

Abstract
The lasonolides are novel polyketides that have displayed remarkable biological activity in vitro against a variety of cancer cell lines. Herein we describe our first generation approach to the formal synthesis of lasonolide A. The key findings from these studies ultimately allowed us to go on and complete a total synthesis of lasonolide A. The convergent approach unites two highly complex fragments utilizing a Ru-catalyzed alkene-alkyne coupling. This type of coupling typically generates branched products, however through a detailed investigation we are now able to demonstrate that subtle structural changes to the substrates can alter the selectivity to favor the formation of the linear product. The synthesis of the fragments features a number of atom economical transformations which are highlighted by the discovery of an engineered enzyme to perform a dynamic kinetic reduction of a β-ketoester to establish the absolute stereochemistry of the southern tetrahydropyran ring with high levels of enantioselectivity.
Graphical Abstract
INTRODUCTION
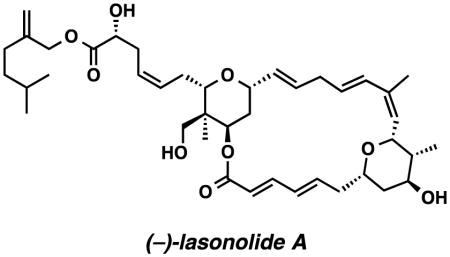
Lasonolide A was discovered in 1994 by McConnell and coworkers in an effort to identify new and diverse antitumor agents from marine organisms.1 The connectivity of the lasonolides and relative stereochemistry of each tetrahydropyran were determined by NMR correlation spectroscopy from isolated materials. In 2002, the correct fully elucidated structure of lasonolide A was disclosed in Lee’s total synthesis.2 This seminal work established the unknown relative stereochemistry of the C28 stereocenter and corrected the C17–C18 and C25–C26 olefin geometries, which had been incorrectly assigned (Figure 1). Importantly, the synthesis also established the absolute stereochemistry of the natural product and revealed that the levorotatory or (−)-lasonolide A was the biologically active enantiomer, contrary to what had been reported in the isolation paper. Despite the remarkable activity lasonolide A displayed in the NCI’s 60-cell line screen,3 very few analogs have been prepared.4,5
Figure 1.

Reassigned Lasonopyran Skeleton – Lasonolides A – G
RESULTS AND DISCUSSION
Our pursuit of the target molecule was not only inspired by its unique biological activity, but also its novel molecular architecture that presented us with the opportunity to execute and expand on many of the organic transformations that had previously been developed in our laboratory. We aimed at developing a more concise and efficient synthesis compared to those previously known. 2,6,7 A preliminary report of our efforts has been communicated.7a
We recognized that the main point of diversification between lasonolides A-F occurred at the C28 and C30 positions (Figure 1. Subtle structural differences had dramatic effects on the biological profiles between each of the six natural analogs. While a comprehensive understanding of the structure activity relationships remains unclear, we felt that it was important to devise a synthesis plan (Scheme 1) that could allow for the installation of a range of diverse structural elements at these positions. A late stage Wittig olefination could install the required Z-olefin geometry present in all lasonolides and enable a flexible strategy for analog synthesis. The macrocycle can then be disconnected into two equally complex tetrahydropyrans, which could be joined together by an esterification/macrolactonization and a Ru-catalyzed alkene-alkyne coupling. These disconnections of the lasonopyran skeleton allowed us to identify three sub-targets 9, 10, and 11 for synthesis.
Scheme 1.

Synthesis Plan for (−)-Lasonolide A
The hallmark of our synthetic plan, the Ru-catalyzed alkene-alkyne coupling, was unique in the fact that we aimed to obtain a linear 1,4-diene product. Previously we demonstrated that branched selectivity is generally observed, as shown in eq.1.8 However, in a few cases, the linear product dominated (eq. 2). We have put forth a mechanistic rationale to understand this behavior. The proposed mechanism for the transformation, via ruthenacyclopentene formation, is depicted in Figure 2. The initial oxidative cycloisomerization with the Ru-catalyst is believed to be reversible and can form one of two possible regioisomers (A or B). A subsequent β-hydride and reductive elimination, from the metallacycles, can afford the linear or branched isomers. Steric factors suggest that intermediate B is more stable than intermediate A which accounts for the preferential formation of the branched product. However, the tautomerization of the initial alkene-alkyne complex C is thought to be faster than that of complex D then forms the new C-C bond between the sterically less-hindered terminus of each unsaturated partner. If the rate of β-hydride elimination of complex A can outcompete its cycloreversion to complex C, a linear product would result. Indeed, introduction of a tetrasubstituted propargylic center inhibits cycloisomerization of complex D to the point that the reaction is now favored via complex A.
