


Abstract
RNase H cleaves RNA in RNA–DNA duplexes. It is present in all domains of life as well as in multiple viruses and is essential for mammalian development and for human immunodeficiency virus replication. Here, we developed a sequencing-based method to measure the cleavage of thousands of different RNA–DNA duplexes and thereby comprehensively characterized the sequence preferences of HIV-1, human and Escherichia coli RNase H enzymes. We find that the catalytic domains of E. coli and human RNase H have nearly identical sequence preferences, which correlate with the efficiency of RNase H-recruiting antisense oligonucleotides. The sequences preferred by HIV-1 RNase H are distributed in the HIV genome in a way suggesting selection for efficient RNA cleavage during replication. Our findings can be used to improve the design of RNase H-recruiting antisense oligonucleotides and show that sequence preferences of HIV-1 RNase H may have shaped evolution of the viral genome and contributed to the use of tRNA-Lys3 as primer during viral replication.
INTRODUCTION
RNase H enzymes specifically hydrolyze RNA hybridized to DNA (1) and are present in all domains of life. They are essential for mammalian embryonic development (2,3) and proteins with RNase H activity are also found in retroelements and viruses, especially reverse transcribing viruses such as Retroviridae, Hepadnaviridae or Caulimoviridae. In these viruses, the RNase H activity helps remove the RNA template from a newly synthesized DNA strand (4). Humans have two enzymes exhibiting RNase H activity, RNase H1 and RNase H2, which differ in their structure and substrate specificity (5). RNase H1 is a monomer, composed of a facultative mitochondrial targeting sequence, a catalytic domain, a connection domain and a hybrid-binding domain (HBD) and is required for mitochondrial replication (6). RNase H2 is a heterotrimer and is, unlike RNase H1, able to cleave at single ribonucleotides embedded in DNA, which is crucial for preserving genome integrity (3).
Apart from these important cellular functions, the RNase H enzymes are also essential for single-stranded oligonucleotide therapeutics, which function by hybridization to target RNA molecules, recruitment of RNase H and cleavage of the target RNA (7). Therapeutic RNase H-recruiting oligonucleotides are often referred to as gapmers, because they typically have a central DNA gap flanked by sugar modified nucleotides, such as locked nucleic acids (LNAs) and methoxyethyl (MOEs), to increase the binding affinity to the target RNA. The requirement of RNase H1 for gapmer activity in vivo is supported by overexpression and depletion experiments (8–10).
To optimize the design of therapeutic gapmers, parameters such as hybridization energy (11), toxic potential (12,13), RNA secondary structure (14) and the existence of off-targets (15,16), are analyzed in silico prior to experimental testing (17). Human RNase H1 is known to cleave certain sequences more efficiently than others (5), but the lack of quantitative information on these preferences makes it impossible to use them for gapmer design. Sequence preferences of RNase H domains of retroviral reverse transcriptases are better understood and are recognized to be functionally important (18). Apart from general degradation of the RNA strand of its genome, human immunodeficiency virus (HIV)-1 RNase H has to perform precise cleavages around polypurine tracts (PPT) and at the last bond of the tRNA-Lys3 primer during viral replication (19–21). The fold of the catalytic domain of human RNase H1 is remarkably similar to the fold of the Escherichia coli and HIV RNase H catalytic domain (22), making it very difficult to develop specific HIV-1 RNase H inhibitors. Recently, progress has been made, but so far no drug that targets RNase H has made it to the clinic (23). Thus, an improved understanding of the RNase H sequence preferences could facilitate both gapmer and antiviral drug design. Here, we present a novel method called RNase H Sequence Preference Assay (H-SPA), which we have used to comprehensively characterize sequence preferences of human RNase H1, E. coli RNase HI and the RNase H domain of HIV-1 reverse transcriptase.
MATERIALS AND METHODS
Oligonucleotides used in the study
Names and sequences of all the oligonucleotides used in the study are shown in Supplementary Table S2.
DNA–RNA–DNA/DNA duplex preparation
For the reverse transcription reaction, 500 pmol of oligonucleotides R7_FS, R4A_FS or R4B_FS were mixed with 1 nmol of reverse transcription primer (DSP for R7_FS, DSP2 for R4A_FS and R4B_FS) in the presence of 1 mM Tris–HCl pH 7 and 0.1 mM ethylenediaminetetraacetic acid (EDTA) in the volume of 100 μl and heated to 65°C for 5 min, followed by incubation at 21°C for 5 min and subsequently kept on ice. Next, 100 μl of enzyme-mix was prepared by mixing four volumes of 5× PrimeScript buffer, four volumes of H2O, one volume of PrimeScript reverse transcriptase (Takara) and one volume of 10 mM dNTP mix and added to the template-primer mixture, followed by incubation at 42°C for 30 min and transfer to ice. Reactions were stopped by addition of 50 μl 50 mM EDTA and 250 μl native elution (NE) Buffer (20 mM Tris–HCl pH 7.5, 20 mM KCl and 1 mM EDTA) and concentrated down to ∼75 μl using Amicon Ultra 3K filter devices (EMD Millipore). Electrophoresis loading buffer (15 μl; 10 mM Tris–HCl pH 7.5, 60% glycerol and 60 mM EDTA) was added to the samples, which were loaded on 20% native polyacrylamide gel (Acrylamide/Bis-acrylamide = 19:1; 1× Tris/Borate/EDTA (TBE); two lanes for each sample) and run for 21 h at constant voltage (150 V). The samples were visualized with blue-light transiluminator and the dominant bands (corresponding to DNA–RNA–DNA/DNA duplex) were cut out, crushed, soaked with 500 μl NE buffer, frozen at −80°C for 10 min and incubated overnight at 21°C with constant shaking (1000 rpm) in the presence of 200 μl phenol. Following elution from the gel, the buffer was extracted with chloroform, concentrated on Amicon Ultra 3K filter devices (elution from the second band added to the same filter unit and centrifuged again) and further purified on illustra S-200HR column (GE Healthcare) pre-equilibrated with NE buffer. The duplex structure of the representative substrate preparations was confirmed using native 20% electrophoresis by comparing to non-hybridized and hybridized oligonucleotides and DSP primer (but not reverse transcribed). Finally, concentration of each duplex was adjusted with NE buffer to 0.1 absorbance units at 260 nm with 10 mm path (which is approximately equivalent to 0.1 μM).
Duplex hydrolysis with RNase H enzymes
Hydrolysis reactions with recombinant human RNase H1 (Creative BioMart cat. RNASEH1-433H) were performed at 37°C in a buffer RNH1 (20 mM Tris–HCl pH 7.5, 20 mM KCl, 20 mM 2-mercaptoethanol, 2 mM MgCl2 and 0.1 mM EDTA (modified from Lima et al., 2001 (24)). Preheated solution containing 0.3 pmol of prepared duplex in 16.5 μl was mixed with preheated 16.5 μl of 0.5 mU/μl RNase H1 dilution to initiate the hydrolysis reaction to give a final enzyme concentration of 0.25 mU/μl. After 10 min, reactions were terminated by transferring them into a tube containing 160 μl precipitation mix (375 mM NaOAc pH 8, 1.25 mM EDTA and 62.5 ng/μl glycogen) and 200 μl phenol and vigorously shaking. Reactions were chloroform extracted, ethanol precipitated and resuspended in 10 μl TE (pH 7) buffer, out of which 2 μl were mixed with 10 μl formamide loading buffer (10 mM EDTA in formamide), resolved with denaturing polyacrylamide (8% PAA, 7M Urea, 1× TBE) electrophoresis and visualized using Typhoon FLA 7000 scanner (GE Healthcare; detection of 6-carboxyfluorescein (FAM) fluorescence). Reactions with E. coli RNase H from Thermo Scientific (cat. EN0201) were performed at 37°C in a buffer composed of 50 mM Tris–HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol (DTT), 0.4 mg/ml bovine serum albumin and 0.1 mM EDTA using the same protocol as for the reactions with human-derived enzyme, but with a final enzyme concentration 0.5 mU/μl. Reactions with HIV-1 Reverse Transcriptase from Worthington Biochemical Corporations (cat. LS05003) were performed as human RNase H1, with the final concentration of the enzyme 0.1 U/μl. Control hydrolysis reactions labeled ‘EDTA’ were performed using identical conditions, but in the presence of 10 mM EDTA, whereas control reactions performed in ‘cold’ conditions were incubated for 30 min at 16°C.
