

Key Points
Regadenoson did not reduce iNKT cell activation to a prespecified level when administered to patients with SCD.
Because iNKT cell activation was not reduced, the benefit of iNKT cell-based therapies in SCD cannot be determined.
Abstract
Adenosine A2A receptor (A2AR) agonists have been shown to decrease tissue inflammation induced by hypoxia/reoxygenation in mice with sickle cell disease (SCD). The key mediator of the A2AR agonist’s anti-inflammatory effects is a minor lymphocyte subset, invariant natural killer T (iNKT) cells. We tested the hypothesis that administration of an A2AR agonist in patients with SCD would decrease iNKT cell activation and dampen the severity of vaso-occlusive (VO) crises. In a phase 2, randomized, placebo-controlled trial, we administered a 48-hour infusion of the A2AR agonist regadenoson (1.44 μg/kg per hour) to patients with SCD during VO crises to produce a plasma concentration of ∼5 nM, a concentration known from prior studies to suppress iNKT cell activation in SCD. The primary outcome measure was a >30% reduction in the percentage of activated iNKT cells. Ninety-two patients with SCD were randomized to receive a 48-hour infusion of regadenoson or placebo, in addition to standard-of-care treatment, during hospital admission for a VO crisis and had analyzable iNKT cell samples. The proportion of subjects who demonstrated a reduction of >30% in activated iNKT cells was not significantly different between the regadenoson and placebo arms (43% vs 23%; P = .07). There were also no differences between regadenoson and placebo groups in length of hospital stay, mean total opioid use, or pain scores. These data demonstrate that a low-dose infusion of regadenoson intended to reduce the activity of iNKT cells is not sufficient to produce a statistically significant reduction in such activation or in measures of clinical efficacy. This trial was registered at www.clinicaltrials.gov as #NCT01788631.
Visual Abstract
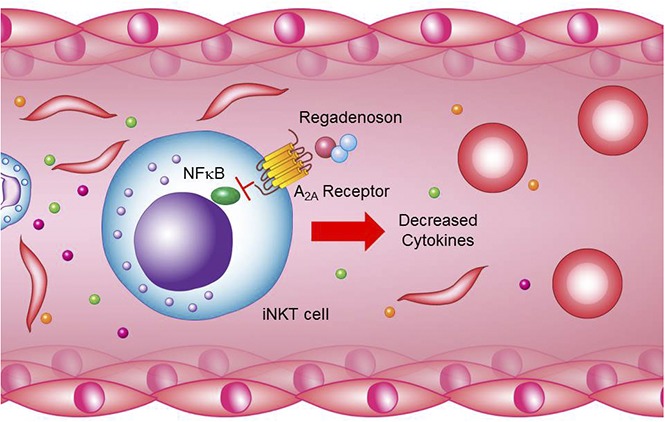
Introduction
Invariant natural killer T (iNKT) cells are implicated in the pathogenesis of sickle cell disease (SCD).1-3 In patients with SCD, iNKT cells are increased in number and activation state compared with controls.1 In a SCD mouse model, depletion of iNKT cells or blockage of their activation reduced pulmonary inflammation and injury.1,2 One strategy used to decrease iNKT cell activation in SCD mice was treatment with a selective adenosine A2A receptor (A2AR) agonist.2 Based on these data, we previously conducted a phase 1 study of the A2AR agonist, regadenoson, in adults with SCD.3 In that study, we established a safe and effective infusion schedule for regadenoson (1.44 μg/kg per hour) that produced a steady-state plasma level of 5 nM and appeared to depress the activity of iNKT cells in SCD patients during a vaso-occlusive (VO) crisis. Although no dose-limiting toxicity was noted, we decided not to examine higher infusion rates to avoid the side effects that are commonly observed during myocardial imaging when regadenoson is administered as a bolus, which achieves a plasma concentration of ∼16 nM,4 and because the cost of higher doses would be prohibitive.
The current study is a phase 2, multicenter, randomized, placebo-controlled trial of regadenoson to determine its efficacy for the treatment of an acute, VO episode. A 48-hour continuous infusion of 1.44 μg/kg per hour regadenoson or placebo was administered to determine whether the drug could decrease the percentage of highly activated iNKT cells, as measured by binding of a fluorescent antibody to the 65-kDa subunit of NF-κB phosphorylated on serine 536 (pNF-κB), and decrease the severity of VO crises, as measured by length of hospital stay, mean parenteral opioid use, and pain and respiratory scores.
