


Abstract
Micro-RNAs (miRNAs) are potent regulators of gene expression and cellular phenotype. Each miRNA has the potential to target hundreds of transcripts within the cell thus controlling fundamental cellular processes such as survival and proliferation. Here, we exploit this important feature of miRNA networks to discover vulnerabilities in cancer phenotype, and map miRNA-target relationships across different cancer types. More specifically, we report the results of a functional genomics screen of 1280 miRNA mimics and inhibitors in eight cancer cell lines, and its presentation in a sophisticated interactive data portal. This resource represents the most comprehensive survey of miRNA function in oncology, incorporating breast cancer, prostate cancer and neuroblastoma. A user-friendly web portal couples this experimental data with multiple tools for miRNA target prediction, pathway enrichment analysis and visualization. In addition, the database integrates publicly available gene expression and perturbation data enabling tailored and context-specific analysis of miRNA function in a particular disease. As a proof-of-principle, we use the database and its innovative features to uncover novel determinants of the neuroblastoma malignant phenotype.
INTRODUCTION
Micro-RNAs (miRNAs) are a class of cellular small non-coding RNAs that act to regulate protein synthesis through messenger RNA (mRNA) destabilization and inhibition of translation (1). Mature miRNAs associate with RNA-binding proteins into the RNA-induced silencing complex (RISC) where they exert their effects through hybridization to mRNA targets. Many microRNAs bind mRNA via complementarity between the 6–8 nt seed of the miRNA with regions typically in the 3′ untranslated region (UTR) of the mRNA.
miRNAs control a large diversity of cellular processes, from differentiation and proliferation to cell death (2–4) and their deregulation can contribute to human disease aetiology. A role for miRNA in cancer is well described (5), and miRNA are proving valuable as biomarkers and a novel class of cancer therapeutics (6). Global miRNA expression decreases with increasing grade in numerous cancers, suggesting a role for some miRNA as tumor-suppressors (7).
The literature contains many reports of miRNA deregulation and function in many cancer types, but these studies are typically small scale, lack systematic functional data, are based on variable selection criteria and are scattered across a multitude of paradigms and models. As such, there are many miRNA that remain uncharacterized, and it is difficult to assess the relative importance of miRNAs to transformation and tumor-suppression. To address this issue we performed a comprehensive and unbiased reverse genetic screen to identify miRNAs important for cancer survival in cell line models of three solid cancer types, breast, prostate and neuroblastoma. We then developed a novel web-based data interface to integrate this functional data with publicly available algorithms for miRNA target prediction, and large-scale datasets of cell-specific gene expression and gene perturbation experiments. This database streamlines the process of identifying the most relevant miRNAs and their targets for cancer cell survival.
Our screen identifies many well-described tumor-suppressor miRNAs such as miR-34 and multiple members of the let-7 family. In addition, we discover numerous other miRNA with both widespread and disease-restricted roles in tumor-suppression. Many of these miRNA converge on overlapping sets of targets, which were over-represented for members of the FOXO signaling, PI3K-Akt signaling and stem cell pluripotency pathways.
As a proof of principle, we used our database to search for novel lethal miRNA and their targets in neuroblastoma, an aggressive childhood cancer of the sympathetic nervous system, composed of primitive neural cells with a high metastatic potential. Only a few oncogenes such as MYCN, LIN28B or ALK have been directly linked to neuroblastoma development and maintenance (8). As the molecular drivers of neuroblastoma proliferation, differentiation and survival remain largely unexplored, we used this integrated miRNA functional database to search for novel determinants of neuroblastoma aetiology. We discovered several miRNAs with a previously unrecognized role in survival of neuroblastoma. Furthermore, using our database we uncover novel factors necessary for neuroblastoma growth and survival.
MATERIALS AND METHODS
Cell lines
Breast cancer cell lines (MDA-MB-231, MDA-MB-468, SK-BR-3; BT474) and prostate cancer cell lines (PC3, DU145) were obtained from American Type Culture Collection. Neuroblastoma cell lines (KELLY, SHEP) were received from the Children’s Cancer Institute. All breast and prostate cancer cell lines were cultivated in Roswell Park Memorial Institute (RPMI) media supplemented with 10% Fetal Calf Serum (FCS), HEPES and insulin. KELLY and SHEP cell lines were grown in 10% FCS/RPMI and 10% FCS/Dulbecco’s modified Eagle’s medium (DMEM), respectively. MCF10A cells were cultured in DMEM F12 containing 5% horse serum, Epidermal Growth Factor (EGF), cholera toxin, hydrocortisone and insulin. IMR-90 cells were grown in 10% FCS/MEM containing sodium pyruvate and non-essential amino acids.
High-throughput micro-RNA screening
All cell lines were reverse transfected in 384-well plate format with 25 nM of Dharmacon miRNA mimic or inhibitor libraries (version 16.0) using a Calliper Sciclone ALH3000 liquid-handling robot according to the conditions listed in Supplementary Table S1. After 24 h, media was changed using a BioTek 406 automated plate washer/dispenser, and 6 days posttransfection, cell viability was determined using the CellTiter-Glo (CTG) luminescent assay (Promega) at 1:2 dilution; luminescence measurements were taken on the Synergy H4 (BioTek) high-throughput multimode micro-plate reader. All screens were performed in technical duplicate.
On each library plate we included a large number of positive and negative technical controls, including non-targeting ON-TARGETplus siRNA SMARTPool (NT), lipid-only mock transfection, and mimic and inhibitor non-targeting controls. For positive controls we selected a panel of siRNA SMARTpools that when knocked down induce different levels of cell death including siPLK1, siTOX, siKIF11 or siCOPB2 (Dharmacon RNAi Technologies). Sample values were normalized to the average of the Nontargeting control (NT) wells on a per plate basis, and then the duplicate plate values were averaged. To define screen hits, normalized viability scores were binned in the following manner: CV1 (cell viability) bin included candidates with viability fold-change scores of ≥0.8; CV2 bin included candidates with viability fold-change scores of <0.8 and >0.5; and low cell (LC) bin was assigned to candidates with viability fold-change scores of ≤0.5. Only LC candidates were defined as screen hits.
To assess the technical performance of each screen, the viability values of the positive and negative controls normalized to NT were used to calculate Z′ factor, a quantitative measurement of the dynamic range of an assay (9). The formula entails:
 |
where, SD indicates standard deviation; h.v. indicates high value; and l.v. indicates low value.
Z′-values were binned in the following manner: 0.5–1 indicates excellent score; 0.3–0.5 indicates good score; 0–0.3 indicates acceptable score; <0 indicates too much overlap between the positive and negative controls.
Robust z-score is a normalization strategy that takes into account all values on a plate and calculates the deviation from the median of these values. The formula is
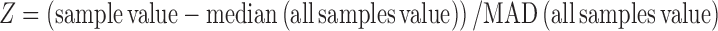 |
where, MAD indicates median absolute deviation.
For alternative inhibitor analysis and hit selection, we set a threshold of ≤−2.5, corresponding to at least 20% cell killing.
