trib1-induced AML at similar levels to those in Trib1-induced AML, but at lower levels in a few nonleukemic mice (Figure 4D). In contrast, MLF1 was strongly expressed in all nonleukemic mice but was more weakly expressed in a few cases of MLF1/Trib1-induced AML. Statistical analyses with the ratio of MLF1 transcripts to Trib1 transcripts suggested that the relative expression balance of MLF1 and Trib1 influences whether an MLF1/Trib1-transduced mouse develops AML. All the mice that did not develop AML showed a significantly lower ratio of Trib1 to MLF1 than those that developed AML (Figure 4E).
Figure 4.

Overexpression of MLF1 partly overcomes Trib1-induced AML. (A) Myeloid leukemia–free survival curves of mice transplanted with MLF1/Trib1-cotransduced BM cells compared with Trib1 alone. Results are derived from 3 independent transfer experiments. The P value between Trib1 and MLF1/Trib1 mice was calculated with the log-rank test. P < .01. (B) May-Grünwald Giemsa–stained peripheral blood (PB) smears and cytospins of BM cells from leukemic Trib1 (left panel), leukemic MLF1/Trib1 (middle panel), and nonleukemic MLF1/Trib1 (right panel) mice. Original magnification ×400. (C) A fluorescence-activated cell sorter (FACS) analysis of BM and spleen (SP) cells for immature (Mac-1+Gr-1lo) and differentiated (Mac-1+Gr-1hi) granulocytes. The population of GFP-positive cells in BM and SP is shown in the upper panels. (D-E) A qRT-PCR analysis of Trib1 and MLF1 expression in BM cells from Trib1 (n = 6), leukemic MLF1/Trib1 (n = 5), and nonleukemic MLF1/Trib1 (n = 5) mice. ΔCt values for each sample were standardized by the ΔCt values of β-actin followed by normalization with the percentage of GFP-positive cells. The level of expression in normal BM was set to 1.0 (D). The Trib1/MLF1 ratio of each sample in panel D. P values between leukemic MLF1/Trib1 and nonleukemic MLF1/Trib1 mice were calculated with Student t test. P < .005. (E). Data are presented as scatter diagrams with mean values (horizontal bar). (F-G) Microarray data analysis of the TRIB1 and MLF1 expression in human leukemia. Gene expression data of normal BMs (n = 9) from healthy donors and AML (n = 285) and ALL (n = 284) patient samples were obtained from the GEO data sets (F). Patients with AML were classified into positive groups with C/EBPα mutation (n = 17), EVI1 mutation (n = 20), RAS mutation (n = 34), FLT3 mutation (n = 106), and t(15;17) chromosomal translocation (n = 18). Negative vs positive groups with each gene alteration were analyzed (G). Box plots showed stratification of the TRIB1/MLF1 ratio from each sample. Significance was calculated by the Mann-Whitney U test (*P < .05; **P < .01; ***P < .001; n.s., not significant).
Our results demonstrate that imbalance of MLF1 and Trib1 expression is essential for the AML development in mice. In order to investigate whether this expression pattern reflects a general trend in human leukemia, we analyzed MLF1 and TRIB1 levels in a GEO data sets of 285 cases of AML compared with those of 9 cases of healthy donor and 284 cases of ALL. This analysis showed a significantly higher ratio of TRIB1 to MLF1 (TRIB1/MLF1 ratio) in AML compared with normal BM and ALL (***P < .001; Figure 4F). We also examined whether the TRIB1/MLF1 ratio is associated with specific mutational status and cytogenetic abnormalities appeared in AML. Interestingly, the TRIB1/MLF1 ratio is significantly lower in AML with C/EBPα and EVI1 mutations and higher in AML with RAS mutation compared with negative groups of these mutations in AML (*P < .05, ***P < .001, and **P < .001, respectively; Figure 4G). The cells carrying C/EBPα mutation may not require higher TRIB1/MLF1 ratio that promotes degradation of C/EBPα for AML development. These results with clinical samples confirm our findings in the mouse model.