Figure 2.

Known Ru-Catalyzed Alkene-Alkyne Couplings Favoring Linear and Branched Products – Proposed Catalytic Cycle
In all our prior total syntheses involving terminal alkyne partners, only branched products have been formed – alternaric acid,9 amphidinolides A10 and P,11 and laulimalide.12 The particular efficiency of the Ru catalyzed macrocyclization in the laulimalide synthesis stimulates exploration of the Ru catalyzed process making linear rather than branched products. To be applied to the total synthesis of lasonolide A, there are two possible approaches that could afford the desired product (Scheme 2). The first would feature an intramolecular reaction to generate the macrocycle (14) of lasonolide A. The second would be an intermolecular approach to form 19. Theoretically, in each scenario the linear isomer can be formed using either functional group orientation (i.e. 12 vs. 13 and 15/16 vs. 17/18).
Scheme 2.

Proposed Ru-catalyzed Alkene-Alkyne Couplings
We, therefore, initiated studies on representative model systems. Tetrahydropyrans 2013 and 21 were subjected to several reaction conditions to test the feasibility of the intermolecular coupling and to gain insight concerning the levels of linear:branched selectivity. We screened several solvents that have been previously utilized in alkene-alkyne couplings. No reaction was observed in non-coordinating solvents such as CH2Cl2 and DCE (Table 1, entries 1 and 2), likely due to catalyst decomposition. Next we screened coordinating solvents (acetone and DMF) (Table 1, entries 3–5) in an effort to stabilize the coordinatively unsaturated Ru-catalyst. To our delight, reactions run in DMF and acetone delivered the desired coupled products. The linear:branched ratios were much higher in acetone (3.5:1) than in DMF (1:1), suggesting that DMF promotes a reversible cycloisomerization and therefore Curtin-Hammett conditions. Increasing the amount of alkene 21 (5 equiv.) delivered the desired 1,4-diene (22) as a 4:1 mixture of linear:branched isomers in 56% yield.14 Importantly, 74% of the excess alkene was recovered at the end of the reaction.
Table 1.
Model Studies for Alkene-Alkyne Coupling
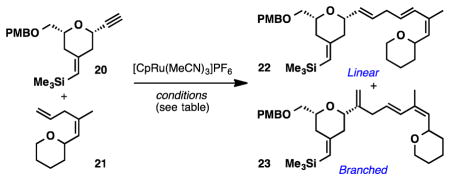
| ||||
|---|---|---|---|---|
| Entrya | Solvent (Conc.) | 21 (equiv)b | Convc | 22:23c |
| 1 | CH2Cl2 (5 M) | 1.2 | 0% | - : - |
| 2 | DCE (5 M) | 1.2 | 0% | - : - |
| 3 | DMF (5 M) | 1.2 | 33% | 1 : 1 |
| 4 | Acetone (5 M) | 1.2 | 37% | 3.5 : 1 |
| 5 | Acetone (1 M) | 5.0 | 56%d | 4.0 : 1 |
All reactions were run using 0.1 mmol (20), and 10 mol% [CpRu(MeCN)3]PF6 at rt for 14 h.
21 was added as a solution drop-wise over 1h using a syringe pump.
Determined by 1H NMR.
Isolated yield.
The alternative substitution pattern was also explored on a similar model system using the optimal conditions discovered in Table 1 - entry 5 (Scheme 3). In this case, the branched isomer (27) was exclusively formed in 33% yield.
Scheme 3.