Validation screen
To confirm the reproducibility of the screen setup, the lead screen hits in the neuroblastoma cell lines (SHEP and KELLY) were rescreened under the same conditions as described above. The hit validation was performed in another facility using a Zephyr liquid-handling robot, and the raw data were analyzed as described above. The viability values normalized to the NT control from the primary and secondary screens were then subjected to the correlation analysis to assess the reproducibility of the assay.
Transfection and cell confluence assays
KELLY and SHEP cells were reverse transfected in 384-well format using liquid-handling robotics described above according to the conditions listed in Supplementary Table S1. For quantitative polymerase chain reaction (qPCR) and Propidium iodide (PI) uptake experiments, KELLY cells were transfected in 6-well plates using 2 μl DF1 at 1.4 × 105 cells/well initial density.
Cell confluence was measured using an IncuCyte ZOOM, an automated microscopy platform able to take images in regular intervals over the timespan of the experiment. Images were taken with a 10× objective at 2 h time intervals. The software was then trained to recognize live cells and exclude the dead ones on a collection of images, after which the cell confluence was calculated automatically in all wells and at all time points. All the graphs display cell confluence normalized to the NT well at the end point of the experiment.
Propidium iodide (PI) uptake
KELLY cells were transfected in a 6-well plate format as described above. Twenty-four hours later media was changed, and 96 h post-transfection, cells were collected and fixed with 70% ethanol overnight. The following day, cells were stained with 1 mg/ml propidium iodide (10 mg/ml final concentration) and RNAse A for up to 5 h. The cells were then collected using Canto I, and cell cycle profiles were constructed using FlowJo.
qPCR
KELLY cells were transfected in a 6-well plate format as described above. Twenty-four hours later media was changed, and 96 h post-transfection, cells were collected for RNA extraction using the miRNeasy isolation kit (Qiagen). RNA was then subjected to the Reverse transcription (RT) reaction to generate cDNA, which was subsequently quantified using qPCR and the appropriate Taqman probes. All values were normalized to two housekeeping genes B2M and GAPDH. The results were graphed as fold-change compared to the NT control. All experiments were performed in biological triplicate.
Micro-RNA luciferase-based reporter gene assay
The 3′ UTR sequences of ISL1, HAND1 and RGS16 were synthesized by Integrated DNA Technologies (IDT) with the corresponding miR-binding sites either left as wild-type (WT) sequence (WT 3′ UTR) or mutated to abolish miR-binding (MUT 3′ UTR). Generated DNA fragments were then cloned in the psiCHECK-2 vector using NotI and XhoI restriction enzymes downstream of hRluc gene (renilla luciferase). The psiCHECK-2 contains also hluc+ gene (firefly luciferase), which enables signal normalization to correct for variable transfection efficiency between samples.
To perform the luciferase assay, HEK293T cells were seeded at 2 × 104 cells per well in a 96-well plate, and the next day, co-transfected with 12.5 ng of the reporter plasmid and 50 nM of the respective miRNA mimic or NT control using Lipofectamine 3000. MDA-MB-231 cells were seeded at 2.4 × 104 and co-transfected with 75 ng of the reporter plasmid and 50 nM of the respective miRNA inhibitor or NT control using Lipofectamine 3000. Twenty-four hours later, cells were lysed and subjected to the Dual–Luciferase Reporter 1000 assay (Promega). Briefly, measurements were recorded at 0.5 s intervals for a total of 26 s with reagent injections of 100 μl of Luciferase Assay Reagent II (firefly luciferase) at 2 s and 100 μl of Stop and Glo Reagent (renilla luciferase) at 14 s. Renilla luciferase activity was normalized to the firefly luciferase signal for a readout of relative miRNA activity. The results were graphed as fold-change compared to the NT control. All experiments were performed in biological triplicate.
Small RNA sequencing
For small RNA profiling, the cells were cultured in 6-well plates for 72 h after which the RNA was extracted using the miRNeasy kit and sent for sequencing. The analysis of the FastQ files was performed using Oasis 1.0 sRNA detection tool (10). Raw count files of filtered miRNAs were then converted to counts per million.
RESULTS
Experimental design
To systematically identify miRNAs that control the survival of cancer cells, we performed functional miRNA screens in breast, prostate and neuroblastoma cancer cells. For each disease, we selected multiple cell lines, which were transfected with libraries containing mimics and inhibitors against 1280 known miRNAs amounting to 2560 constructs in total (Figure 1A). miRNA mimics act as mature miRNAs and are fully functional within a cell while single-stranded inhibitors efficiently block the activity of endogenous miRNAs through competitive binding. To monitor cellular viability, we used CellTiter-Glo (CTG, Promega), an assay highly sensitive to changes in cellular adenosine triphosphate (ATP) content.
Figure 1.

miRNA functional genomics screening in cancer cell lines. (A) The overview of the screen performed across different cancer cell lines. (B) A summary of the screen results in MDA-MB-231 breast cancer cell line. Y-axis displays CellTiter-Glo (CTG) values normalized to the negative control (NT) and the X-axis plots miRNA libraries (mimics or inhibitors) as well as the positive death controls (PLK1 and TOX) and additional negative controls (non-coding inhibitor control (Inhibitor NT), non-coding mimic control (Mimic NT)). (C) An overview of the primary screen hits across different cancer cell lines. Hits were defined as miR mimics or inhibitors that killed at normalized viability cut-off value ≤0.5. Venn diagram represents an overall number of hits across three diseases and their intersection, and the table lists the hit breakdown between the mimics and inhibitors. (D) Hit validation in KELLY and SHEP neuroblastoma cell lines. The graphs plot normalized viability values for selected miRNA mimics or inhibitors in the primary screen against their values in the secondary validation screen. These were subjected to the correlation analysis, and Pearson correlation coefficient is displayed for each cell line. The screens were performed in different facilities and on different transfection platforms (see ‘Materials and Methods’ section).
A representative box plot of the entire screen results in the MDA-MB-231 breast cancer cell line is shown in Figure 1B, with data from other cell lines shown in Supplementary Figure S1. In total, over 500 miRNA mimics or inhibitors decreased cellular viability >50% in at least one cell line compared to the NT control demonstrating that around 40% of miRNAs screened are putatively able to regulate survival-associated pathways in cancer cells (Figure 1C). Differences in miRNA mimic or inhibitor activities were apparent between different cancer types and even subtypes of the same cancer (Figure 1C), supporting the conclusion that the function of many miRNAs is strongly context dependent (11). To validate the reproducibility of our screen, we performed secondary screens in a different laboratory, using KELLY and SHEP cell lines with selected mimics and inhibitors. These results demonstrate high reproducibility of our assay (Figure 1D).