Methods
Study conduct
A phase 2, multicenter, double-blinded, randomized, placebo-controlled trial of regadenoson was conducted in children and adults with sickle cell anemia (HbSS/HbSβ0-thalassemia), 10 to 60 years of age, who were admitted to the hospital for an acute VO crisis (see supplemental Methods for specific inclusion/exclusion criteria). Patients were randomized using a permuted blocks algorithm for each participating site. Treatment was assigned centrally by registrars in the Office of Data Quality at the Dana-Farber Cancer Institute, with unblinded assignment transferred only to the investigational pharmacy at the treating institution. No treatment assignment was unblinded during the trial. After randomization, infusional regadenoson (1.44 μg/kg per hour) or placebo was started within 36 hours of the first dose of parenteral opioids and continued for 48 hours. A 36-hour window was selected, as opposed to 24 hours, to facilitate accrual; a 48-hour infusion was selected, as opposed to 24 hours as was done in phase 1, in order to have a more significant clinical effect on inflammation. The primary outcome measure was a >30% decrease in the percentage of circulating iNKT cells expressing high levels of pNF-κB at 48 hours compared with predrug levels. Secondary outcome measures were: length of hospital stay, parenteral opioid use, visual analog pain scale, and composite respiratory score, defined as: (1) respiratory rate decreased by 25% from baseline or normalized (≤20 bpm) or (2) degree of hypoxia (SpO2) on room air increased by 10% from baseline or normalized (≥92%) or (3) thoracic pain improved by 3 points from baseline on a 10-point visual analog scale. If subjects experienced any of these outcomes, they were classified as having an improvement in respiratory symptoms. The trial was performed at 9 collaborating hospitals (see supplemental Methods for participating sites); blood samples were shipped to a central laboratory in La Jolla, CA, at 4°C to 6°C for the performance of flow cytometry. Institutional review boards approved the study protocol at participating sites, and informed consent was obtained from all participants.
Laboratory methodology
iNKT cells were identified by flow cytometry as live, CD19− (SJ25-C1; Invitrogen), CD3+ (UCHT1; Invitrogen and BD Biosciences), and Vα24-Jα18 TCR+ (6B11; eBioscience) cells. Conventional T cells were identified as live, CD19−, CD3+, and Vα24-Jα18 TCR− cells. The active phosphorylated form of NF-κB was identified with anti-pNF-κB (Ser536, 93H1; Cell Signaling Technology).
To determine the activation state of iNKT cells, we measured by flow cytometry their expression of pNF-κB relative to conventional CD3+ T cells, which are minimally activated by VO. The lower boundaries of activation gates were set to exclude expression levels of 95% of conventional CD3+ T cells. The data were expressed as the percentage of iNKT cells in the activation gate and were reproducible when repeated on different days in steady-state patients.
Statistical analysis
For data analysis, to achieve a 30% reduction in pNF-κB–activated iNKT cells at 48 hours, compared with predrug levels, 100 subjects (50 in each arm) provide 80% power to distinguish a 50% success rate in the regadenoson arm compared with a 20% success rate in the placebo arm, using a 2-sided test with an α of 0.05. Although we hypothesized that regadenoson treatment would decrease pNF-κB activation, we nevertheless chose to evaluate the data using a 2-sided analysis.
Laboratory results were analyzed with robust methods (median, range). Comparisons of pNF-κB parameters between placebo and regadenoson arms pretreatment and during treatment were performed. The 2-sided Fisher exact test or the Wilcoxon rank sum test, as appropriate, was used to compare randomized treatment groups. Analysis of iNKT cell activation markers was reported as the percentage change from the value at time 0 to the value at 48 hours from the start of the infusion ([% time 0 − % time 48 hours]/% time 0). If a 48-hour sample was not available, a 24-hour sample was substituted.
Results
Four thousand nine hundred forty hospital admissions were screened in order to enroll and randomize 100 subjects with HbSS/HbSβ0-thalassemia, 53 to the regadenoson arm and 47 to the placebo arm (Figure 1; Table 1). Ninety-two subjects were treated and provided blood samples for the assessment of the primary end point. There were no differences in age, sex, or hydroxyurea use between the regadenoson and placebo group. The majority of patients were adults, and most were prescribed hydroxyurea. Study enrollment began in June 2012 and ended in November 2016.
Figure 1.

Subject flow through the study.
Table 1.