An MLF1 deficiency caused more immature AML
We generated MLF1 knockout mice (Figure 5A-B), which were fertile, grew normally, and did not develop AML (Figure 5C). As a minor anomaly, they showed a slight increase in immature erythrocytes 2 years after birth (data not shown). We examined the effects of an MLF1 deficiency on Trib1-induced AML in mouse models. We infected WT or Mlf1−/− BM cells with retroviruses expressing Trib1 (Trib1-IRES-GFP) or control (MigR1) and transplanted them into sublethally irradiated WT mice. Although mice transplanted with Mlf1−/− cells expressing control GFP (Mlf1−/− MigR1 mice) did not develop AML, all the mice transplanted with either WT cells or Mlf1−/− cells expressing Trib1 (WT Trib1 mice or Mlf1−/− Trib1 mice) developed AML within 240 days. The MLF1 deficiency did not appear to affect the onset of Trib1-induced AML (Figure 5C-D). However, a flow cytometric analysis revealed that the Mlf1−/− background initiated Trib1-induced leukemogenesis at more immature progenitors of differentiation. As shown in Figure 4C and our previous study, most GFP-positive leukemic cells in WT Trib1 mice were Mac-1+Gr-1lo immature granulocytic cells (Figure 5E), and the granulocyte-macrophage–committed immature myelocyte population (Lin*negSca-1−c-Kit+Mac-1+, Lin*; lineage marker without Mac-1 and Gr-1) markedly expanded with a decrease in lineage-negative populations including LSK cells and multipotent progenitors (CMP, GMP, and MEP) (Figure 5F-G).15 In contrast, the Mac-1+Gr-1lo cell population in Mlf1−/− Trib1 mice decreased with an increase in lineage-negative populations, mostly including the GMP population (Figure 5E-F), suggesting that Mlf1−/− Trib1 leukemia-initiating cells (LICs) arise from more immature progenitors than WT LICs.
Figure 5.

An MLF1 deficiency causes more immature AML in Trib1 mice. (A) Schematic representation of the Mlf1 gene targeting strategy. The coding exon 1 is shown as a filled box; an open box denotes a noncoding portion. The probe used for a Southern blot analysis is shown as a gray box. Primers a, b, and c used for genomic PCR are indicated as arrows with the amplified direction. Neo, the neomycin phosphotransferase gene; TK, thymidine kinase gene. A restriction site of HindIII is shown as a single letter, H. (B) A Southern blot analysis of the targeted Mlf1 locus (left panel). Genomic DNA was extracted from ES cell clones, digested with HindIII, and hybridized with the probe. The sizes of WT (7.4 kb) and disrupted (3.2 kb) alleles are indicated. A PCR analysis of genomic DNA isolated from WT and knockout mouse tails (right panel). The positions of WT (primers a and b, 409 bp) and mutant (KO, primers b and c, 819 bp) amplification products are indicated. (C) Myeloid leukemia–free survival curves of mice transplanted with Trib1-transduced Mlf1−/− BM cells (Mlf1−/− Trib1 mice) compared with Trib1-transduced WT BM cells (WT Trib1 mice) and MigR1 control vector-transduced Mlf1−/− BM cells (Mlf1−/− MigR1 mice). Results are derived from 5 independent transfer experiments. The P value between WT Trib1 and Mlf1−/− Trib1 mice was calculated with the log-rank test. P = .14. (D) May-Grünwald Giemsa–stained PB smears and cytospins of BM cells from WT Trib1 (left panel), Mlf1−/− Trib1 (middle panel), and Mlf1−/− MigR1 (right panel) mice. Original magnification ×400. (E) A FACS analysis of BM and SP cells for immature (Mac-1+Gr-1lo) and differentiated (Mac-1+Gr-1hi) granulocytes. The population of GFP-positive cells in BM and SP is shown in the upper panels. (F) A detailed FACS analysis of GFP-positive BM cells for the lineage negative cell (Lin−: CD3−B220−TER-119−Mac-1−Gr-1−), stem cell (LSK cells), and multipotent progenitor (CMP: Lin−c-Kit+Sca-1−FcγRloCD34+; GMP: Lin−c-Kit+Sca-1−FcγRhiCD34+; MEP: Lin−c-Kit+Sca-1−FcγRloCD34−). (G) A detailed FACS analysis of GFP-positive BM cells for the lineage negative cell excluding Mac-1 and Gr-1 (Lin*−: CD3−B220−TER-119−), committed myeloid progenitor (Lin*−Sca-1−c-Kit+Mac-1+), and differentiated myeloid cell (Lin*−Sca-1−c-Kitlo/−Mac-1+). (H) Total RNA extracted from GFP-positive BM cells was analyzed by semi-qRT-PCR using a pair of primers specific to COP1, Trib1, and β-actin.