Interestingly, a global overview of the screen results reveals profound difference between the effects of miRNA mimics and inhibitors—only 10 inhibitors reduced viability more than 50% in at least one cell line (Figure 1B and C; Supplementary Figure S2). To understand this observation, we performed small RNA sequencing in the breast and neuroblastoma cell lines, and on average only 180 (∼12%) miRNAs showed expression at >100 reads per million in each of the cell lines, an expression threshold shown to be correlated with miRNA suppressive activity (12) (Supplementary Figure S3a). We conclude that the low expression levels of the majority of endogenous miRNAs in part explains the low frequency of inhibitors with activity. We postulated that inhibitors would most likely display modest lethal activity, so we reanalyzed our data using robust z-score statistics and a less stringent threshold of >20% viability reduction for hit selection. This led to a higher rate of inhibitor hit detection (Supplementary Figure S3b). Among these hits, we identified inhibitors against miR-92a-3p, previously shown to induce potent killing effect in glioma cells (13); miR-125a-5p inhibitor, which induces apoptosis across different cancer cells (14); and miR-27a-3p inhibitor, known to decrease viability of renal cell carcinoma cells (15). Importantly, these miRNAs were highly expressed in the target cell lines. Because of the more frequent and stronger lethal effects, however, we focused on miRNA mimics in all subsequent analyses.
Database overview
To streamline the search for essential miRNAs and the mechanisms they use to suppress viability of cancer cells, we constructed a database that combines screen results with mRNA and miRNA expression data, published siRNA screen data, established miRNA target prediction algorithms and a variety of pathway enrichment and visualization tools (Figure 2B and Supplementary Figure S4). This database is available online (http://microrna.garvan.unsw.edu.au/mtp/search/index).
Figure 2.

miRNA function database structure and metrics. (A, i) miR Selection: mining of the screening data gives a list of miRNAs with a lethal effect in one or more cancer cell lines, or a custom list can be used. (A, ii) miR filtering: there is an option of excluding miRNAs that affect normal cells, for example cardiomyocytes and normal breast cells. (A, iii) Target filtering: the miRNAs are further subjected to five different target prediction algorithms and the targets are ranked according to the strength of predictions and the number of miRNAs that target them. The targets are prioritized based on their expression in the relevant cell line as well as their impact on the survival of the same cell line (siRNA screens). (A, iv) Interaction analysis: the final list of targets can be further analyzed using internal and external gene enrichment tools. (B) A list of all external data incorporated in the miRNA function database. Detailed summary can be found in the Supplementary Methods.
When searching for potential novel cancer therapy targets, it is important to consider effects on non-cancerous tissues and resultant toxicity in patients. We therefore conducted an additional screen in the non-transformed breast cell line MCF10A as well as the lung fibroblasts IMR-90. We incorporated these with a published dataset generated in cardiomyocytes (16) into the database (Figure 2A, ii). These data can be used to filter for miRNA with lethality restricted to cancer cells.
Considering that each miRNA can target hundreds of genes simultaneously, predicting targets for a single miRNA can be a challenging task. However, our screen uncovered large numbers of miRNAs inducing the same cellular phenotype and we expect these to have overlapping targets, allowing the ranking and prioritization of targets through integrated analysis. A series of filters can be applied to assist in target identification. First is the choice of miRNA-target prediction algorithm and thresholds (Figure 2A, iii). We incorporated several widely used computational algorithms including TargetScan, DIANA and MiRDB. Importantly, we also included already validated miRNA–mRNA interactions from high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP) and crosslinking, ligation and sequencing of hybrids (CLASH) experiments stored in StarBase and MirTarBase databases. A statistical weighting can be applied to any one of these datasets to suit the research question.
Although high-throughput methods for experimental target identification greatly improve miRNA target prediction, they are typically performed in one or few cell lines. Context dependency of miRNA actions, however, limits their broader use and requires additional cell- or tissue-specific filters. We therefore incorporated into the database RNA sequencing data for both coding and non-coding targets in all cell lines allowing for the option to remove targets that are expressed below a specified threshold (Figure 2A, iii). Finally, to increase the confidence of finding targets that mediate miRNA tumor-suppressor activity, we incorporated the results of large siRNA essential screens from the DPSC-cancer database (Figure 2A, iii). This database provides data on the requirement for cellular survival of over 16 000 genes in cancer cells and can be used to determine the requirement for each predicted miRNA target in cell survival.
After selecting preferred filtering options, an analysis reaches the ‘Target analysis’ page (Supplementary Figure S4). A summary is provided of how many miRNAs and targets remain after filtering and the number of each biotype of predicted targets (e.g. protein-coding, lincRNA, snoRNA). The data tab displays a table with all the targets and their unique score, a metric that takes into account the number of miRNAs targeting a specific gene as well as the number of databases providing evidence for that relationship. The ‘Heatmap’ tab gives an immediate overview of the relationship between miRNAs and targets for a specified number of targets. Finally, the ‘Expression’ tab provides unsupervized clustering of all the cell lines in the database according to target expression levels.
The final filtered set of miRNAs and targets can be further analyzed and visualized using custom internal and external methods, including pathway enrichment tools. The database also provides summary heatmaps to visualize miRNAs, targets and pathways. Alternatively, a selected target list can be analyzed using Reactome API or three external tools Enrichr, DAVID and String DB. Finally, we included a Biodalliance genome browser track (http://www.biodalliance.org/), which automatically displays the selected target and their location in the genome. For further details about site construction, data origin and database structure see Supplementary Data. Also, to fully explore options within the database, follow the page-specific tours and help buttons.
Data summary across cancer types
We next used the database and its inbuilt features to take a closer look at lethal miRNA mimics in each of the analyzed cancer types. As noted previously, differences in miRNA mimic activities were evident not only between diseases (Figure 1C) but also subtypes or cell lines of the same cancer type (Figure 3A). Breast cancer cell lines, for instance, displayed widely different profiles with triple-negative cell lines MDA-MB-231 and MDA-MB-468 showing significantly higher number of hits and a different set of lethal miRNA compared with ER+ and HER+ cell lines BT474 and SK-BR-3, respectively. Prostate cancer cell lines PC3 and DU145, which are both castrate-resistant and androgen signaling independent showed less divergence, and hundreds of mimics acted as potent tumor-suppressors in each of the cell lines. Similarly, neuroblastoma cell lines KELLY (MYCN amplified) and SHEP (MYCN WT) displayed comparably high number of lethal miRNA mimic hits. These results reflect at least in part the heterogeneous biology of different tumor types and subtypes.
Figure 3.

Data summary across different diseases. (A) Venn diagrams depict number of hits and their intersection across different cell types in each of the diseases. (B) For each cancer type, the graphs listing the most enriched miR families (three or more members) among the screen hits (at a viability cut-off ≤0.5) are presented. The white portion of the bars shows the number of lethal miR family members irrespective of the cancer type (general) and the black portion shows the members that were disease specific (disease-specific). The graphs are adapted from the database (enriched families with two members were omitted for better legibility). (C) The disease-specific miR family members were subjected to target prediction analyzed for the enrichment of KEGG pathways. The enrichment was regarded as significant at False Discovery Rate (FDR) ≤0.05.