Sequencing library construction
RNase H-hydrolyzed samples (5 μl in total; some samples combined at this step) were mixed with 5 μl of 0.4 μM RTP primer and reverse transcribed using the same protocol as used for duplex preparation (volumes scaled down 10×). A total of 5 μl of the reverse transcription (RT) reaction was used as a template for polymerase chain reaction (PCR) amplification with Phusion polymerase (New England Biolabs), RP1 and RPIx primers (x corresponds to index number) performed in 50 μl volume with thermal cycling protocol of (98°C for 3 min), (98°C for 80 s; 60°C for 30 s; 72°C for 15 s) × 8, (72°C, 10 min; 4°C, hold). The PCR reactions were quantified with the Bioanalyzer DNA 1000 kit (Agilent Technologies) on an Agilent 2100 Bioanalyzer, pooled in the presence of the excess of EDTA and Ampure XP purified (Beckman Coulter). The prepared library was sequenced on an Illumina HiSeq Rapid Flow Cell (1 × 50 bp protocol).
RNase H cleavage assays
The designed RNase H cleavable substrates (One of FAM-labeled oligonucleotides: gapR4b_der1, gapR4b_der2, gapR4b_der3 or one of Cy5 labeled oligonucleotides: gap_O1_der3_Cy5; gap_O5_der3_Cy5) were mixed with 10-fold excess of complementary DNA in 1× annealing buffer (20 mM Tris–HCl pH 7.5, 20 mM KCl, 20 mM 2-mercaptoethanol; RNA containing strand = 0.2 μM, DNA complement = 2 μM). The mixtures were incubated at 95°C for 2 min, transferred to 55°C for 10 min and to 30°C for 10 min, before 0.5 volume of 1× Mg-annealing buffer (20 mM Tris–HCl pH 7.5, 20 mM KCl, 20 mM 2-mercaptoethanol and 6 mM MgCl2) was added, followed by incubation at 30°C for 10 min and transfer to ice. These annealed duplexes were mixed with another duplex (prepared in the same way), but labeled with different fluorophore. The combined duplexes were pre-heated for 10 min at 30°C, mixed with an equal volume of 2 mU/μl human RNase H1 in RNH1 buffer (preheated to 30°C). Reactions were terminated at time points: 10 and 40 min by transferring 30 μl reaction to an equal volume of formamide-EDTA (99% formamide and 5 mM EDTA). The setup of the undigested control reaction was identical, but with RNH1 buffer instead of enzyme. The hydrolysis reactions were heat denatured (95°C, 2 min; transferred on ice) and resolved on 15% polyacrylamide, 7M Urea, 1× TBE gel and visualized on Typhoon FLA 7000 scanner (detection of Cy5 followed by detection of FAM). Signal intensities of different bands were background subtracted and quantified using ImageJ software. Results of 40 min incubation are presented in the manuscript.
Hydrolysis of duplexes using molar excess of the human RNase H1 enzyme was performed as described above except: (i) the annealed duplexes were not mixed with a reference substrate; (ii) the final enzyme concentration was 4 U/μl (∼7× molar excess over the duplex in this experiment); (iii) the hydrolysis reactions were assembled in the cold room using pre-chilled pipette tips and incubated for 5 s at 4°C, before they were terminated by addition of an equal volume of urea stop solution (8M Urea, 1× TBE and 5 mM EDTA); (iv) the reactions were denatured and resolved on a 10 well Novex™ TBE–Urea gel (15%) and the gel was visualized with the ChemiDoc Touch Imaging System (Bio-Rad) using a Blue Tray.
Cleavage assays with nicked dumbbell inhibitors
RHase H hydrolysis reactions were performed with either substrate A (gapR7_der3 + gapR7_der3_Cy5) or substrate B (gapR4b_der3 + gapR4b_der3_Cy5). Each substrate was a mixture of two RNA-containing strands with two different fluorophores (FAM or Cy5). Oligonucleotides were mixed with 10-fold excess of complementary DNA (gapR7_der3_comp or gapR4b_der3_comp) in 1× annealing buffer (20 mM Tris–HCl pH 7.5, 20 mM KCl, 20 mM 2-mercaptoethanol; RNA containing strand with FAM = 0.1 μM, RNA containing strand with Cy5 = 0.1 μM, DNA complement = 2 μM), incubated at 95°C for 2 min, transferred to 55°C for 10 min, transferred to 30°C for 10 min, added 0.5 volume of 1× Mg-annealing buffer (20 mM Tris–HCl pH 7.5, 20 mM KCl, 20 mM 2-mercaptoethanol and 6 mM MgCl2), incubated at 30°C for 10 min and transferred on ice. Nicked dumbbells (gapR4b_der1_nickcirc and gapR4b_der2_nickcirc) were prepared by mixing 8 μl of 10 μM oligonucleotide, 2.5 μl 10× annealing buffer and 14.5 μl H2O. They were incubated at 95°C for 2 min and transferred on ice. A total of 12.5 μl 1× Mg-annealing buffer was added. Serial dilutions of the nicked dumbbells were prepared by diluting 6× with equal volume of a dilution buffer (10 μl 10× annealing buffer, 90 μl H2O and 50 μl 1× Mg-annealing buffer). The no-dumbbell control was only dilution buffer. For the cleavage reactions, 7.5 μl of the substrate A or substrate B was mixed with 7.5 μl of a dilution of one of the nicked dumbbells, yielding 32 reactions in total. Human RNase H1 was diluted to 5 mU/μl in 10× RNH1 buffer. Equal volumes (15 μl each) of the substrate-nicked dumbbell and of the RNase H1 dilution (both preheated to 30°C) were combined and incubated for 40 min, before the reactions were terminated by addition of 30 μl formamide-EDTA. Samples were gel analyzed as described above. For the dumbbells design, previously described stable terminal tetraloops were utilized (25).
Data pre-processing
The raw sequencing reads were filtered to contain only those that perfectly matched the sample index (default illumina processing workflow allows for single mismatch, which we have noticed slightly decreased our data quality). The data from pooled samples prepared with DSP (R7) and DSP2 (R4a, R4b) primers were divided using the cutadapt utility (26) by matching the sequence immediately after the randomized part of the constructs (12% error rate allowed, with quality filtering ‘–q 20’) and requiring the preceding sequence to match the length of the designed randomized fragment. Using an awk script, the reads containing ambiguous nucleotides ‘N’ in the randomized fragment or not matching the design of the library, were removed (e.g. if the designed fragment contained S nucleotides at a given position, all reads having A or T there were discarded). In the R7 experiment for the HIV-1 enzyme, we found no reproducibility of the counts of the sequencing reads starting with the sequence ‘ATTA’ and all the reads starting with this tetramer were left out of the analysis. Next, a shell script was used to split the sequences into 1–8 nt long stretches starting at each possible position of the randomized fragment and the number of occurrences of unique sequence was counted. The data frames containing these raw counts were read into R (27) to perform the subsequent data analysis.
Calculation of log2 fold changes from the sequencing data
Fold-changes of different motifs were calculated by comparing the fractions of a given k-mer at a given position within the assayed construct before and after RNase H treatment (across replicates), using edgeR package (28). This analysis takes the advantage of the count nature of the dataset and models the dispersion based on the observed data. See Supplementary Data for the full list of samples used for the calculations.
Prediction of log2 fold changes for human RNase H1
To predict the log2 fold change of a hexamer using the single nucleotide model, we first calculated the position-specific log2 fold changes of single nucleotides (see above), and then we summed the log2 fold changes for the nucleotides, which made up a given hexamer. For the dinucleotide model, a similar procedure was used, but before summing the log2 fold changes of the dinucleotides that made up a given hexamer, they were multiplied by 0.5 to adjust for the fact that a given nucleotide is covered by two dinucleotides. Likewise, the flanking dinucleotides were multiplied by 0.75 to correct for one of the positions being part of two dinucleotides.