Discussion
In the present study, we demonstrated that MLF1 functions as an inhibitory protein for the E3 ubiquitin ligase complex, Trib1-COP1, which targets one of the key transcription factors of myeloid differentiation, C/EBPα, for degradation. We found that MLF1 interacts directly with COP1 in the nucleus and inhibits the polyubiquitination of the C/EBPα protein induced by the Trib1-COP1 ligase complex. The overexpression of MLF1 prevents the formation of the Trib1-COP1 ligase complex by competing with Trib1 for binding to COP1. In order to interpret our results in the context of hematopoiesis and leukemogenesis, we examined the precise distribution of MLF1 in normal hematopoiesis. MLF1 is expressed in hematopoietic stem cells (HSCs) and myeloid progenitors (CMP and GMP) but is more weakly expressed in MEP and lineage-positive cells. MLF1 expression gradually increases from CD34-LSK to CMP and starts decreasing in GMP. The distribution of C/EBPα overlaps with that of MLF1; C/EBPα is expressed in HSCs and increases in CMP and GMP (most abundant in GMP) but is almost absent in MEP.31,32 Because C/EBPα knockout mice fail to produce GMP, C/EBPα is essential to lineage specification programs, differentiating from stem cells to myeloid progenitors (GMP commitment).33 Once myeloid differentiation reaches the stage of GMP, C/EBPα is no longer required for further maturation. The timing of the expression of MLF1 parallels that of C/EBPα, suggesting that MLF1 stabilizes C/EBPα by blocking Trib1-COP1 activity until the differentiation program is properly completed at this stage.
We further showed a role for MLF1 in Trib1-COP1 complex–mediated leukemogenesis using mouse models. As expected from the results obtained by the in vitro culture system, in which MLF1 counteracted Trib1-COP1 activity, MLF1 significantly suppressed Trib1-induced AML development in mice doubly transduced with MLF1 and Trib1. However, half the MLF1/Trib1 doubly transduced mice still developed AML, and analyses of GFP-positive cells isolated from these mice with the quantitative real-time RT-PCR method revealed that a balance between Trib1 and MLF1 expression levels is the key factor influencing whether cells differentiate normally or develop AML. If the amount of MLF1 is in excess, the formation of the active Trib1-COP1 complex is suppressed, which allows progenitor cells to reach the GMP stage at which C/EBPα is no longer required, whereas a lower amount of MLF1 is not sufficient to inhibit the leukemogenic action of the Trib1-COP1 complex.
We initially expected the MLF1 deficiency to accelerate the onset of Trib1-induced AML because MLF1 exhibits growth-suppressing activity when introduced into proliferating primary cells in culture. However, the MLF1 deficiency conferred a more immature phenotype on Trib1-induced AML development without accelerating the onset of AML, implying that MLF1 affects lineage differentiation rather than proliferation in the C/EBPα-Trib1-COP1 pathway in hematopoiesis. LICs in p42C/EBPα-deficient mice and Trib1-transduced mice reside in Mac-1+c-Kit+ populations (granulocyte-macrophage commitment),15,34 whereas the Mlf1−/− background enforces the initiation of Trib1-induced leukemogenesis at more immature progenitors (GMP). The MLF1 deficiency may allow for the rapid access of the active Trib1-COP1 complex to C/EBPα for degradation in an earlier phase than that in normal myelopoiesis. These results from mouse models support MLF1 acting as a stabilizer of the C/EBPα protein to properly complete the transition from the HSC to GMP phase.
As for other inhibitory proteins for ubiquitin ligases, Arf for Mdm2, a specific ligase for p53, and CAND1 for Cullin-RING ubiquitin ligases (CRLs) have been studied in detail. In response to oncogenic stress, Arf directly inhibits Mdm2 ligase activity by sequestering it to the nucleolus.26 CAND1 dissociates the Skp1 and F box proteins from Cullin to promote the assembly of a new SCF ligase complex through the exchange of the F box protein. CAND1 was originally considered to be an inhibitor of the SCF complex; however, CAND1 binding to Cullin has recently been recognized as a part of the reactivation program of the SCF complex.35,36 In the case of MLF1, both the subcellular localization of MLF1 and the ability of MLF1 to interfere with Trib1-COP1 complex formation are crucial for the inhibitory activity of MLF1. MLF1 binding to COP1 in the nucleus appears to be a part of the differentiation program of normal hematopoiesis regulating the expression of specific transcription factors.