To get a broad overview of each of the cancer types’ biology, we searched for miR candidates that were lethal (viability cut-off ≤0.5) when overexpressed in cell lines representing a particular cancer type. The database displays a list of all miRNA mimics and inhibitors acting at a specified cut-off and groups them according to their membership of sequence-related miRNA families. Figure 3B displays part of this data and shows miR families that were enriched for lethal miR candidates in a particular disease. Most of the candidates were hits in more than one cancer type (white bars), but in each of the diseases there were miR family members potently killing only the cells of the particular type (Figure 3B, black bars). For instance, members of the miR-515, miR-506 and miR-154 families were active across different diseases, but other members showed specificity for a particular cancer type, suggesting the existing of unique targets between members of these families.
We next used the database to look at the predicted targets of the most enriched miRNA families specific for each of the cancer types. The targets were ranked according to their database score and the top 100 targets were analysed for the enrichment of Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways (Figure 3C).
In breast cancer cell lines, the only significantly enriched pathways were FoxO and TGF-β, and the predicted targets mediating these pathways included among others TGFBR2, THBS1, BMPR2, SP1 as well as several apoptosis regulators such as BCL2L11, DDX3X and MCL1 (Supplementary figures S5a and S6). The miRNAs predicted to regulate targets in these pathways included among others miR-181b-5p and miR-181d-5p (miR-181 family) as well as miR-99b-5p (miR-10 family), which have been shown to operate within the TGF-β signaling axis in normal breast cells and breast cancer cells (17,18). Additional miR families with the predicted activity in the pathway were miR-25 (miR-25-3p and miR-92a-3p) and miR-515 family (miR-525-3p, miR-524-3p and miR-520f-3p) as well as the additional member of the miR-10 family (miR-125a-5p) (Supplementary Figure S5a).
In neuroblastoma cells, where numerous disease-specific lethal miRNAs clustered into miRNA families, the top enriched KEGG pathways included some of the same pathways appearing in the breast cancer cell line (Figure 3C and Supplementary Figure S5c), however the targets driving enrichment of these pathways were different and over-represented for those known to be neuroblastoma specific or important in this disease. Enriched pathways include the insulin-like growth factor pathway, which regulates proliferation and survival in many pediatric cancers including neuroblastoma (19) (Supplementary Figure S6c). Foxo pathway and Foxo transcription factors, in addition, regulate differentiation of neuroblastoma cells (20). This target signature was largely driven by the let-7 family of miRNAs, established tumor-suppressors in neuroblastoma aetiology (21), but other miRNAs families such as miR-506 (miR-507, miR-508-3p, miR-511), miR-27 (miR-27a-3p, miR-27b-5p) and miR-15 (miR-15b-5p, miR-195-5p) were strongly predicted to regulate these targets (Supplementary Figures S5c and 6c). Interestingly, melanoma and glioma KEGG pathways were also enriched, perhaps reflecting their shared neural (glioma) and neural crest (melanoma) origin with neuroblastoma.
Prostate cancer cell lines displayed the least disease-specific lethal miRNAs, but we observed the miR-500 family to be enriched among these hits with three members potently killing up to 80% of cells (miR-501-5p, miR-500a-5p and miR-500b-5p). Interestingly, none of the miR-500 candidates showed activity in normal MCF10A and cardiac cells. This family has not been described before in the context of prostate disease.
Although the enrichment analysis of predicted miRNA targets did not show any significantly enriched KEGG pathways after adjusting for multiple testing (Figure 3C), pathways related to fatty acid metabolism showed enrichment trends driven by gene targets such as ACADSB, ACSL6, SCP2 and PEX2 (Supplementary Figures S5b and 6b). Fatty acid metabolism is a dominant bioenergetics pathway in prostate cancer cells and increased lipid synthesis is associated with cell growth, survival and drug resistance (22). Based on our data and database predictions, miR-500 family members and their targets may be novel candidates to be investigated in prostate cancer.
Identification of lethal microRNA and their targets in neuroblastoma
To validate that our dataset and database can identify previously known biology as well as generate new biological insights, we focused on miRNAs acting potently as tumor-suppressors in neuroblastoma. For this purpose, we queried screen results in the KELLY cell line, which models poor-prognosis MYCN-amplified disease and selected only miRNAs that killed at a very stringent normalized viability of ≤0.3 relative to controls. The resulting list of 184 miRNA mimics contained 13 miRNA already shown to act as tumor-suppressors in neuroblastoma and to supress MYCN expression (23), including nine members of the let-7 family in addition to miR-19b-1-5p, miR-34a-3p, miR-101-3p and miR-202-3p (Figure 4A and Supplementary Figure S7a). Interestingly, let-7 family members were only lethal in neuroblastoma cell lines. Next, we looked at the most commonly shared predicted targets between these miRNAs and filtered them according to their expression levels in KELLY cells (RPKM >1 for protein coding genes). Remarkably, numerous genes were predicted to be targeted by a majority of these miRNA, suggesting the existence of a highly interconnected network of miRNA and their targets (Supplementary Figure S7b). For instance, out of the top 100 predicted targets, 7 are known to be major cancer-related genes and are targeted by at least 70% of these miRNAs.
Figure 4.

Validation of the screening and data analysis pipeline. (A) A heatmap showing the lethal effect (normalized viability values) of the miRNAs known to affect neuroblastoma survival through targeting MYCN. Red color signifies strong lethal effect and white color no effect on cell survival. (B) The list of top 20 targets most commonly shared between these miRNAs as computed by our database. The targets were ranked according to the strength of predictions and filtered based on their expression levels in KELLYs (RPKM > 1). (C) KELLY cells were transfected with let-7b-5p, miR-101-3, miR-202-3p or NT, and changes in mRNA levels of MYCN and LIN28B were assessed using qPCR. B2M was used as a housekeeping gene. The graph plots gene expression (dCt expression) relative to B2M and is representative of three independent experiments. The significance was determined using one-way ANOVA with Dunnett’s multiple comparisons test (n = 3, *P-value ≤ 0.05).
These targets were enriched for genes with well-described roles in oncology (KEGG enrichment tab; data not shown) including DICER1, DUSP1, TGFBR1 and EZH2 (Figure 4B). Importantly, MYCN was one of the top targets, agreeing with previously published data. In addition, several other bona fide oncogenes in neuroblastoma appeared among the top targets. LIN28B is a well-known target of the let-7 family of miRNAs, and has been recently shown to play a major role in neuroblastoma development through a feedback loop involving let-7 miRNAs and MYCN (24). IGF2BP1, another top candidate in our list, is highly expressed in neuroblastoma, frequently associated with MYCN amplification and shown to strongly promote MYCN expression levels (25). To check whether these genes are truly miRNA targets in our system, we chose three representative miRNA, let-7b-5p, miR-101-3p and miR-202-3p, transfected the KELLY cell line with miRNA mimics and examined the expression of MYCN and LIN28B by reverse transcriptase PCR. Both MYCN and LIN28B mRNA levels were significantly reduced as predicted by our database (Figure 4C). Therefore our database effectively identifies previously validated lethal miRNA and their targets ab initio.