Prediction of log2 fold changes for HIV-1 RNase H
To model sequence preferences of the HIV-1 RNase H, each heptamer was associated with two values: preferred cleavage position and the observed cleavage extent. First, the log2 fold changes of each possible heptamer sequence were calculated for each of the seven possible locations within the R7 construct. Assuming that heptamers are cleaved most efficiently when located central in the RNA part of the R7 construct (as we observe for GCGCAA), the preferred location of cleavage can be assigned using the following algorithm. For ∼70% of the heptamers, we find that the two most downregulated heptamer locations within R7 were neighboring. If the strongest downregulation was observed for a given heptamer starting at positions 1 and 2 of R7, then preferred cleavage site within the heptamer was assigned to be after nucleotide 7. If the strongest downregulation was observed for a given heptamer starting at positions 2 and 3 of R7, then preferred cleavage site within the heptamer was assigned to be after nucleotide 6. Likewise, if the strongest downregulation was observed for heptamers starting at positions 3 and 4, 4 and 5, 5 and 6 or 6 and 7, then preferred cleavage site within the heptamer was assigned to be after nucleotide 5, 4, 3 or 2, respectively. If the two most downregulated locations were not neighboring, only the most downregulated was considered—and the cleavage site was assigned after 8-n (for n being the most downregulated position) nucleotide. To calculate the observed cleavage extent, the value of the position having the most efficient downregulation was used.
Calculation of relative processing rate constants (krel)
The relative processing rate constant (krel) is defined as a ratio of kcat/Km of a compound of interest to the reference compound. The use of krel for analysis of enzymatic reactions with multiple competing substrates has previously been described Guenther et al. (29). Here, we have calculated krel by comparing the processing rate to the average processing rate observed in our pool of substrates rather than to a specific sequence. To calculate krel for a given nucleotide at a given position for each of the three sequenced constructs, krel value for nucleotide i was calculated according to:
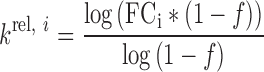 |
Here, FCi is a fold change of a given nucleotide as calculated using edgeR and f is a cleaved fraction of the pooled duplex as quantified from scans of the gel. Since the analysis used replicates to estimate FC’s, the value of f is an average of the replicates.
Values of krel for substrates relative to the reference compound were calculated according to:
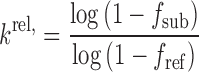 |
Here, fsub is a cleaved fraction of a substrate of interest and fref is a cleaved fraction of a reference substrate.
Comparison with RNA knockdown data
The knockdown efficiencies of microtubule-associated protein tau (MAPT) (ENSG00000186868) and ANGPTL3 (ENSG00000132855) after treatment with the different gapmers were extracted from the published patents (30,31). For MAPT, we used the data from the SH-SY5Y cell line transfected using electroporation with 8 μM gapmer and having a 5-8-5 design. For ANGPTL3, knockdown efficiencies originated from the HepG2 cell line transfected using electroporation with 4.5 μM gapmers with a 5-10-5 design. In addition, only gapmers having a perfect reverse complementary match to the relevant pre-mRNA were used in the analysis. For the ANGPTL3 dataset, many gapmers have too low affinity to function efficiently (11) and we therefore only used the data obtained with gapmers having six or more G or C nucleotides for the analysis. When a gapmer was tested in replicates, the results from the individual replicates were averaged and included once for the correlation analysis. Knockdown data were compared with human RNase H1 cleavability predictions based on the results of the R4b construct probing, considering 8 dinucleotides starting from position 7 of the randomized gap.
Mapping cleavage scores to viral genomes
Each heptamer in the viral genome was modeled to direct cleavage to a particular bond with an associated cleavage efficiency (based on the HIV-1 RNase H model described above). Some bonds were not modeled to be cleaved at all, if all heptamers in their vicinity directed the cleavage to other bonds. When multiple heptamers directed cleavage to the same position, the minimal cleavage score (corresponding to the most efficient cleavage) was assigned.
Random sampling of the viral genomes
The HIV-1 (GenBank: K03455.1), HIV-1 vector pNL4-3 (GenBank: AF324493.2, positions 1-9709) and HIV-2 (GenBank: KX174311.1) genomes were randomly sampled while statistically preserving the local dinucleotide content. For each sampled position, a dinucleotide was randomly selected so that: (i) its first nucleotide matches the last nucleotide of the preceding dinucleotide [does not apply to the first dinucleotide] and (ii) sampling probabilities are proportional to the counts of the dinucleotides in the local, 65 nt wide window centered on sampled dinucleotide. The procedure was repeated 10 000×, followed by mapping cleavage scores to viral genomes (see above) and calculating distances between nearest 10% best cleaved bonds.
RESULTS
RNase H sequence preference assay (H-SPA)
In the H-SPA method, an engineered DNA duplex carrying a central randomized cleavable RNA–DNA sequence (Figure 1A) is subject to limited digestion with RNase H. The fixed flanks of the construct allow subsequent quantification of the uncleaved molecules by massive parallel sequencing (Figure 1B). We used three different construct designs to evaluate the effect on the enzyme’s sequence preferences (Figure 1A). Constructs R4a and R4b contain a central stretch of four ribonucleotides flanked by 2′-O-Me modified nucleotides, thereby restricting hydrolysis to one specific position (22,32). The 2′-O-Me modified nucleotides block cleavage and have a preference for C3′-endo puckering at the cleaved strand (33,34) similar to the structure of RNA in a regular RNA–DNA hybrid. The R7 construct contains a stretch of seven ribonucleotides flanked by DNA and is therefore more similar to native RNase H substrates, but can be cleaved at several different positions. After limited cleavage with three type 1 RNases H enzymes: human RNase H1, E. coli RNase HI and RNase H of the HIV-1 reverse transcriptase (Figure 1C and Supplementary Figure S1) followed by massive parallel sequencing, we compared counts of k-mers at the same position of the randomized sequence between control and treated samples. We observed a very high correlation between replicated controls and between replicated cleaved samples, whereas the correlation between control and cleaved samples was reduced (Figure 1D). This demonstrates that the H-SPA method is highly reproducible and accurately quantifies the cleavage of different sequences in our duplex libraries. The decreased correlation between control and cleaved samples shows that the RNase H treatment results in the biased hydrolysis of different sequences (P-value ∼ 0 for difference between correlation coefficients; Fisher r-to-z transformation).
Figure 1.

Overview and validation of the H-SPA method. (A) Sequences of the cleavable randomized gaps used in this study. (B) Schematic representation of the experimental strategy. (C) Denaturing gel electrophoresis showing the limited digestion of double stranded substrates. The uncleaved duplexes are indicated with the asterisk and the cleaved fragment with the tilde. The reaction conditions are indicated: w: 37°C; c: 16°C and EDTA: 0.1 mM. (D) Correlation of heptamer counts for the construct R4b (positions 9–15) between two untreated controls (top left), two human RNase H1 treated samples (top right) and a control and a treated sample (bottom).
Sequence preferences of different RNase H enzymes
To better understand the sequence preferences of the three RNase H enzymes, we used our sequencing data to determine the (i) position-specific changes in nucleotide content after RNase H treatment, (ii) importance (information content) of different positions of the recognized sequence and (iii) best-cleaved motifs (Figure 2A and Supplementary Figure S2a). The sequence preferences of the E. coli and human enzymes are nearly identical. Both enzymes efficiently cleave the RNA strand in the sequence context CA↓AG and prefer a G-rich sequence upstream of the cleavage site. Importantly, for each enzyme the observed preferred sequences are very similar for the three different duplexes tested, demonstrating that they are not dependent on the context of the specific constructs and that H-SPA is reproducible. The HIV-1 RNase H yielded a different sequence preference than the human and E. coli enzymes (Figure 2A and Supplementary Figure S3) and did not efficiently cleave the R4b construct (Supplementary Figure S1). In the case of all three enzymes, our data demonstrate that they exhibit clear sequence preferences. This supports the notion previously observed for E. coli RNase P that even apparently non-specific nucleic acid-interacting enzymes display inherent specificity when studied in-depth (29).