Demographics and medications by treatment arm
| Regadenoson, n = 53 | Placebo, n = 47 | P | |
|---|---|---|---|
| Age, median (range), y | 25 (10-54) | 26 (13-56) | .29 |
| Pediatric, <18 y (%) | 6 (11) | 8 (17) | .57 |
| Sex (%) | .42 | ||
| Female | 22 (42) | 24 (51) | |
| Male | 31 (58) | 23 (49) | |
| Race (%) | 1.0 | ||
| Black or African American | 49 (92) | 44 (94) | |
| Other | 4 (8) | 3 (6) | |
| Currently taking hydroxyurea (%) | 38 (72) | 33 (70) | 1.0 |
There was no significant effect of 5 nM regadenoson on pNF-κB iNKT cell activation. When expressed as percentage of baseline, median end-of-study (48 hours) pNF-κB iNKT cell activation was only modestly reduced compared with baseline (pretreatment) levels for both the regadenoson (7.5% reduction) and placebo groups (1.3% reduction). And, although there was a trend, there was also not a significant difference in the primary outcome measure, a >30% reduction of pNF-κB p65-activated iNKT cells at 48 hours compared with baseline. In the regadenoson and placebo arms, respectively, 21 of 49 (42.9%) and 10 of 43 (23.3%) experienced such a reduction (P = .07) (Figure 2).
Figure 2.

Baseline vs end-of-infusion p65-phospho NF-κB iNKT cell activation. Points below the black line represent patients who achieved the primary outcome measure of a p65-phospho-NF-κB iNKT cell activation at 48 hours that was a >30% reduction from baseline. Subjects in the regadenoson and placebo arms are represented by red and white circles, respectively.
Differences in measures of clinical efficacy were not detected. Median length of hospital stay was similar in both arms (3.96 days vs 3.99 days; P = .80). There was also no difference between the regadenoson and placebo arms in the amount of parenteral opioids used during infusion (median morphine equivalent dose, 0.03 mg/kg per hour vs 0.04 mg/kg per hour; P = .34), mean reduction in visual analog pain score (−2.68 vs −2.80; P = .91), or improvement in composite respiratory score (20.83% vs 21.43%; P = 1.0). When we compared clinical outcomes within the regadenoson arm between those who did and did not respond (48-hour pNF-κB iNKT cells with a decline of >30% from baseline), there were no differences in length of stay (4.41 days vs 3.74 days; P = .47), amount of parenteral opioids used during infusion (median morphine equivalent dose, 0.04 mg/kg per hour vs 0.03 mg/kg per hour; P = .63), mean reduction in visual analog pain score (−2.80 vs −2.67; P = .86), or improvement in composite respiratory score (26.67% vs 18.33%; P = .42).
Finally, regadenoson was not associated with an increased rate of adverse events or serious adverse events, including alterations to blood pressure and heart rate (see supplemental Table 1).
Discussion
This study did not achieve its primary outcome measure of a reduction in iNKT cell activation in subjects treated with regadenoson compared with placebo. We also did not observe an improvement in clinical outcomes and, because we examined several measures of clinical efficacy, the likelihood we falsely accepted the null hypothesis is low. Obviously, one reason for the negative result is that the null hypothesis is true and regadenoson does not have a significant effect on iNKT cell activation or clinical outcomes. There are other potential explanations, however, as to why we did not see a biological or clinical response.
The dose of regadenoson used in our trial may have been too low to reduce iNKT cell activation. For its US Food and Drug Administration (FDA)-indicated use during myocardial stress imaging, regadenoson is administered as a 400-μg bolus, achieving peak plasma concentrations of ∼16 nM and then redistributing over time to lower levels.5 Potential side effects are hypotension, reflex tachycardia, flushing, and bronchospasm. Because we would be administering regadenoson during a VO crisis, we were concerned about the effect of these toxicities on an already-ill patient. Other challenges were regadenoson’s 5-minute half-life, which necessitated that we delivered it as a continuous infusion in order to decrease the severity of a VO crisis and cost.6 A dose was chosen that appeared to maximize the effect on iNKT cells, while minimizing toxicities and not being cost prohibitive. In the phase 1 study, we extrapolated the target dose, 1.44 μg/kg per hour, from animal studies.2,7 Using this dose, we achieved peak plasma concentrations of ∼5 nM, or about one-third of the concentrations seen in stress imaging,3 and encountered no dose-limiting side effects.3 Still, in the 6 patients who received regadenoson infusion for 24 hours in the phase 1 study, there was 50% median reduction in the fraction of circulating pNF-κB–high iNKT cells. On the basis of these data, we concluded that the drug concentration (1.44 μg/kg per hour, resulting in 5 nM steady-state plasma regadenoson concentration) was sufficient to strongly inhibit iNKT cells. It is possible that we might have attained a more significant effect on iNKT cell activation, while still avoiding unwanted side effects, if we had used a higher infusional dose of regadenoson.
A reduction in activated circulating iNKT cells could be the result of a drug effect or the exodus of activated cells from the circulation. In the current study, we found that 23% of patients who received placebo for 48 hours during VO crisis displayed a 30% or greater reduction in the fraction of activated circulating iNKT cells. A possible explanation for this reduction in circulating activated iNKT cells during VO may be that many activated iNKT cells extravasate into inflamed tissues or become adhered to activated endothelial cells during VO.