Identification of novel micro-RNA targets essential for neuroblastoma survival
Experimental techniques for miRNA target identification unequivocally demonstrate that individual miRNAs can interact with many mRNAs within the cell. We therefore searched for additional targets of these validated lethal miRNA that might play a significant and specific role in neuroblastoma aetiology. The database-predicted targets of MYCN-targeting miRNAs (Supplementary Figure S7) were significantly enriched for two KEGG pathways, ‘miRNAs in cancer’ and ‘signaling pathways regulating pluripotency of stem cells’ (Supplementary Figure S8a). When we clustered these predicted targets according to their expression level across the panel of eight cell lines (‘Expression’ tab), we noticed a number of targets in addition to MYCN and LIN28B with high expression in the KELLY cell line but very low expression in breast and prostate cancer (Supplementary Figure S8b; first 50 targets displayed). We chose four such factors (HAND1, RGS16, RBFOX2 and ACVR2B) that exhibited high expression levels in other neuroblastoma cell lines (data derived from Oncomine) and were not previously implicated in neuroblastoma biology for further analysis. We again transfected KELLY cells with miR-101-3p, miR-202-3p and let-7b-5p mimics and measured the mRNA levels of HAND1, RGS16, RBFOX2 and ACVR2B. In agreement with the predictions, miRNA overexpression downregulated the majority of targets (Figure 5A). Mining of high-throughput CLASH and HITS-CLIP experiments revealed multiple lines of experimental evidence for direct binding between these miRNA–mRNA pairs (26,27). We confirmed these results using luciferase-based reporter assays for HAND1 and RGS16 (Figure 5B). Transfection of let-7b-5p significantly reduced the luciferase signal when co-transfected with a vector carrying WT 3′ UTR of both HAND1 and RGS16. This effect was completely abolished in the presence of RGS16 3′ UTR with a mutated let-7b-5p-binding site, and only partially rescued in the presence of the mutated HAND1 3′ UTR, suggesting additional binding sites for this miRNA. To determine whether endogenous Let-7b can target HAND1 and RGS16, we selected the MDA-MB-231 cell line, which expresses high levels of endogenous let-7b-5p miRNA and co-transfected it with let-7b-5p inhibitor and WT or mutant HAND1 and RGS16 reporter vectors. In both cases, the presence of let-7b-5p inhibitor increased the luciferase signal of the WT let-7b-5p reporter but not the mutant reporter (Figure 5B). These data validate our discovery approach by demonstrating that Let-7b can directly target HAND1 and RGS16 through binding to specific sites in the 3′ UTR.
Figure 5.

Novel let-7 targets regulate the survival of neuroblastoma cancer cells. (A) KELLY cells were transfected with let-7b-5p, miR-101-3, miR-202-3p or NT, and changes in mRNA levels of HAND1, RGS16, ACVR2B and RBFOX2 were assessed using qPCR. B2M was used as a housekeeping gene. The graph plots gene expression (dCt expression) relative to B2M and is representative of three independent experiments. The significance was determined using one-way ANOVA with Dunnett’s multiple comparisons test (n = 3, *P-value ≤ 0.05). (B) HEK293T and MDA-MB-231 cells were co-transfected with NT or let-7b-5p mimic or inhibitor, and HAND1 or RGS16 reporter plasmid encoding either WT or mutated 3′ UTR. Twenty-four hours later, the cell lysates were prepared and subjected to dual luciferase assay to measure both firefly (control) and renilla luciferase (experimental signal). The graph plots fold-change relative to NT control and is representative of three independent experiments. The significance was determined using two-way ANOVA with Sidak’s multiple comparisons test (n = 3, *P-value ≤ 0.05). (C) KELLY cells were transfected with siRNAs against MYCN, LIN28B, HAND1, RGS16, ACVR2B, RBFOX2 or NT control in 384-well format. The images of the wells were taken every 2 h for 3 days using IncuCyte ZOOM system. Cell viability was determined using CellTiter-Glo luminescent assay. Both cell confluence and cell viability were plotted as values normalized to the NT control, and each graph is a representative of three independent experiments. The significance was determined using one-way ANOVA with Dunnett’s multiple comparisons test (*P-value ≤ 0.05).
We next sought to investigate whether these factors have an impact on neuroblastoma survival. Knockdown of all four of the novel factors reduced either cell confluence and/or ATP content, similar to knockdown of MYCN or LIN28B (Figure 5C). In particular, RGS16 and RBFOX2 siRNAs led to a dramatic decrease of KELLY cell viability in all biological replicates and assay types, suggesting that these previously unrecognized factors play a fundamental role in neuroblastoma cell viability. These data suggest that miRNAs can be used as a novel perturbation to discover protein-coding genes important in disease.
Identification of novel microRNAs with a role in neuroblastoma aetiology
We next mined our functional screening data to uncover previously unrecognized miRNAs important for neuroblastoma biology. We queried the database for other miRNA families that effectively kill KELLY or SHEP cells at the original cut-off of normalized viability ≤0.5. The most enriched family besides let-7 miRNAs was the miR-515 family with 14 members exhibiting a potent tumor-suppressor effect, which has not been previously described in neuroblastoma (Figure 3B and Figure 6A). Seven of these were selectively lethal to neuroblastoma, particularly MYCN-amplified KELLY cells (Figure 6A). To investigate the underlying mechanism of their lethality, we applied target prediction algorithms to the 14 miR-515 family members and obtained a list of ranked high-confidence targets clustered based on their expression in our panel of cancer cell lines (Supplementary Figure S9a). Many of the targets were expressed across all cell lines, but several were specifically expressed in KELLY cells. One such mRNA was the transcription factor SOX11 (database score 0.29; targeted by 5/14 miRNAs). While a role for SOX11 in neuroblastoma is not described, it is essential for the development of sympathetic neural ganglia, a likely cell of origin for some neuroblastoma (28). Another top candidate predicted to be targeted by 5/14 miRNAs (database score 0.36) was ISL-1, a LIM homeobox transcription factor highly expressed in primary neuroblastoma tumors and cell lines compared to other tumor types (Supplementary Figure S9b). In agreement with the database prediction, transfection with miR-517a-5p mimic downregulated Isl1 levels in KELLY cells (Figure 6B). In addition, mining of HITS-CLIP data (29) demonstrates direct binding of miR-517a-5p to Isl1 mRNA. We confirmed this finding by performing a reporter assay for ISL1. Transfection of miR-517a significantly reduced the luciferase signal when co-transfected with a vector carrying WT 3′ UTR and this affect was completely abolished in the presence of a vector carrying the 3′ UTR with a mutated miR-517a-binding site (Figure 6C). Finally, we wanted to determine whether ISL1 plays a role in neuroblastoma survival as this has not been previously shown. Indeed, transfection with siRNA against ISL1 markedly reduced the confluence of KELLY cell cultures (Figure 6D and Supplementary Figure S9c). ISL1 knockdown also reduced proliferation based on a reduced S-phase fraction in DNA histograms (Figure 6E). In addition, ISL1 knockdown led to a dramatic change in cellular morphology into tightly packed clusters of rounded cells (Supplementary Figure S9c) suggesting changes in differentiation. We measured two differentiation markers of the neuronal lineage previously reported to increase in KELLY cells upon differentiation, NTRK1 and NEFL. Knockdown of ISL1 reproducibly increased the levels of NTRK1, a neurotrophic tyrosine kinase receptor known to mediate neuroblastoma differentiation in different models (30,31), suggesting a role for ISL1 in neuroblastoma differentiation (Figure 6F). Taken together, our functional screen and miRNA target analysis uncovered a previously unrecognized role for the miR-515 family and its novel targets in neuroblastoma.