Figure 2.

Sequence preferences of Escherichia coli, Homo sapiens and HIV-1 RNase H (A) The heatmaps display the changes in nucleotide composition at different positions for the R7 construct (left) and the R4b construct (right) after cleavage with the three different RNase H enzymes. The intensity of the red and blue color indicates the krel of having given nucleotide at a given position fixed relative to the average hydrolysis rate of the randomized pool. The barplots below the heatmaps show the overall information content at each position and the sequence logos are based on the 1% most downregulated pentamers. Note that only the randomized parts of the probed duplexes is displayed. (B) Cleavage of sequences predicted to be preferred (‘P’), avoided (‘A’) and neutral (‘N’) with respect to cleavage with human RNase H1 compared to the cleavage of a reference substrate. With respect to the reference substrate, the krel of the preferred substrate is 3.7, of the avoided is 0.26 and of the neutral it is 1.4. (C) The design of the dumbbell substrate mimics. The gray box indicates the region having either the preferred (‘P’) or avoided (‘A’) sequence. (D) The cleavage of a reference substrate in the presence of increasing concentrations of a preferred or avoided dumbbell substrate mimic.
To validate the observed preferences of the human RNase H1, we synthesized three duplexes with the sequences found to be the most preferred, the most avoided or the closest to the average for cleavage by the RNase H, respectively (Supplementary Figure S4). A convenient metric to compare kinetics of enzymatic reaction with internal competition is a relative processing rate constant (krel), described in ‘Materials and Methods’ section. In agreement with our predictions, the ‘preferred’ substrate was cleaved considerably more efficiently and the ‘avoided’ less efficiently than a reference, whereas the extent of the cleavage of a ‘neutral’ substrate was close to the reference (Figure 2B and Supplementary Figure S2b). Unexpectedly, the overall amount of hydrolyzed material was reduced in the reaction with the ‘preferred’ substrate, indicating that it stays associated with the enzyme longer than the other two substrates either by having a higher melting temperature (Supplementary Table S1) or because of a more energetically favorable inter-molecular interaction with the enzyme (Supplementary Discussion). We therefore tested nicked dumbbell versions of the preferred and avoided substrates, designed in such a way that the cleaved strand cannot dissociate from the duplex (Figure 2C). We find that the hydrolysis of a reference substrate in the presence of the ‘preferred’ nicked dumbbell is potently inhibited, whereas the ‘avoided’ nicked dumbbell has a negligible effect (Figure 2D and Supplementary Figure S2c), indicating that the preferred substrate has high affinity for the enzyme and this could be the molecular basis for both the efficient cleavage of this sequence and its ability to inhibit the cleavage of the reference sequence. Moreover, when using a molar excess of enzyme over the substrates (Supplementary Figure S2d), we find that the ‘preferred’ substrate is cleaved more efficiently than the ‘avoided’ substrate, which supports our H-SPA findings and the interpretation that the ‘preferred’ substrate can cause product inhibition.
Sequence preference model for human RNase H1
To further validate our data and explore the importance of the Human RNase H1 sequence preferences, we used our comprehensive dataset for duplex R4a to construct a position weight matrix (PWM), which for each nucleotide at each position quantifies the relative concentration change after RNase H cleavage of the subset of molecules having this nucleotide at this position change (Supplementary Data). Using the PWM constructed from the result of the R4a experiment to predict the outcome of the R4b experiment, we can explain 69% of the changes occurring (Figure 3A). Interestingly, the predictive power increased to 78% when we constructed a model based on dinucleotide content and used this to predict the R4b experiment outcome (Figure 3B). This finding suggests that features such as stacking or bending of the duplex, which are affected by dinucleotide dependencies, are involved in RNase H1 cleavage efficiency. More complex models using 3, 4 or 5 nt words as the input did not lead to further improvements in the performance of the prediction (Supplementary Figure S5), indicating that long-range dependencies for the substrate recognition by the RNase H1 catalytic domain are negligible. To further validate the predictive power of the dinucleotide model for human RNase H1, we predicted cleavage of the DNA/RNA duplex that was used to crystallize the catalytic domain of human RNase H1 (Figure 3C) (22). When requiring the previously reported interaction with 11 bp of the duplex, we find that out of the eight possible interaction modes, RNase H1 contacts the enzyme at exactly the position predicted to have the highest preference for cleavage (Figure 3C). Since the structure was solved using a catalytic deficient mutant, this result supports the idea that the RNase H sequence preferences at least partially depend on the binding of the duplex substrate.
Figure 3.

RNase H Sequence Preferences correlate with gapmer efficiency. (A) Correlation between the log2 fold changes of different hexamers observed for the R4b construct in the experiment and the corresponding log2 fold changes as predicted by a single nucleotide model prepared from the data obtained in the R4a experiment. (B) As in (A), but with prediction with a dinucleotide model. (C) Prediction of RNase H1 mediated downregulation of the different 11-mers present in RNA sequence used for the RNase H RNA–DNA heteroduplex crystal structure [PDB: 2QK9]. Each bar corresponds to the cleavage site of a potential binding mode of RNase H1. The filled bar corresponds to the RNase H1 binding mode observed in the crystal structure and is also indicated in the drawing below the plot. (D) Correlation between the change of target RNA level for MAPT (30) after treatment with 1518 different gapmers and the corresponding downregulation predicted by the dinucleotide model for the different binding modes of RNase H1 on each gapmer target duplex. The error bars show the 99% confidence intervals. The drawing below the plot indicates the RNase H1 binding mode associated with the best-observed correlation. (E) The same analysis as in (D), but with 1581 different gapmers targeted against ANGPTL3 (31).
RNase H1 is central to the therapeutic action of gapmers and we therefore wanted to investigate whether our sequence preference model could predict gapmer potency. We used two large published datasets of in vitro activity screening of gapmers targeted against different positions of the MAPT (30) and Angiopoetin-like 3 (ANGPTL3) (31) mRNAs. For the target site of each of the gapmers tested in these studies, we calculated the predicted cleavage using the H-SPA 9 nt wide dinucleotide model and compared this value to the observed reduction of mRNA level. For both the MAPT and ANGPTL3 knockdown experiments, we find a similar correlation pattern. The predictions for positions in the central part, which can be cleaved by RNase H, significantly correlated with the observed gapmer activity, but predictions for the flanks, which cannot be cleaved, did not (Figure 3D and E), indicating that the H-SPA dinucleotide model has the potential to improve gapmer design.
Sequence preference model for HIV-1 RNase H
The HIV-1 RNase H enzyme activity of the viral reverse transcriptase is essential for the viral life cycle, but so far it has not been possible to make a general prediction of cleavage positions in the viral genome. In our HIV-1 experiment, we observe that the fold changes of different RNA heptamers had a non-Gaussian distribution (Figure 4A), showing that the HIV-1 enzyme has distinct sequence preferences for cleavage. This agrees with the findings from a previous study, which analyzed a limited number of sequences for in vitro cleavage efficiency by HIV-1 RNase H (18), but our dataset allows a much more comprehensive characterization of the sequence preferences (Figure 2A and Supplementary Figure S6). For the HIV-1 enzyme, we only observed efficient cleavage of the construct with the seven RNA positions, which allows the HIV-1 RNase H to cleave at several different positions, thereby making the interpretation of the results more difficult. We therefore reasoned that the sequence preferences observed in Figure 2A could be further improved by aligning sequence words by their position of cleavage. First, we aligned the hexamer (‘GCGCAA’), which is cleaved between its last 2 nt (18), at different positions of the R7 substrate, and noted that the level of downregulation depended on the location (Figure 4B). For optimal cleavage of this motif by HIV-1 RNase H, at least four ribonucleotides upstream and at least two ribonucleotides downstream of the cleavage site are required. In addition, we find that having less than three ribonucleotides upstream of the cleavage site completely inhibits the cleavage. Understanding these requirements, we could assign two values to each heptamer sequence, one describing the efficiency of cleavage and another describing at which bond the heptamer was predominantly cleaved (for details see ‘Materials and Methods’ section; for values see Supplementary Data). This allowed us to split all the heptamers into seven groups depending on the cleavage location. For each set, we used the most downregulated quartile to create sequence logos (Figure 4C). The logos from the different groups largely resemble each other when aligned by the assigned cleavage location. The analysis shows that the preferred sequence relative to the cleavage site is (C/G) at −5, (C/G/U) at −4, (C/G) at −2 and (A/U) at −1 and +1; the most important positions within the assayed region are −5, −4, −2 and +1, agreeing with the results obtained by the evaluation of a limited number of single sequences (18).