COP1 has a number of substrates for degradation in the cell, with some (c-Jun, ETV1, and TORC2) being directly recognized,12,13,16 and others with the aid of a specific adaptor protein such as Tribbles for C/EBPα and ACC1.7,15 These substrates include an oncoprotein and tumor suppressor, and findings obtained from COP1 knockout mice revealed that COP1 functions as a tumor suppressor,13,28 suggesting that targeting and inhibiting the catalytic activity of COP1 ubiquitin ligase is not a realistic approach for cancer therapy. Alternatively, it is feasible to target particular functions of COP1 by inhibiting the specific interaction between COP1 and a limited set of adaptor proteins such as the Trib-COP1 complex. Most conventional anticancer molecular medicines target the catalytic activities of oncoproteins such as protein kinases and transcription factors; however, the recent development of small chemicals interfering with protein-protein interaction37 may be applied to the regulation of COP1 activity; for example, inhibiting the interaction between COP1 and Trib1 by specifically blocking the COP1-binding site of Trib1. In this study, we found that MLF1 is a natural inhibitor of the COP1-Trib1 interaction, implying that the transcriptional enhancement of MLF1 expression and/or increased nuclear compartmentalization of MLF1 has potential as a novel strategy for cancer therapy.
Acknowledgments
The authors thank C. J. Sherr and M. F. Roussel for the NIH3T3 cell line.
This work was partly supported by Grants-in-Aid for Scientific Research and for Cancer Research from the Ministry of Education, Science, and Culture of Japan.
Authorship
Contribution: N.Y.-K. designed the experiments; I.N., J.-y.K., and N.Y.-K. performed the experiments; I.N., J.-y.K., and N.Y.-K. analyzed the data; T.Y. and H.I. performed the bioinformatics analysis; and J.-y.K. and N.Y.-K. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Noriko Yoneda-Kato, Graduate School of Biological Sciences, Nara Institute of Science and Technology, 8916-5 Takayama, Ikoma, Nara 630-0101, Japan; e-mail: noriko-k@bs.naist.jp.
References
- 1.Koschmieder S, Halmos B, Levantini E, Tenen DG. Dysregulation of the C/EBPalpha differentiation pathway in human cancer. J Clin Oncol. 2009;27(4):619-628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pabst T, Mueller BU, Harakawa N, et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med. 2001;7(4):444-451. [DOI] [PubMed] [Google Scholar]
- 3.Radomska HS, Bassères DS, Zheng R, et al. Block of C/EBP alpha function by phosphorylation in acute myeloid leukemia with FLT3 activating mutations. J Exp Med. 2006;203(2):371-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keeshan K, He Y, Wouters BJ, et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell. 2006;10(5):401-411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin G, Yamazaki Y, Takuwa M, et al. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood. 2007;109(9):3998-4005. [DOI] [PubMed] [Google Scholar]
- 6.Rørth P, Szabo K, Texido G. The level of C/EBP protein is critical for cell migration during Drosophila oogenesis and is tightly controlled by regulated degradation. Mol Cell. 2000;6(1):23-30. [DOI] [PubMed] [Google Scholar]
- 7.Qi L, Heredia JE, Altarejos JY, et al. TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism. Science. 2006;312(5781):1763-1766. [DOI] [PubMed] [Google Scholar]
- 8.Eyers PA. TRIBBLES: a twist in the pseudokinase tail. Structure. 2015;23(11):1974-1976. [DOI] [PubMed] [Google Scholar]
- 9.Yokoyama T, Kanno Y, Yamazaki Y, Takahara T, Miyata S, Nakamura T. Trib1 links the MEK1/ERK pathway in myeloid leukemogenesis. Blood. 2010;116(15):2768-2775. [DOI] [PubMed] [Google Scholar]
- 10.Dedhia PH, Keeshan K, Uljon S, et al. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood. 2010;116(8):1321-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yi C, Deng XW. COP1—from plant photomorphogenesis to mammalian tumorigenesis. Trends Cell Biol. 2005;15(11):618-625. [DOI] [PubMed] [Google Scholar]
- 12.Wertz IE, O’Rourke KM, Zhang Z, et al. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science. 2004;303(5662):1371-1374. [DOI] [PubMed] [Google Scholar]