Figure 6.

Novel determinants of neuroblastoma cell survival. (A) A heatmap showing the lethal effect (normalized cell viability) of the miR-515 family members in all screened cancer cell lines. ‘Count’ on the histogram represents the frequency of miRNAs showing a particular lethal effect. Seven family members are hits (normalized viability cut-off ≤0.5) only in KELLY cells. Red color signifies strong lethal effect and white color no effect on cell survival. (B) KELLY cells were transfected with miR-517a-3p or NT control, and changes in mRNA levels of ISL1 were assessed using qPCR. B2M was used as a housekeeping gene. The graph plots gene expression (dCt expression) relative to B2M and is representative of three independent experiments. The significance was determined using a t-test (n = 3, *P-value ≤ 0.05). (C) HEK293T cells were co-transfected with NT or miR-517a and reporter plasmid encoding either WT or mutated ISL1 3′ UTR. Twenty-four hours later, the cells lysates were prepared and subjected to dual luciferase assay to measure both firefly (control) and renilla luciferase (experimental signal). The graph plots fold-change relative to NT control and is representative of three independent experiments. The significance was determined using two-way ANOVA with Sidak’s multiple comparisons test (n = 3, *P-value ≤ 0.05). (D) KELLY cells were transfected with a SMARTpool siRNA against ISL1 or NT control in a 384-well format. The images of the wells were taken every 2 h over 3 days using the IncuCyte ZOOM system. Cell confluence was plotted as values normalized to the NT control and is representative of three independent experiments. The significance was determined using a t-test (*P-value ≤ 0.05). (E) KELLY cells were transfected with a SMARTpool siRNA against ISL1 or NT control, and 96 h later, cells were fixed and stained with PI. Cell cycle profiles were determined using Canto I and FlowJo for data analysis. Histograms are representative of three independent experiments. (F) KELLY cells were transfected with a SMARTpool siRNA against ISL1 or NT control, and changes in mRNA levels of differentiation markers NEFL and NTRK1 were assessed using qPCR. B2M was used as a housekeeping gene. The graph plots gene expression levels relative to B2M and is representative of three independent experiments. The significance was determined using two-way ANOVA with Sidak’s multiple comparisons test (*P-value ≤ 0.05).
DISCUSSION
miRNA target hundreds of transcripts within a cell and regulate the most fundamental cellular processes including tumorigensis and cancer cell survival. The large majority of studies rely on miRNA expression changes to identify the most relevant miRNAs in this context, which does not necessarily identify functionally important miRNAs. One solution to this problem is to use high-throughput functional screening approaches, and in the last several years, there have been a number of reports that applied this methodology to systematically explore miRNA function in human cancers (32–35). All these studies, however, focused on one specific context or cancer type (and typically only one cell line model) and analysed a limited subset of miRNA, typically <400. They all have limited integration with other datasets such as target predictions, miRNA and mRNA expression and siRNA screens. Here, we greatly expand these efforts by exploring the function of 1280 miRNA across eight cell lines representing three cancer types. In addition, we provide an easy-to-use environment to interrogate this data and perform downstream analyses. The result is the identification of hundreds of miRNAs with previously unknown roles in cancer proliferation and survival and the discovery of novel miRNA targets required in disease. Two important caveats of our study are worth noting: the first is that proliferation in 2D cell culture was used as our screening endpoint, and we did not measure the impact of miRNAs on other phenotypes important to cancer, such as self-renewal or invasion. These endpoints may be incorporated into the database through subsequent studies. Second, due to the nature of the screen, increases in proliferation or survival following miRNA mimic or inhibitor transfection may not have been detected.
One of the difficulties in exploring miRNA biology is identification of direct targets. Experimental methods for verifying direct interactions are laborious and time-consuming while computational algorithms generate numerous false positives and negatives. An important feature of our database is the integration of multiple orthogonal datasets to allow users to parse the list of predicted targets and rank them according to the strength of predictions and additional information available for that particular target. We based these filtering steps on several assumptions. First, mRNAs containing canonical seed matches—which are therefore more likely to be predicted by multiple computational algorithms—are also more likely to be genuine mRNA targets. Indeed, recent reports demonstrate more significant repression by miRNAs of targets containing 8 and 7 mer seed sites compared to the ones containing only 6 mer matches or non-canonical binding sites (36). Predicted targets in our database are initially assigned a score based on how many algorithms recognized them as targets. Importantly, the users are able to complement this by giving more weight to experimentally derived targets. A second important assumption is that miRNAs exerting similar lethal effects will function through overlapping targets. Indeed, miRNA co-targeting has been found to be ubiquitous among miRNAs, which frequently function together to regulate target-gene expression (37). For a given set of miRNAs, our database can be used to search for the most commonly shared targets, which are assigned a higher database score and therefore rank higher then the less connected targets. The list of targets can be additionally customized by using gene expression data to eliminate low abundance mRNA. This is an important feature of the database considering the context-dependency of miRNA regulatory activity (38). It should be noted that a limitation of our database is that targets repressed via non-seed based mechanisms will be poorly represented in predictions.
Previous findings demonstrated that malignant tissues display widespread downregulation of miRNAs related to a less differentiated phenotype (5), suggesting that miRNA on the whole are tumor-suppressive. Consistent with this, more than 40% of miRNA mimics were active in at least one cell line, and in most cell lines we observed hundreds of miRNA mimics with a lethal effect. One could argue, however, that such dramatic effects result from an overexpression of miRNA mimics upon transfection, causing non-physiological effects that would not be otherwise present in these tissues. We note two important points of discussion here. First, transfection with candidate miRNA mimics at markedly lower concentrations recapitulated the potent lethal effects observed at higher concentrations (data not shown). Furthermore, it has been previously shown that upon transfection a large portion of miRNA mimic molecules ends up in vesicles and is therefore not functional while the functional fraction, associated with the RISC, is comparable to the most abundant endogenous miRNAs (39).
Second, only a fraction of lethal miRNAs from our screens are likely to function as tumor-suppressors under physiological conditions, and this screen data and associated analytical tools should be seen as a starting point for further analysis of candidate miRNA networks arising from this perturbation screen. Regardless of their endogenous role, miRNAs with potent activity in this screen may be important candidate therapeutics for miRNA replacement therapy.
An important observation from our analyses is the diverse and specific activity of miRNA family members across disease models. While family members would be predicted to act similarly due to their sequence homology, some members of the miR-515 family, for instance, showed activity in all three diseases, while other members had activity restricted to breast cancer or neuroblastoma models. For these miRNAs, the predicted targets are different, and are enriched for adhesion signaling molecules and the RAS-MAPK/PI3K-Akt signaling axis, respectively (data not shown and Figure 3C). Although these predictions and their importance for the disease would have to be experimentally validated, this example illustrates a complex relationship between miRNAs and their targets, and the underlying effect of the local transcriptional landscape.