Figure 4.

Refining the HIV-1 RNase H sequence preference model. (A) Distributions of the observed log2 fold changes of RNA heptamers in R7 for human RNase H1 (right) and HIV-1 RNase H (left). (B) The observed log2 fold changes after cleavage with HIV-1 RNase H for an efficiently cleaved hexamer (GCGCAA) located at different positions of R7. The position of the arrow indicates the cleavage site as aligned to the picture of scissors in the box and the arrow length represents the efficiency of cleavage. (C) Sequence logos of the best cleaved quartile of sets of heptamers predicted to have the same cleavage site. The arrows indicate the predicted cleavage site, with the length proportional to the observed cleavage efficiency.
Functional significance of HIV-1 RNase H sequence preferences
To learn more about the function of HIV-1 RNase H in the viral lifecycle, we assigned RNase H cleavage scores to each bond in the HIV-1 genome (Figure 5A). In HIV-1 replication, the reverse transcription of the DNA minus strand is primed with human tRNA-Lys3 at the primer binding site (PBS) of the viral RNA genome (35). The newly synthesized minus strand DNA fragment is transferred to the RNA genome’s 3′ end and DNA minus strand synthesis is continued (Figure 5B). The HIV-1 RNase H is responsible for degradation of the HIV-1 RNA genome upon synthesis of the DNA minus strand. Biochemical experiments suggest that after the initial nicking concurrent with DNA polymerization, the genomic RNA degradation predominantly occurs by RNA 5′-end-directed cleavage with cleavages occurring about 18 nt from the RNA 5′ end (36) and in a progressive manner with the 5′ end created by the previous cleavage facilitating the next cleavage (37). We therefore calculated the distances between sites in the HIV-1 genome that were predicted by H-SPA to be well cleaved. Strikingly, we find that HIV-1 has a highly significant over-representation (P < 10−5) of well-cleaved sites separated with a distance of 13–19 nt, as compared to the cleavage distances we obtain when we repeatedly perform the same analysis on randomized versions of the HIV-1 sequence that preserve the local dinucleotide content (see ‘Materials and Methods’ section) (Figure 5C and Supplementary Figure S7a). This finding supports that degradation of the HIV-1 RNA occurs by a 5′-end-directed mechanism and indicates that there is a selective pressure for the HIV-1 genomic sequences to be efficiently cleaved by the viral RNase H.
Figure 5.

Functional significance of predicted HIV-1 RNase H cleavage sites. (A) Predicted RNase H cleavage efficiency of the HIV-1 genome, shown as log2(fold change) (log2FC). (B) Schematic of the HIV reverse transcription. White scissors at the black circle indicate specific areas zoomed-in in subsequent panels. (C) Comparison of distances (in nucleotides) between well-cleaved sites in the HIV-1 genome and in the randomized HIV-1 genomes. The red rhombi shows the observed count of distances between positions predicted to be efficiently cleaved in HIV-1 genome that fall into the indicated distance intervals. The violin plots show the density of the distributions that resulted from the same analysis, but repeated 10 000× on HIV-1 genome sequences that were randomized with preserving the local dinucleotide content; Predicted cleavage efficiency of (D) the sequence surrounding the 3′PPT, (E) of the terminal 18 nt of the tRNA-Lys3 primer and (F) it is reverse complement (primer binding site). (G) Predicted cleavage efficiency of the best-cleaved site in the terminal 18 nt of the different human tRNAs (plus CCA) and of the corresponding reverse complement. The tRNA-Lys3 is indicated in red.
During minus-strand DNA synthesis, HIV-1 RNase H is known to cleave specifically at the 5′ and 3′ ends of the cleavage-resistant 3′PPT region, which forms a primer for the plus-strand DNA synthesis. H-SPA predicts the precise location of specific cleavage at the 5′ end of the PPT (21) (Figure 5D, bond 9064), but not the 3′ cleavage (Figure 5D; bond 9083). This is in agreement with the 3′ cleavage being precise, but not kinetically favored (19) and the PPT adopting an untraditional structure with the base pairing out of register (38), which is unlikely to be recaptured in our assay. The plus-strand DNA is synthesized from the 3′PPT primer and forms local RNA–DNA hybrid with tRNA-Lys3 primer located at the DNA minus strand terminus (Figure 5B). The tRNA primer is cleaved by HIV-1 RNase H at the last bond of the tRNA, leaving rA at the 5′ end of the (−)DNA strand (20), which is essential for the plus-strand transfer to occur (39) and facilitates the integration process (40). According to the sequence preferences determined by H-SPA, cleavage at the last bond is indeed the most preferred location within the tRNA 3′ terminal 18 nt (Figure 5E). The last part of the RNA genome to be reverse transcribed is the PBS, which remarkably contains the sequence predicted to be the fifth best RNase H-cleavage site within the entire HIV-1 genome (Figure 5F). Clearing the DNA minus strand complement of the PBS from any hybridized RNA fragments is necessary to allow plus-strand DNA transfer to take place during viral replication (Figure 5B) and we therefore propose that the presence of this previously unrecognized efficient cleavage site in the PBS sequence is important for the HIV-1 life cycle. Since the PBS is the reverse-complement of the tRNA primer, our findings show that compared to other human tRNAs and their potential PBSs (41), the tRNA-Lys3 sequence is the best substrate for RNase H cleavage in both directions (Figure 5G).
DISCUSSION
We present the H-SPA method, which allows comprehensive mapping of RNase H sequence preferences by detecting changes in frequencies of specific sequences after RNase H cleavage within a randomized pool of RNA–DNA substrates. Conceptually, our strategy resembles the strategy previously used to detect the sequence preference of RNase P (29), but in our method, we have introduced a double stranded substrate with a mixed chemistry and increased the length of the randomized gap.
To deal with the increased sequence complexity of our experimental set-up, which exceeded the number of obtained sequencing reads, we also developed a novel data analysis approach focusing on enrichment of k-mers at specific positions of the randomized sequence. The computational strategies and methods developed for H-SPA data analysis use robust count-based RNA-seq analysis (28), which explicitly takes experimental variability into account and could be adapted for the analysis of other types of SPAs. To ensure the reproducibility and to facilitate future studies, we have implemented and released an R package ‘spaseq’ and a vignette illustrating each step of the data analysis workflow (see ‘Materials and Methods’ section).
Using H-SPA, we comprehensively characterized sequence preferences of three type 1 RNase H enzymes from phylogenetically distant hosts: human RNase H1, E. coli RNase HI and RNase H domain of HIV-1 reverse transcriptase. We were particularly interested in the preferences of the human enzyme, which is necessary for gapmer-mediated target knockdown (8–10) and therefore influences the pharmaceutical activity of an entire class of human drugs. We demonstrate that human RNase H prefers to cleave the sequence context CA↓AG, downstream of a G-rich sequence. Moreover, these preferences significantly correlate with the efficiency of antisense oligonucleotides (ASOs) in cell culture, strongly suggesting that our results can be used to improve their design. Interestingly, our findings also explain previous anecdotal findings that ASOs with the TCCC motif (complementary to GGGA on RNA) yielded more potent gene knockdown (42).