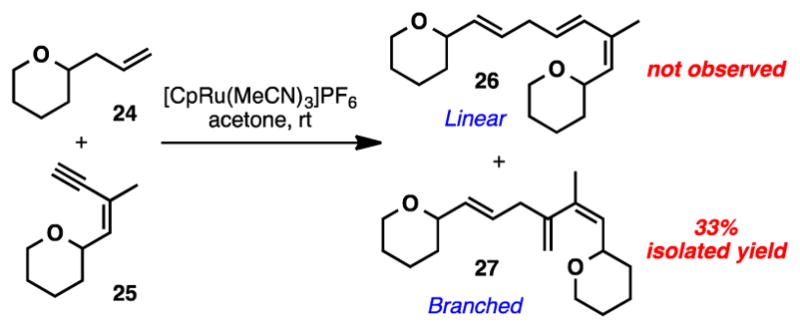
Additional Model Studies for Alkene-Alkyne Coupling
The synthesis plan for alkyne 28 is presented in Scheme 4. A dinuclear Zn-catalyzed aldol reaction between ynone 32 and aldehyde 31 is envisioned to establish the absolute stereochemistry of fragment 30. It is important that high enantioselectivity is attained, since the C21 hydroxyl group will dictate the formation of each subsequent stereocenter. The C22 quaternary stereocenter will be created from the thermodynamic formation of benzylidene acetal 29 via transacetilization. An analogous approach was previously reported by the Kang group in their total synthesis of (+)-lasonolide A.7d Finally, Horner-Wadsworth-Emmons (HWE) olefination with subsequent oxy-Michael addition should favor formation of the desired equatorial C23 stereocenter to complete the synthesis of alkyne 28.
Scheme 4.
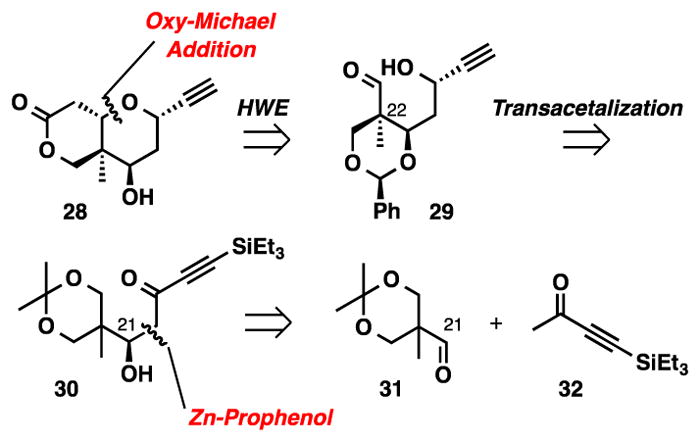
Synthesis Plan for Alkyne 28
The synthesis of alkyne 28 commenced with an asymmetric aldol reaction between ynone 32 and aldehyde 31.15 Under our standard set of conditions significant elimination was observed when reactions were run at room temperature (Table S2, entry 1), and at 4°C (Table S2, entry 2).16 The elimination could be suppressed by running the reaction at −20°C (Table S2, entry 3) for 72 h. This affords the desired β-hydroxy ketone (30) in a moderate 54% yield but with high levels of enantioselectivity (99% ee). Elimination of the β-hydroxy group could be entirely suppressed by modifying the work-up procedure. Switching from an aqueous work-up to a direct filtration through celite improved the isolated yield of 30 to 78% (Table S2, entry 4).
With an optimized protocol in hand, we then investigated a 1,3-syn reduction of β-hydroxy ketone 30 (Scheme 5). Under standard conditions (Et2BOMe and NaBH4)17 the reduction of 30 only afforded a ~4:1 mixture of diastereomers favoring the syn-diol. Switching to Kiyooka’s conditions (DIBAL-H, −78°C) increased the diastereoselectivity to 17:1 and the desired diol (33) was isolated in 98% yield.18 Selective protection of the less sterically hindered secondary propargyl alcohol with TBDPSCl gave 34.
Scheme 5.