We used neuroblastoma to test the value of our miRNA functional database in identifying not just new miRNA, but also novel protein-coding targets controlling cancer aetiology. We identified numerous miRNA previously associated with neuroblastoma to also be lethal in our screen, thus validating our screening approach. This included the let-7 family of miRNAs, which interestingly we find to be lethal specifically in MYCN-amplified neuroblastoma (KELLY cell line) and not in breast or prostate cancer cell lines (data not shown). We identified novel let-7 targets that might be involved in neuroblastoma initiation or maintenance in addition to the well-characterized MYCN and LIN28B oncogenes. RGS16 belongs to a family of G-protein signaling regulators with some members already implicated in human cancer (40). Furthermore, RBFOX2 acts as a splicing regulator involved in stem cell differentiation as well as regulation of cancer-specific splicing signatures (41,42). More detailed investigation of the role for these two genes in neuroblastoma cell differentiation, proliferation and survival in long-term assays is warranted.
We also identified several members of the miR-515 seed family with a novel role in neuroblastoma. Interestingly, five of these miRNA target ISL-1, a LIM homeobox transcriptional coactivator, which is expressed by cardiac neural crest derivatives and is important for cardiac progenitor survival during development (43,44). ISL1 is expressed by pancreatic well-differentiated neuroendocrine neoplasms, but in extrapancreatic tumors, it appears to be restricted to neuroblastomas (45). Profiling of 48 stage four neuroblastoma tumors revealed ISL1 to be a reliable marker of minimal residual disease (46). For the first time, we show here a functional role for ISL1 in neuroblastoma cell survival most likely through regulating the switch between cellular proliferation and differentiation. Further experiments are needed to pinpoint ISL1 transcriptional targets and clarify its relationship with other known determinants of neuroblastoma proliferation and differentiation.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the assistance of Gillian Lehrbach and Jess Yang with certain experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Kid’s Cancer Project (to A.S.); National Breast Cancer Foundation (to A.S.); McMurtrie family (to A.S.); Prostate Cancer Foundation Australia (to A.S.). AS is the recipient of a career development award from the National Health and Medical Research Council. ED is the recipient of an Australian Postgraduate Award. The Victorian Centre for Functional Genomics (K.J.S.) was funded by the Australian Cancer Research Foundation (ACRF); Victorian Department of Industry, Innovation and Regional Development (DIIRD); Australian Phenomics Network (APN); Australian Government’s Education Investment Fund through the Super Science Initiative; Australasian Genomics Technologies Association (AGTA); Brockhoff Foundation and the Peter MacCallum Cancer Centre Foundation. Funding for open access charge: NBCF Grant funding.
Conflict of interest statement. None declared.
REFERENCES
- 1. Bushati N., Cohen S.M.. microRNA functions. Ann. Rev. Cell Dev. Biol. 2007; 23:175–205. [DOI] [PubMed] [Google Scholar]
- 2. Bernstein E., Kim S.Y., Carmell M.A., Murchison E.P., Alcorn H., Li M.Z., Mills A.A., Elledge S.J., Anderson K.V., Hannon G.J.. Dicer is essential for mouse development. Nat. Genetics. 2003; 35:215–217. [DOI] [PubMed] [Google Scholar]
- 3. Yi R., Poy M.N., Stoffel M., Fuchs E.. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature. 2008; 452:225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cimmino A., Calin G.A., Fabbri M., Iorio M.V., Ferracin M., Shimizu M., Wojcik S.E., Aqeilan R.I., Zupo S., Dono M. et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lu J., Getz G., Miska E.A., Alvarez-Saavedra E., Lamb J., Peck D., Sweet-Cordero A., Ebert B.L., Mak R.H., Ferrando A.A. et al. MicroRNA expression profiles classify human cancers. Nature. 2005; 435:834–838. [DOI] [PubMed] [Google Scholar]
- 6. Ling H., Fabbri M., Calin G.A.. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013; 12:847–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dvinge H., Git A., Graf S., Salmon-Divon M., Curtis C., Sottoriva A., Zhao Y., Hirst M., Armisen J., Miska E.A. et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature. 2013; 497:378–382. [DOI] [PubMed] [Google Scholar]
- 8. Molenaar J.J., Koster J., Zwijnenburg D.A., van Sluis P., Valentijn L.J., van der Ploeg I., Hamdi M., van Nes J., Westerman B.A., van Arkel J. et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012; 483:589–593. [DOI] [PubMed] [Google Scholar]
- 9. Birmingham A., Selfors L.M., Forster T., Wrobel D., Kennedy C.J., Shanks E., Santoyo-Lopez J., Dunican D.J., Long A., Kelleher D. et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat. Methods. 2009; 6:569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capece V., Garcia Vizcaino J.C., Vidal R., Rahman R.U., Pena Centeno T., Shomroni O., Suberviola I., Fischer A., Bonn S.. Oasis: online analysis of small RNA deep sequencing data. Bioinformatics. 2015; 31:2205–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Erhard F., Haas J., Lieber D., Malterer G., Jaskiewicz L., Zavolan M., Dolken L., Zimmer R.. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014; 24:906–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mullokandov G., Baccarini A., Ruzo A., Jayaprakash A.D., Tung N., Israelow B., Evans M.J., Sachidanandam R., Brown B.D.. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat. Methods. 2012; 9:840–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song H., Zhang Y., Liu N., Zhao S., Kong Y., Yuan L.. miR-92a-3p exerts various effects in glioma and glioma stem-like cells specifically targeting CDH1/beta-catenin and notch-1/Akt signaling pathways. Int. J. Mol. Sci. 2016; 17:1799–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tong Z., Liu N., Lin L., Guo X., Yang D., Zhang Q.. miR-125a-5p inhibits cell proliferation and induces apoptosis in colon cancer via targeting BCL2, BCL2L12 and MCL1. Biomed. Pharmacother. 2015; 75:129–136. [DOI] [PubMed] [Google Scholar]
- 15. Nakata W., Uemura M., Sato M., Fujita K., Jingushi K., Ueda Y., Kitae K., Tsujikawa K., Nonomura N.. Expression of miR-27a-3p is an independent predictive factor for recurrence in clear cell renal cell carcinoma. Oncotarget. 2015; 6:21645–21654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eulalio A., Mano M., Dal Ferro M., Zentilin L., Sinagra G., Zacchigna S., Giacca M.. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012; 492:376–381. [DOI] [PubMed] [Google Scholar]