One of the limitations of the H-SPA method is the unnatural structure of the probed substrates. For example, the R4b construct contains both DNA and 2′-O-Me modified nucleotides in the cleaved strand, which will change the structure compared to a conventional RNA–DNA heteroduplex. Nevertheless, only one of the modified nucleotides directly interacts with the RNase H1 enzyme (22) (Supplementary Figure S8) and reassuringly, for the human and E. coli RNase H, we obtained similar results with all three constructs having different modification patterns (Figure 2A and Supplementary Figure S2a), suggesting that the different modifications in the substrates did not distort the results.
Human RNase H1 has two nucleic acid binding domains: the catalytic domain, which cleaves RNA and is structurally similar to the E. coli RNase H1 (22,43,44) and the HBD, which allows for the enzyme’s processive action (45). We observe strikingly similar sequence preferences of the human and E. coli enzymes, indicating that in our experiments the HBD did not affect the preferences of the human enzyme. Thus, although we cannot rule out that HBD binding preferences affect ASO efficiency in vivo, our results indicate that the catalytic domain is central for human RNase H1 target preferences.
Interestingly, our results show that preferred sequences, apart from having a higher relative processing rate, also have slower dissociation rate of the enzyme from the cleaved substrate, which in some of our biochemical assays resulted in the depletion of the active enzyme. Depending on the exact cellular conditions these two counteracting mechanisms will have opposite effects on the knockdown efficiency. It has been reported that the concentration of RNase H is a limiting factor to the antisense activity (46). Given the relatively low copy number of majority of mRNAs in cells (47), we do not expect the use of ASOs with RNase H preferred sequences will lead to substantial RNase H depletion, which would suggest that taking the preferences of RNase H into account when designing ASOs sequences will lead to more efficient cleavage for the large majority, if not all mRNAs.
For the three RNase H enzymes we tested, the most preferred trinucleotide around the cleavage site is CA↓A, which highlights the structural conservation of the catalytic center (22). In contrast, we observed pronounced differences in the preferences of HIV-1 and other tested RNases H upstream from the cleavage site, with HIV-1 having a propensity to cleave after GC-rich sequences, as opposed to G-rich for the two other enzymes. Moreover, the sequence preferences of the HIV-1 RNase H seem to differ fundamentally from the human or E. coli enzymes for which the affinity for different substrates can be understood as coming from a continuous spectrum (29), allowing them to cleave all sequences albeit with varying kinetics, whereas the viral enzyme can truly discriminate between different substrates and prefers (C/G) at −5, (C/G/U) at −4 and (C/G) at −2. The sequence preferences that we observe for positions −5 to +2 agree with the previously described preferences by Champoux and co-workers (18,48–51). In these studies, additional significant preferences were observed for positions −10 and −14 (18). Because of the design of our R7 substrate with randomized positions not fully extending into the polymerase domain, these preferences and any other preferences that may be present between positions −19 and −7 will not be detected by our method. Future studies are needed to resolve to what degree the polymerase domain dictates the overall sequence preferences.
The difference in preferences between the HIV-1 and the human RNase H probably reflects the structural differences of these two enzymes. The HIV-1 RNase H lacks the so-called basic protrusion, which forms a DNA-binding channel in the human and E. coli enzyme and has an important role in substrate binding (22) and it has been shown that the HIV-1 RNase H activity depends on substrate binding in the polymerase domain and is inactive in isolation (52). Our R7 substrate is sufficiently long to simultaneously bind both the HIV-1 polymerase and RNase H domains and is efficiently cleaved in our experiments, indicating that the constant dsDNA part of the R7 substrate binds the polymerase domain, even though a RNA–DNA heteroduplex have been shown to bind with higher affinity to the polymerase domain than dsDNA (53). Possibly, the preference for G or C in positions −2, −4 and −5 may be related to the requirement of substrate untwisting in order to reach the RNase H active site while simultaneously binding the polymerase domain (54,55).
The R7 substrate used for the HIV-1 experiments differs from a typical HIV-1 RNase H substrate by having the RNA–DNA hybrid region embedded within the long dsDNA structure. This is similar to the natural substrate after plus strand transfer when HIV-1 RNase H cleaves off the tRNA primer (Figure 5B), but the large majority of cleavages by HIV-1 RNase H during the viral lifecycle is on DNA–RNA duplexes. We observe a preference for G or C in RNA positions −5 and −6 relative to the scissile phosphate. Those two positions are located at the border of the DNA:DNA and RNA:DNA duplex of the R7 construct and the helical conformation of this region is therefore likely to be more B-like than a pure RNA:DNA substrate. This difference may influence the preference that we observe, however, in a previous study with a limited set of tested RNA–DNA duplexes, G has been reported to be preferred in position −5 (18). Additionally, in this region the enzyme contacts exclusively the non-cleaved DNA strand, which has the same sugar chemistry in our R7 substrate as in the natural substrate (38,54).
The comprehensive nature of our analysis allowed us to create a preference motif sufficiently rich in information that we could perform global prediction of RNase H cleavage sites in the HIV genome. We correctly predict the known cleavage sites, 5′of the PPT and the tRNA primer (Figure 5E and F), indicating that the identified preferences are biologically relevant. Interestingly, we not only found that our sequence preferences match the known cleavage of the tRNA-Lys3 primer, but also that the reverse complement of the tRNA-Lys3 sequence (PBS sequence) is effectively cleaved by the HIV-1 RNase H, which may facilitate the DNA plus-strand transfer during replication. Despite millions of years of extremely rapid evolution (56) the same host molecule (tRNA-Lys3) is utilized by almost all lentiviruses to prime reverse transcription (35). A number of different mechanisms likely contribute to the strong dependence of HIV-1 on the tRNA-Lys3, such as: specific packaging of lysine tRNAs (57,58) and interactions between the HIV-1 genome and the TΨC arm of tRNA-Lys3 (59–61), hLysRS (62,63) and the tRNA anticodon (64,65). We suggest that the efficient Rnase H cleavage of tRNA-Lys3 and of the complementary PBS sequence may also contribute to the conserved use of tRNA-Lys3 as primer in HIV-1 replication.
During HIV-1 replication, cleavage of the HIV-1 RNA genome occurs occasionally at a specific distance from the growing DNA 3′ end to produce RNA oligomers of variable length, some of which stay associated with the newly synthesized DNA strand (66,67). Next, the RNA oligomers are further cleaved 13–19 nt from the RNA 5′ end by a RNA 5′-end-directed mechanism (36,51). Previously, these end-directed HIV-1 RNase H cleavage modes have been shown to have sequence preferences very similar to the preference of internal cleavage on long DNA–RNA duplexes (50). Cleavage of the R7 substrate resembles the internal mode of cleavage and we assume the observed preferences are relevant also for HIV-1 RNases H end-directed cleavages. In support of this, we find that HIV-1 genomic positions predicted by our cleavage model to be efficiently cleaved are preferentially separated by 13–19 nt (Figure 5C). This finding indicates that the HIV-1 genome is under selective pressure for efficient processing by its own RNase H. Interestingly, similar analysis performed for HIV-2 genome did not show significant enrichment (Supplementary Figure S7b), which may reflect that the role of viral RNase H in the reverse transcription of HIV-2 is less important, as indicated by much lower RNase H activity (68). Finally, we note that the two sites in the HIV-1 genome predicted to be the most efficiently cleaved are located within the Rev response element, suggesting a functional relevance.
In conclusion, this study uncovers sequence preferences of the important RNase H enzymes and thereby provides valuable information for the future design of antisense oligonucleotides and contributes to an improved understanding of HIV-1 biology. The presented method, both experimental and computational, can be readily applied to other enzymes acting on duplexed oligonucleotides, such as the human RNase H2 or restriction endonucleases.