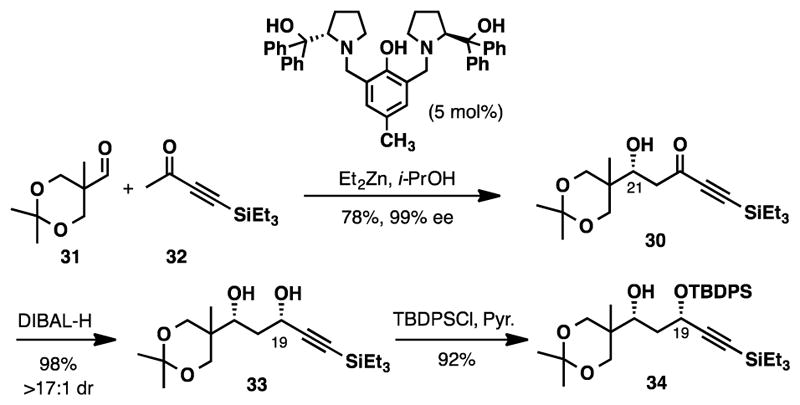
Preparation of 34
We then aimed to establish the C22 quaternary stereocenter via transacetalization of 34 to form a thermodynamically favored benzylidene acetal.7d Utilization of Kang’s conditions (PhCHO, TFA, PhMe −20°C to rt, 3.5 h), led primarily to the formation of isomers 35c and 35d (Figure 3). Only trace amounts of 35a and 35b were detected in the crude 1H NMR spectrum. Although the cyclization favored the incorrect isomer, we were encouraged that these products were isolated in near quantitative yield with little evidence of decomposition that may have arisen from the acidic conditions. Assuming that the observed products were not the thermodynamically preferred ones, we decided to conduct the acetal formation in a more polar solvent (CHCl3) for an extended period of time. Under these new conditions the desired acetal (35a) was strongly favored in good yield, 5:1 chemoselectivity and 10:1 diastereoselectivity. Importantly, the undesired isomers (35b, 35c and 35d) could be separated from the predominant diasteromer (35a) by silica gel chromatography and re-subjected to the reaction conditions. After 2 rounds of recycling the undesired diastereomers, acetal 35a, containing the newly formed C22 quaternary stereocenter, was isolated in 93% yield.
Figure 3.

Selective Formation of Benzylidene Acetal 35a
With 35a in hand we pushed forward towards the completion of the alkyne 28 (Scheme 6). The sequence began with exploring an oxidation of the primary neopentylic alcohol. Surprisingly, 35a was resistant to oxidation using Dess-Martin periodinane, PDC, TEMPO, and under Moffatt-Swern conditions. Fortunately, Ley’s catalytic TPAP/NMO oxidation reliably delivered aldehyde 36 in 76% yield.19 TBAF mediated deprotection of the TBDPS and TES protecting groups followed by concomitant cyclization produced lactol 38 in 59% yield. In addition to lactol 38, carboxylic acid 37 was also isolated in 20% yield. We postulated the undesired side product was arising from a Cannizzaro reaction promoted by the hydroxide typically present in TBAF solutions. Buffering the reaction with acetic acid20 suppressed formation of the carboxylic acid and improved the isolated yield to 94%.
Scheme 6.
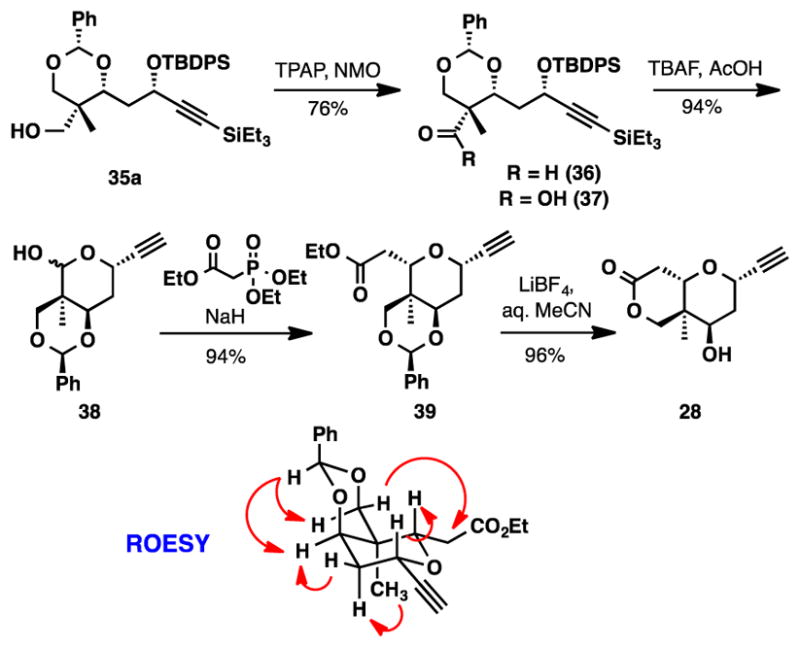
Synthesis of Alkyne 28
Horner-Wadsworth-Emmons olefination and concomitant intramolecular oxy-Michael addition generated tetrahydropyran 39 as a single diastereomer detectable by 1H NMR in 94% isolated yield.21 The stereochemistry of the tetrahydropyran was assigned by ROESY correlation.