- 17. Taylor M.A., Sossey-Alaoui K., Thompson C.L., Danielpour D., Schiemann W.P.. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J. Clin. Invest. 2013; 123:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Turcatel G., Rubin N., El-Hashash A., Warburton D.. MIR-99a and MIR-99b modulate TGF-beta induced epithelial to mesenchymal plasticity in normal murine mammary gland cells. PLoS One. 2012; 7:e31032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang F., Greer A., Hurlburt W., Han X., Hafezi R., Wittenberg G.M., Reeves K., Chen J., Robinson D., Li A. et al. The mechanisms of differential sensitivity to an insulin-like growth factor-1 receptor inhibitor (BMS-536924) and rationale for combining with EGFR/HER2 inhibitors. Cancer Res. 2009; 69:161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mei Y., Wang Z., Zhang L., Zhang Y., Li X., Liu H., Ye J., You H.. Regulation of neuroblastoma differentiation by forkhead transcription factors FOXO1/3/4 through the receptor tyrosine kinase PDGFRA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:4898–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Powers J.T., Tsanov K.M., Pearson D.S., Roels F., Spina C.S., Ebright R., Seligson M., de Soysa Y., Cahan P., Theissen J. et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature. 2016; 535:246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brusselmans K., De Schrijver E., Verhoeven G., Swinnen J.V.. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 2005; 65:6719–6725. [DOI] [PubMed] [Google Scholar]
- 23. Buechner J., Tomte E., Haug B.H., Henriksen J.R., Lokke C., Flaegstad T., Einvik C.. Tumour-suppressor microRNAs let-7 and mir-101 target the proto-oncogene MYCN and inhibit cell proliferation in MYCN-amplified neuroblastoma. Br. J. Cancer. 2011; 105:296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Molenaar J.J., Domingo-Fernandez R., Ebus M.E., Lindner S., Koster J., Drabek K., Mestdagh P., van Sluis P., Valentijn L.J., van Nes J. et al. LIN28B induces neuroblastoma and enhances MYCN levels via let-7 suppression. Nat. Genet. 2012; 44:1199–1206. [DOI] [PubMed] [Google Scholar]
- 25. Bell J.L., Turlapati R., Liu T., Schulte J.H., Huttelmaier S.. IGF2BP1 harbors prognostic significance by gene gain and diverse expression in neuroblastoma. J. Clin. Oncol. 2015; 33:1285–1293. [DOI] [PubMed] [Google Scholar]
- 26. Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H.. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014; 42:D92–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang J.H., Li J.H., Shao P., Zhou H., Chen Y.Q., Qu L.H.. starBase: a database for exploring microRNA-mRNA interaction maps from argonaute CLIP-seq and degradome-seq data. Nucleic Acids Res. 2011; 39:D202–D209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Potzner M.R., Tsarovina K., Binder E., Penzo-Mendez A., Lefebvre V., Rohrer H., Wegner M., Sock E.. Sequential requirement of Sox4 and Sox11 during development of the sympathetic nervous system. Development. 2010; 137:775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xue Y., Ouyang K., Huang J., Zhou Y., Ouyang H., Li H., Wang G., Wu Q., Wei C., Bi Y. et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell. 2013; 152:82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matsushima H., Bogenmann E.. Expression of trkA cDNA in neuroblastomas mediates differentiation in vitro and in vivo. Mol. Cell. Biol. 1993; 13:7447–7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lavenius E., Gestblom C., Johansson I., Nanberg E., Pahlman S.. Transfection of TRK-A into human neuroblastoma cells restores their ability to differentiate in response to nerve growth factor. Cell Growth Differ. 1995; 6:727–736. [PubMed] [Google Scholar]
- 32. Poell J.B., van Haastert R.J., de Gunst T., Schultz I.J., Gommans W.M., Verheul M., Cerisoli F., van Noort P.I., Prevost G.P., Schaapveld R.Q. et al. A functional screen identifies specific microRNAs capable of inhibiting human melanoma cell viability. PLoS One. 2012; 7:e43569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nakano H., Yamada Y., Miyazawa T., Yoshida T.. Gain-of-function microRNA screens identify miR-193a regulating proliferation and apoptosis in epithelial ovarian cancer cells. Int. J. Oncol. 2013; 42:1875–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leivonen S.K., Sahlberg K.K., Makela R., Due E.U., Kallioniemi O., Borresen-Dale A.L., Perala M.. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol. Oncol. 2014; 8:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hatano K., Kumar B., Zhang Y., Coulter J.B., Hedayati M., Mears B., Ni X., Kudrolli T.A., Chowdhury W.H., Rodriguez R. et al. A functional screen identifies miRNAs that inhibit DNA repair and sensitize prostate cancer cells to ionizing radiation. Nucleic Acids Res. 2015; 43:4075–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bracken C.P., Li X., Wright J.A., Lawrence D.M., Pillman K.A., Salmanidis M., Anderson M.A., Dredge B.K., Gregory P.A., Tsykin A. et al. Genome-wide identification of miR-200 targets reveals a regulatory network controlling cell invasion. EMBO J. 2014; 33:2040–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tsang J.S., Ebert M.S., van Oudenaarden A.. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Mol. Cell. 2010; 38:140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mukherji S., Ebert M.S., Zheng G.X., Tsang J.S., Sharp P.A., van Oudenaarden A.. MicroRNAs can generate thresholds in target gene expression. Nat. Genet. 2011; 43:854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thomson D.W., Bracken C.P., Szubert J.M., Goodall G.J.. On measuring miRNAs after transient transfection of mimics or antisense inhibitors. PLoS One. 2013; 8:e55214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hurst J.H., Hooks S.B.. Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochem. Pharmacol. 2009; 78:1289–1297. [DOI] [PubMed] [Google Scholar]
- 41. Venables J.P., Brosseau J.P., Gadea G., Klinck R., Prinos P., Beaulieu J.F., Lapointe E., Durand M., Thibault P., Tremblay K. et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol. Cell. Biol. 2013; 33:396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Venables J.P., Lapasset L., Gadea G., Fort P., Klinck R., Irimia M., Vignal E., Thibault P., Prinos P., Chabot B. et al. MBNL1 and RBFOX2 cooperate to establish a splicing programme involved in pluripotent stem cell differentiation. Nat. Commun. 2013; 4:2480. [DOI] [PubMed] [Google Scholar]
- 43. Cai C.L., Liang X., Shi Y., Chu P.H., Pfaff S.L., Chen J., Evans S.. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell. 2003; 5:877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Engleka K.A., Manderfield L.J., Brust R.D., Li L., Cohen A., Dymecki S.M., Epstein J.A.. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circ. Res. 2012; 110:922–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Agaimy A., Erlenbach-Wunsch K., Konukiewitz B., Schmitt A.M., Rieker R.J., Vieth M., Kiesewetter F., Hartmann A., Zamboni G., Perren A. et al. ISL1 expression is not restricted to pancreatic well-differentiated neuroendocrine neoplasms, but is also commonly found in well and poorly differentiated neuroendocrine neoplasms of extrapancreatic origin. Mod. Pathol. 2013; 26:995–1003. [DOI] [PubMed] [Google Scholar]
- 46. Cheung I.Y., Feng Y., Gerald W., Cheung N.K.. Exploiting gene expression profiling to identify novel minimal residual disease markers of neuroblastoma. Clin. Cancer Res. 2008; 14:7020–7027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.