- 13.Vitari AC, Leong KG, Newton K, et al. COP1 is a tumour suppressor that causes degradation of ETS transcription factors. Nature. 2011;474(7351):403-406. [DOI] [PubMed] [Google Scholar]
- 14.Dornan D, Wertz I, Shimizu H, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429(6987):86-92. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida A, Kato JY, Nakamae I, Yoneda-Kato N. COP1 targets C/EBPα for degradation and induces acute myeloid leukemia via Trib1. Blood. 2013;122(10):1750-1760. [DOI] [PubMed] [Google Scholar]
- 16.Dentin R, Liu Y, Koo SH, et al. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449(7160):366-369. [DOI] [PubMed] [Google Scholar]
- 17.Kato S, Ding J, Pisck E, Jhala US, Du K. COP1 functions as a FoxO1 ubiquitin E3 ligase to regulate FoxO1-mediated gene expression. J Biol Chem. 2008;283(51):35464-35473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marine JC. Spotlight on the role of COP1 in tumorigenesis. Nat Rev Cancer. 2012;12(7):455-464. [DOI] [PubMed] [Google Scholar]
- 19.Yoneda-Kato N, Look AT, Kirstein MN, et al. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12(2):265-275. [PubMed] [Google Scholar]
- 20.Falini B, Mecucci C, Tiacci E, et al. ; GIMEMA Acute Leukemia Working Party. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254-266. [DOI] [PubMed] [Google Scholar]
- 21.Colombo E, Alcalay M, Pelicci PG. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene. 2011;30(23):2595-2609. [DOI] [PubMed] [Google Scholar]
- 22.Matsumoto N, Yoneda-Kato N, Iguchi T, et al. Elevated MLF1 expression correlates with malignant progression from myelodysplastic syndrome. Leukemia. 2000;14(10):1757-1765. [DOI] [PubMed] [Google Scholar]
- 23.Williams JH, Daly LN, Ingley E, et al. HLS7, a hemopoietic lineage switch gene homologous to the leukemia-inducing gene MLF1. EMBO J. 1999;18(20):5559-5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoneda-Kato N, Tomoda K, Umehara M, Arata Y, Kato JY. Myeloid leukemia factor 1 regulates p53 by suppressing COP1 via COP9 signalosome subunit 3. EMBO J. 2005;24(9):1739-1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6(7):493-505. [DOI] [PubMed] [Google Scholar]
- 26.Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6(9):663-673. [DOI] [PubMed] [Google Scholar]
- 27.Yoneda-Kato N, Kato JY. Shuttling imbalance of MLF1 results in p53 instability and increases susceptibility to oncogenic transformation. Mol Cell Biol. 2008;28(1):422-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Migliorini D, Bogaerts S, Defever D, et al. Cop1 constitutively regulates c-Jun protein stability and functions as a tumor suppressor in mice. J Clin Invest. 2011;121(4):1329-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7(8):2745-2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato JY, Yoneda-Kato N. Mammalian COP9 signalosome. Genes Cells. 2009;14(11):1209-1225. [DOI] [PubMed] [Google Scholar]
- 31.Akashi K, Traver D, Miyamoto T, Weissman IL. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404(6774):193-197. [DOI] [PubMed] [Google Scholar]
- 32.Iwasaki H, Mizuno S, Arinobu Y, et al. The order of expression of transcription factors directs hierarchical specification of hematopoietic lineages. Genes Dev. 2006;20(21):3010-3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang P, Iwasaki-Arai J, Iwasaki H, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21(6):853-863. [DOI] [PubMed] [Google Scholar]
- 34.Kirstetter P, Schuster MB, Bereshchenko O, et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell. 2008;13(4):299-310. [DOI] [PubMed] [Google Scholar]
- 35.Pierce NW, Lee JE, Liu X, et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell. 2013;153(1):206-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu S, Zhu W, Nhan T, Toth JI, Petroski MD, Wolf DA. CAND1 controls in vivo dynamics of the cullin 1-RING ubiquitin ligase repertoire. Nat Commun. 2013;4:1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nim S, Jeon J, Corbi-Verge C, et al. Pooled screening for antiproliferative inhibitors of protein-protein interactions. Nat Chem Biol. 2016;12(4):275-281. [DOI] [PMC free article] [PubMed] [Google Scholar]