The completion of the alkyne 28 involved hydrolysis of the benzylidene acetal and cyclization of the resulting primary alcohol. Toward this end, a variety of acids were screened to effect the desired hydrolysis/cyclization sequence. The use of p-toluenesulfonic acid (MeOH)22 or acetic acid (THF, 50 °C)23 gave only recovered starting material. Reactions involving BCl3 (DCM, −78 °C to rt),24 2M HCl (MeOH, 65 °C),25 or Amberlyst A-15 (4 Å MS, MeCN)26 led to complicated mixtures of partially hydrolyzed products and alkyne 28 in <40% isolated yield. This partial conversion to the lactone was not unexpected since the equilibrium to remove the benzylidene requires the presence of both acid and water, whereas the equilibrium to form the lactone requires the presence of acid, but the exclusion of water/alcohol. Along these lines, we discovered that the desired lactol could be obtained in 53–82% isolated yield by utilizing a two-step procedure, which involved hydrolysis of the acetal with HCl in THF at 80 °C and then ring closure using catalytic p-TsOH in refluxing toluene.27 We later discovered that formation of alkyne 28 could be accomplished in a single step using LiBF4 in aq. MeCN in 96% yield.28
The synthesis plan for alkene 11 is presented in Scheme 7. A dynamic kinetic asymmetric reduction of β-ketoester 41 was envisioned to establish the absolute stereochemistry. The C7 stereocenter would be formed through a Michael addition, which could occur after the installation of an appropriate acceptor. Several possible routes were considered for the formation of the dienoate fragment. Our first tier of experiments would focus on incorporating this side chain via Horner-Wadsworth-Emmons olefination. Alternatively, if complications arose from the proposed HWE reaction, we also developed a convenient new strategy to form 2,4-dienoates that evolved from the reaction of terminal alkynes with ethyl diazoacetate29 followed by subsequent phosphine-catalyzed isomerization.30 A series of model studies, as illustrated in the conversion of alkyne 43 to dienoate 46 via intermediate 45, validated this latter approach (see Scheme 7).16
Scheme 7.
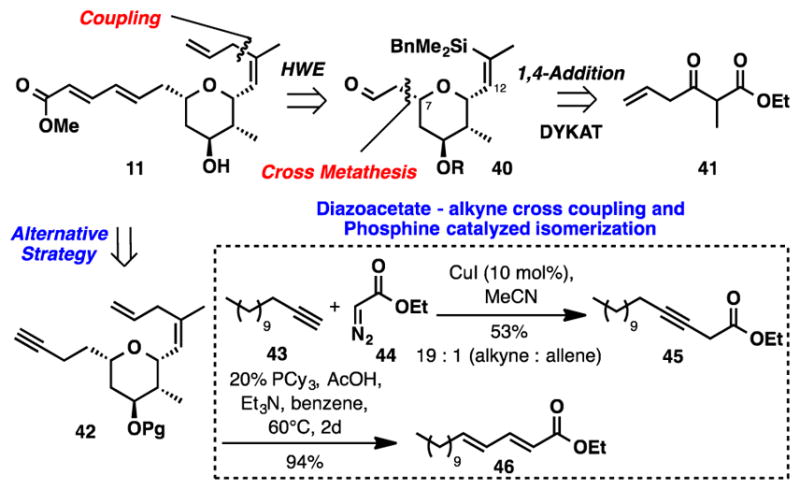
Synthesis Plans for Alkene 11
Initially, we were intrigued by the prospect of utilizing a transition metal catalyzed hydrogenation reaction to establish the absolute stereochemistry of fragment 11.31 Although these highly atom economical32 methods proceed with high enantio- and diastereoselectivities for a range of substrates, the chiral catalysts developed so far exhibit poor diastereoselectivities for methyl substituted β-ketoesters such as 41.33 As a result, we believed it would be worthwhile to investigate a microbial transformation that could establish the necessary absolute and relative stereochemistry via dynamic reduction of a racemic β-ketoester. After surveying the literature, we discovered that Baker’s yeast reductions of methyl-substituted β-ketoesters could provide the requisite products.16,34