AVAILABILITY
An R package ‘spaseq’ together with a vignette describing the full analysis workflow can be downloaded from https://github.com/lkie/spaseq. Sequencing data are available from European Nucleotide Archive (ENA) at http://www.ebi.ac.uk/ena/data/view/PRJEB20181.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Søren Ottosen and Sandro Bottaro for helpful discussions, and Peter Brodersen and Lars Jønson for critical reading and commenting on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Roche Postdoc Fellowship Program; Danish Council for Strategic Research (Center for Computational and Applied Transcriptomics) [DSF-10–092320]. Funding for open access charge: Roche Innovation Center Copenhagen A/S.
Conflict of interest statement. L.J.K., P.H.H. and M.L are employees of Roche Innovation Center Copenhagen A/S, which develops LNA-modified therapeutic oligonucleotides.
REFERENCES
- 1. Stein H., Hausen P.. Enzyme from calf thymus degrading the RNA moiety of DNA-RNA Hybrids: effect on DNA-dependent RNA polymerase. Science. 1969; 166:393–395. [DOI] [PubMed] [Google Scholar]
- 2. Cerritelli S.M., Frolova E.G., Feng C., Grinberg A., Love P.E., Crouch R.J.. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell. 2003; 11:807–815. [DOI] [PubMed] [Google Scholar]
- 3. Reijns M.A.M., Rabe B., Rigby R.E., Mill P., Astell K.R., Lettice L.A., Boyle S., Leitch A., Keighren M., Kilanowski F. et al. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012; 149:1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malik H.S., Eickbush T.H.. Phylogenetic analysis of ribonuclease H domains suggests a late, chimeric origin of LTR retrotransposable elements and retroviruses. Genome Res. 2001; 11:1187–1197. [DOI] [PubMed] [Google Scholar]
- 5. Cerritelli S.M., Crouch R.J.. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009; 276:1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holmes J.B., Akman G., Wood S.R., Sakhuja K., Cerritelli S.M., Moss C., Bowmaker M.R., Jacobs H.T., Crouch R.J., Holt I.J.. Primer retention owing to the absence of RNase H1 is catastrophic for mitochondrial DNA replication. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:9334–9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walder R.Y., Walder J.A.. Role of RNase H in hybrid-arrested translation by antisense oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 1988; 85:5011–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. ten Asbroek A.L.M.A., van Groenigen M., Nooij M., Baas F.. The involvement of human ribonucleases H1 and H2 in the variation of response of cells to antisense phosphorothioate oligonucleotides. Eur. J. Biochem. 2002; 269:583–592. [DOI] [PubMed] [Google Scholar]
- 9. Wu H., Lima W.F., Zhang H., Fan A., Sun H., Crooke S.T.. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J. Biol. Chem. 2004; 279:17181–17189. [DOI] [PubMed] [Google Scholar]
- 10. Lima W.F., Murray H.M., Damle S.S., Hart C.E., Hung G., De Hoyos C.L., Liang X.-H., Crooke S.T.. Viable RNaseH1 knockout mice show RNaseH1 is essential for R loop processing, mitochondrial and liver function. Nucleic Acids Res. 2016; 44:5299–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pedersen L., Hagedorn P.H., Lindholm M.W., Lindow M.. A kinetic model explains why shorter and less affine enzyme-recruiting oligonucleotides can be more potent. Mol. Ther. Nucleic Acids. 2014; 3:e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hagedorn P.H., Yakimov V., Ottosen S., Kammler S., Nielsen N.F., Høg A.M., Hedtjärn M., Meldgaard M., Møller M.R., Orum H. et al. Hepatotoxic potential of therapeutic oligonucleotides can be predicted from their sequence and modification pattern. Nucleic Acid Ther. 2013; 23:302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burdick A.D., Sciabola S., Mantena S.R., Hollingshead B.D., Stanton R., Warneke J.A., Zeng M., Martsen E., Medvedev A., Makarov S.S. et al. Sequence motifs associated with hepatotoxicity of locked nucleic acid–modified antisense oligonucleotides. Nucleic Acids Res. 2014; 42:4882–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vickers T.A., Wyatt J.R., Freier S.M.. Effects of RNA secondary structure on cellular antisense activity. Nucleic Acids Res. 2000; 28:1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Woolf T.M., Melton D.A., Jennings C.G.. Specificity of antisense oligonucleotides in vivo. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:7305–7309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hagedorn P.H., Hansen B.R., Koch T., Lindow M.. Managing the sequence-specificity of antisense oligonucleotides in drug discovery. Nucleic Acids Res. 2017; 45:2262–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindow M., Vornlocher H.-P., Riley D., Kornbrust D.J., Burchard J., Whiteley L.O., Kamens J., Thompson J.D., Nochur S., Younis H. et al. Assessing unintended hybridization-induced biological effects of oligonucleotides. Nat. Biotechnol. 2012; 30:920–923. [DOI] [PubMed] [Google Scholar]
- 18. Schultz S.J., Zhang M., Champoux J.J.. Multiple nucleotide preferences determine cleavage-site recognition by the HIV-1 and M-MuLV RNases H. J. Mol. Biol. 2010; 397:161–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rausch J.W., Le Grice S.F.J.. ‘Binding, bending and bonding’: polypurine tract-primed initiation of plus-strand DNA synthesis in human immunodeficiency virus. Int. J. Biochem. Cell Biol. 2004; 36:1752–1766. [DOI] [PubMed] [Google Scholar]
- 20. Furfine E.S., Reardon J.E.. Human immunodeficiency virus reverse transcriptase ribonuclease H: specificity of tRNA(Lys3)-primer excision. Biochemistry. 1991; 30:7041–7046. [DOI] [PubMed] [Google Scholar]
- 21. Huber H.E., Richardson C.C.. Processing of the primer for plus strand DNA synthesis by human immunodeficiency virus 1 reverse transcriptase. J. Biol. Chem. 1990; 265:10565–10573. [PubMed] [Google Scholar]
- 22. Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W.. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007; 28:264–276. [DOI] [PubMed] [Google Scholar]
- 23. Kankanala J., Kirby K.A., Liu F., Miller L., Nagy E., Wilson D.J., Parniak M.A., Sarafianos S.G., Wang Z.. Design, synthesis, and biological evaluations of hydroxypyridonecarboxylic acids as inhibitors of HIV reverse transcriptase associated RNase H. J. Med. Chem. 2016; 59:5051–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lima W.F., Wu H., Crooke S.T.. Human RNases H. Methods Enzymol. 2001; 341:430–440. [DOI] [PubMed] [Google Scholar]
- 25. Nakano M., Moody E.M., Liang J., Bevilacqua P.C.. Selection for thermodynamically stable DNA tetraloops using temperature gradient gel electrophoresis reveals four motifs: d(cGNNAg), d(cGNABg), d(cCNNGg), and d(gCNNGc). Biochemistry. 2002; 41:14281–14292. [DOI] [PubMed] [Google Scholar]
- 26. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10–12. [Google Scholar]
- 27. R Core Team R: a Language and Environment for Statistical Computing. R Foundation for Statistical Computing. 2016. [Google Scholar]
- 28. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guenther U.-P., Yandek L.E., Niland C.N., Campbell F.E., Anderson D., Anderson V.E., Harris M.E., Jankowsky E.. Hidden specificity in an apparently nonspecific RNA-binding protein. Nature. 2013; 502:385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kordasiewicz H., Swayze E., Freier S., Bui H.-H.. Compositions for Modulating Tau Expression. 2015; World Intellectual Property Organization, WO2015010135. [Google Scholar]
- 31. Freier S., Graham M., Crooke R.. Modulation of angiopoietin-Like 3 expression. 2015; World Intellectual Property Organization, WO2015100394. [Google Scholar]
- 32. Hogrefe H.H., Hogrefe R.I., Walder R.Y., Walder J.A.. Kinetic analysis of Escherichia coli RNase H using DNA-RNA-DNA/DNA substrates. J. Biol. Chem. 1990; 265:5561–5566. [PubMed] [Google Scholar]