Treatment of a VO crisis is likely more challenging than prevention. Regadenoson’s short half-life and low oral bioavailability necessitated that it be administered as an infusion during a VO crisis.5 By the time a patient with SCD presents to the hospital for pain, the VO crisis has likely been ongoing for days, with the pathogenesis of VO already widespread. Historically, therapeutic approaches to attenuate this process have been unsuccessful,8,9 whereas there have been successful approaches to prevent VO, such as transfusion,10 hydroxyurea,11 and crizanlizumab.12
Despite murine data suggesting that iNKT cells are critical to the initiation and propagation of VO,1,2,13 we found that a 48-hour infusion of regadenoson did not significantly decrease pNF-κB iNKT cell activation, or the severity of VO crises, in patients with SCD. Although a higher dose may have provided benefit, further studies of single-agent regadenoson for the management of VO are probably not worth the investment. A better approach, and one that is similar to the crizanlizumab strategy, may be the long-term depletion of iNKT cells with a monoclonal antibody, such as NKTT120, aimed to prevent VO crises.14 Alternatively, iNKT cells may not play a significant role in VO in humans.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Charles T. Quinn (Cincinnati Children's Hospital), Jeffrey Keefer (Johns Hopkins University), Paul Swerdlow (Wayne State University), and Marilyn Telen (Duke University) for their contributions to the study. They also thank research nurse coordinators Debora Nischik and Lisa Garrett for their dedication to the study.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL098526 and P50HL110790, with additional support from Astellas Pharma.
Authorship
Contribution: J.J.F., D.G.N., J.L., and D.N. conceived and designed the studies; J.J.F., E.M., V.R.G., M.G., C.H., M.M.H., M.A., A.G., H.C., and B.S. acquired data; M.P., D.N., and G.L. analyzed the data; J.J.F., D.G.N., J.L., M.P., D.N., and G.L. interpreted the data; J.J.F. wrote the manuscript; and D.G.N., J.L., and D.N. edited the manuscript.
Conflict-of-interest disclosure: J.J.F. declares past or current research support from NKT Therapeutics, Astellas Pharma, and Prolong Pharmaceuticals. D.G.N. declares past or current research support from NKT Therapeutics and Astellas Pharma. The remaining authors declare no competing financial interests.
Correspondence: Joshua J. Field, BloodCenter of Wisconsin, 8733 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: joshua.field@bcw.edu.
References
- 1.Wallace KL, Marshall MA, Ramos SI, et al. . NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114(3):667-676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace KL, Linden J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood. 2010;116(23):5010-5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Field JJ, Lin G, Okam MM, et al. . Sickle cell vaso-occlusion causes activation of iNKT cells that is decreased by the adenosine A2A receptor agonist regadenoson. Blood. 2013;121(17):3329-3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordi T, Blackburn B, Lieu H. Regadenoson pharmacokinetics and tolerability in subjects with impaired renal function. J Clin Pharmacol. 2007;47(7):825-833. [DOI] [PubMed] [Google Scholar]
- 5.Astellas Pharma. Lexiscan [package insert]. Northbrook, IL: Astellas Pharma; 2009.
- 6.Field JJ, Nathan DG, Linden J. The role of adenosine signaling in sickle cell therapeutics [published correction appears in Hematol Oncol Clin North Am 2015;29(2):xv]. Hematol Oncol Clin North Am. 2014;28(2):287-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203(12):2639-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gladwin MT, Kato GJ, Weiner D, et al. ; DeNOVO Investigators. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011;305(9):893-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orringer EP, Casella JF, Ataga KI, et al. . Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease: A randomized controlled trial. JAMA. 2001;286(17):2099-2106. [DOI] [PubMed] [Google Scholar]
- 10.DeBaun MR, Gordon M, McKinstry RC, et al. . Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371(8):699-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charache S, Terrin ML, Moore RD, et al. ; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 1995;332(20):1317-1322. [DOI] [PubMed] [Google Scholar]
- 12.Ataga KI, Kutlar A, Kanter J, et al. . Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017;376(5):429-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin G, Field JJ, Yu JC, et al. . NF-κB is activated in CD4+ iNKT cells by sickle cell disease and mediates rapid induction of adenosine A2A receptors [published correction appears in PLos One 2015;10(2):e0117760]. PLoS One. 2013;8(10):e74664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Field JJ, Majerus E, Ataga KI, et al. . NNKTT120, an anti-iNKT cell monoclonal antibody, produces rapid and sustained iNKT cell depletion in adults with sickle cell disease. PLoS One. 2017;12(2):e0171067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.