Encouraged by this precedent, we embarked on our synthesis of fragment 11 first by preparing the necessary starting material (Scheme 8). β-ketoester 41 was generated through a Blaise reaction with commercially available α-bromoester 47 and allyl cyanide.35 Interestingly, when we investigated the reduction of β-ketoester 41 with Bakers’ yeast (Sigma-Aldrich, YSC2, batch 035K0169) the undesired anti-product was formed with 10:1 diastereoselectivity (30% ee). Without heat treatment or additives, the diastereoselectivity for the reduction decreased to 4:1. The relative stereochemistry was further confirmed by nOe and J-coupling analysis on an advanced intermediate.36 These results were in direct contrast to the trends observed from the literature, therefore we decided to screen a number of isolated enzymes,37 generously donated to us by Codexis Inc., to determine their performance in the dynamic kinetic asymmetric reduction of β-ketoester 41. Two types of enzymes were examined in the reaction; the first was derived from Bakers’ yeast and utilized GDH (glucose dehydrogenase) and NADP with glucose as the stoichiometric reductant.16 The pH of these reactions must be carefully controlled due to the accumulation of gluconic acid. The second type of enzyme utilizes NADPH as the cofactor and 2-propanol as the reductant. Table S5 of the SI provides a comprehensive screening of enzymatic methods.
Scheme 8.
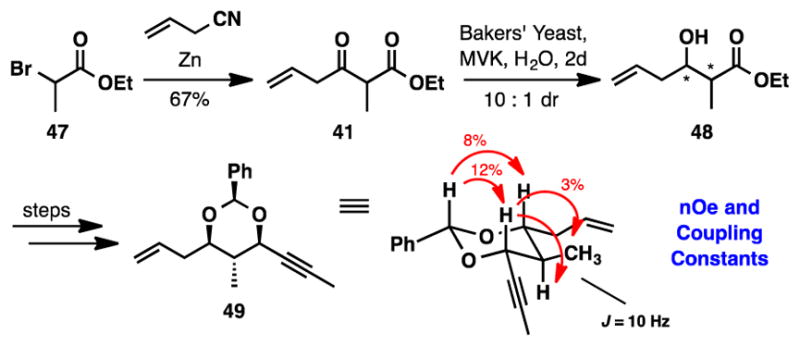
Relative Stereochemical Assignment of 49
NADP dependent ketoreductases, in general, showed enhanced selectivity for the syn product. However, under the standard conditions (Conditions A) using triethanolamine as a buffer, significant olefin isomerization of β-ketoester 41 was observed. After an exhaustive investigation of various buffers (pH 4.5 – 10 screened), we found that a pH 4.5 phosphate buffer completely suppressed the isomerization. CDX-024 demonstrated the highest diastereoselectivity favoring the syn-diastereomer (48) with high levels of enantioselectivity (see SI, Table S5, entries 7 and 8). Using this enzyme we were able to obtain the desired syn product (48) in 75% yield, with a 4 : 1 diastereoselectitivy and greater than 95% ee (Table S5, entry 8).38
The synthesis of fragment 11 was advanced using the synthetic sequence depicted in Scheme 9. It began with the silyl protection of the C9 alcohol, which was subsequently followed by a 2-step Weinreb amide synthesis and Grignard addition to obtain ynone 52/53 in excellent yield.39 (S)-CBS reduction under standard conditions ((S)-CBS, BH3·DMS, THF) delivered the desired propargyl alcohols (54/55) in good yields (50–70%) as a 6.7 : 1 mixture of diastereomers. Yu has reported that the use of nitroethane as solvent has a profound effect on both reaction rate and stereoselectivity.40 Pleasingly, we found that when we made this switch and used freshly distilled catechol borane the diastereoselectivity improved to >20:1.