- 33. Horton N.C., Finzel B.C.. The structure of an RNA/DNA hybrid: a substrate of the ribonuclease activity of HIV-1 reverse transcriptase. J. Mol. Biol. 1996; 264:521–533. [DOI] [PubMed] [Google Scholar]
- 34. Venkateswarlu D., Lind K.E., Mohan V., Manoharan M., Ferguson D.M.. Structural properties of DNA:RNA duplexes containing 2′-O-methyl and 2′-S-methyl substitutions: a molecular dynamics investigation. Nucleic Acids Res. 1999; 27:2189–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leis J., Aiyar A., Cobrinik D.. 3 Regulation of initiation of reverse transcription of retroviruses. Cold Spring Harb. Monogr. Arch. 1993; 23:33–47. [Google Scholar]
- 36. Palaniappan C., Fuentes G.M., Rodríguez-Rodríguez L., Fay P.J., Bambara R.A.. Helix structure and ends of RNA/DNA hybrids direct the cleavage specificity of HIV-1 reverse transcriptase RNase H. J. Biol. Chem. 1996; 271:2063–2070. [PubMed] [Google Scholar]
- 37. Wisniewski M., Balakrishnan M., Palaniappan C., Fay P.J., Bambara R.A.. Unique progressive cleavage mechanism of HIV reverse transcriptase RNase H. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:11978–11983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sarafianos S.G., Das K., Tantillo C., Clark A.D., Ding J., Whitcomb J.M., Boyer P.L., Hughes S.H., Arnold E.. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001; 20:1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith C.M., Smith J.S., Roth M.J.. RNase H requirements for the second strand transfer reaction of human immunodeficiency virus type 1 reverse transcription. J. Virol. 1999; 73:6573–6581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sherman P.A., Dickson M.L., Fyfe J.A.. Human immunodeficiency virus type 1 integration protein: DNA sequence requirements for cleaving and joining reactions. J. Virol. 1992; 66:3593–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lowe T.M., Chan P.P.. tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016; 44:W54–W57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tu G.C., Cao Q.N., Zhou F., Israel Y.. Tetranucleotide GGGA motif in primary RNA transcripts. Novel target site for antisense design. J. Biol. Chem. 1998; 273:25125–25131. [DOI] [PubMed] [Google Scholar]
- 43. Katayanagi K., Miyagawa M., Matsushima M., Ishikawa M., Kanaya S., Ikehara M., Matsuzaki T., Morikawa K.. Three-dimensional structure of ribonuclease H from E. coli. Nature. 1990; 347:306–309. [DOI] [PubMed] [Google Scholar]
- 44. Yang W., Hendrickson W.A., Kalman E.T., Crouch R.J.. Expression, purification, and crystallization of natural and selenomethionyl recombinant ribonuclease H from Escherichia coli. J. Biol. Chem. 1990; 265:13553–13559. [PubMed] [Google Scholar]
- 45. Gaidamakov S.A., Gorshkova I.I., Schuck P., Steinbach P.J., Yamada H., Crouch R.J., Cerritelli S.M.. Eukaryotic RNases H1 act processively by interactions through the duplex RNA-binding domain. Nucleic Acids Res. 2005; 33:2166–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Vickers T.A., Crooke S.T.. The rates of the major steps in the molecular mechanism of RNase H1-dependent antisense oligonucleotide induced degradation of RNA. Nucleic Acids Res. 2015; 43:8955–8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M.. Global quantification of mammalian gene expression control. Nature. 2011; 473:337–342. [DOI] [PubMed] [Google Scholar]
- 48. Schultz S.J., Zhang M., Champoux J.J.. Recognition of internal cleavage sites by retroviral RNases H. J. Mol. Biol. 2004; 344:635–652. [DOI] [PubMed] [Google Scholar]
- 49. Schultz S.J., Zhang M., Champoux J.J.. Sequence, distance, and accessibility are determinants of 5′-end-directed cleavages by retroviral RNases H. J. Biol. Chem. 2006; 281:1943–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schultz S.J., Zhang M., Champoux J.J.. Preferred sequences within a defined cleavage window specify DNA 3′ end-directed cleavages by retroviral RNases H. J. Biol. Chem. 2009; 284:32225–32238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Champoux J.J., Schultz S.J.. Ribonuclease H: properties, substrate specificity and roles in retroviral reverse transcription. FEBS J. 2009; 276:1506–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hostomsky Z., Hostomska Z., Hudson G.O., Moomaw E.W., Nodes B.R.. Reconstitution in vitro of RNase H activity by using purified N-terminal and C-terminal domains of human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bohlayer W.P., DeStefano J.J.. Tighter binding of HIV reverse transcriptase to RNA-DNA versus DNA-DNA results mostly from interactions in the polymerase domain and requires just a small stretch of RNA-DNA. Biochemistry. 2006; 45:7628–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lapkouski M., Tian L., Miller J.T., Le Grice S.F.J., Yang W.. Complexes of HIV-1 RT, NNRTI and RNA/DNA hybrid reveal a structure compatible with RNA degradation. Nat. Struct. Mol. Biol. 2013; 20:230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Figiel M., Krepl M., Poznański J., Gołąb A., Šponer J., Nowotny M.. Coordination between the polymerase and RNase H activity of HIV-1 reverse transcriptase. Nucleic Acids Res. 2017; 45:3341–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gifford R.J. Viral evolution in deep time: lentiviruses and mammals. Trends Genet. 2012; 28:89–100. [DOI] [PubMed] [Google Scholar]
- 57. Kleiman L., Cen S.. The tRNALys packaging complex in HIV-1. Int. J. Biochem. Cell Biol. 2004; 36:1776–1786. [DOI] [PubMed] [Google Scholar]
- 58. Kleiman L., Jones C.P., Musier-Forsyth K.. Formation of the tRNALys packaging complex in HIV-1. FEBS Lett. 2010; 584:359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Beerens N., Groot F., Berkhout B.. Initiation of HIV-1 reverse transcription is regulated by a primer activation signal. J. Biol. Chem. 2001; 276:31247–31256. [DOI] [PubMed] [Google Scholar]
- 60. Beerens N., Berkhout B.. Switching the in vitro tRNA usage of HIV-1 by simultaneous adaptation of the PBS and PAS. RNA. 2002; 8:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abbink T.E.M., Beerens N., Berkhout B.. Forced selection of a human immunodeficiency virus type 1 variant that uses a non-self tRNA primer for reverse transcription: involvement of viral RNA sequences and the reverse transcriptase enzyme. J. Virol. 2004; 78:10706–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Puglisi E.V., Puglisi J.D.. HIV-1 A-rich RNA loop mimics the tRNA anticodon structure. Nat. Struct. Biol. 1998; 5:1033–1036. [DOI] [PubMed] [Google Scholar]
- 63. Jones C.P., Saadatmand J., Kleiman L., Musier-Forsyth K.. Molecular mimicry of human tRNALys anti-codon domain by HIV-1 RNA genome facilitates tRNA primer annealing. RNA. 2013; 19:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yu Q., Morrow C.D.. Essential regions of the tRNA primer required for HIV-1 infectivity. Nucleic Acids Res. 2000; 28:4783–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McCulley A., Morrow C.D.. Nucleotides within the anticodon stem are important for optimal use of tRNA(Lys, 3) as the primer for HIV-1 reverse transcription. Virology. 2007; 364:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. DeStefano J.J., Buiser R.G., Mallaber L.M., Myers T.W., Bambara R.A., Fay P.J.. Polymerization and RNase H activities of the reverse transcriptases from avian myeloblastosis, human immunodeficiency, and Moloney murine leukemia viruses are functionally uncoupled. J. Biol. Chem. 1991; 266:7423–7431. [PubMed] [Google Scholar]
- 67. DeStefano J.J., Mallaber L.M., Fay P.J., Bambara R.A.. Quantitative analysis of RNA cleavage during RNA-directed DNA synthesis by human immunodeficiency and avian myeloblastosis virus reverse transcriptases. Nucleic Acids Res. 1994; 22:3793–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hizi A., Tal R., Shaharabany M., Loya S.. Catalytic properties of the reverse transcriptases of human immunodeficiency viruses type 1 and type 2. J. Biol. Chem. 1991; 266:6230–6239. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.