Scheme 9.
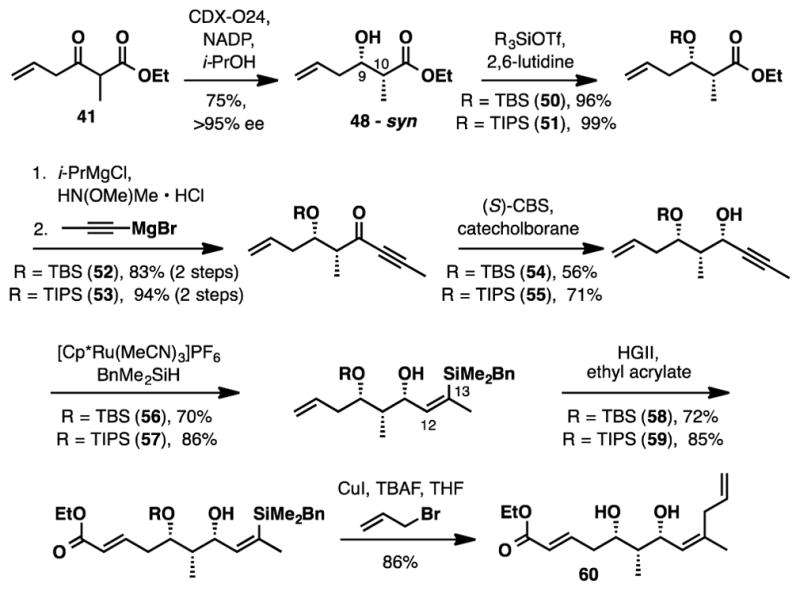
Synthesis of Diol 60
Propargyl alcohol 54/55 was then subjected to a Ru-catalyzed hydrosilylation to generate trisubstituted (Z)-vinylsilane 56/57.41 In each case (R=TBS, or R=TIPS) high selectivities for a single geometric isomer (>15:1) were obtained. In preparation for tetrahydropyran formation via oxy-Michael addition, the enoate acceptor was installed through cross metathesis with ethyl acrylate.42 Finally, the C13 allyl moiety was introduced by a copper mediated cross coupling to form diol 60.43
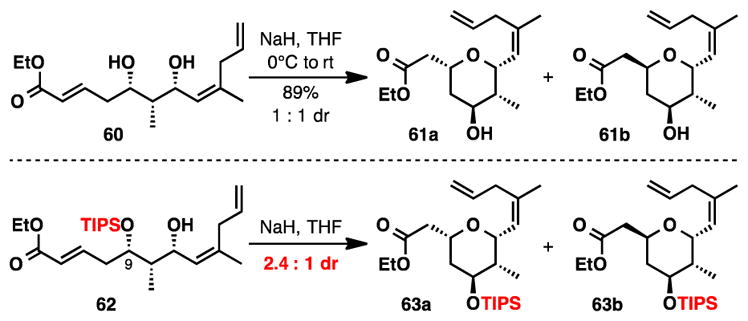
Deprotonation of diol 60 with sodium hydride generated tetrahydropyrans 61a/61b as a 1:1 mixture of diastereomers. Unfortunately, no improvement in diastereoselectvity was observed after screening various bases (t-BuOK and EtONa), solvents (DMF, THF, and DMSO) and reaction temperatures (−78°C, 0°C, rt and 60°C). Furthermore, equilibration of the diastereomeric mixture under basic conditions did not improve diastereoselectivity. We were aware that a similar approach to the southern tetrahydropyran ring was utilized in Kang’s total synthesis of (+)-lasonolide A;7d during their studies, an enhancement in diastereoselectivity was observed upon switching the protecting group on the C9 hydroxyl from TBS to TIPS. Likewise, we observed a modest increase in diastereoselectivity (1:1 to 2.4:1) for the cyclization when a TIPS protecting group was incorporated (62). This experiment supported the notion that this structural feature was important for improving selectivity in the cyclization.
Several reports in the literature have indicated that cyclizations of secondary alcohols onto a proximal α,β-unsaturated aldehyde could enhance diastereoselectivity.44 It is conceivable that the lower pKa of the proton adjacent to the aldehyde would render the reaction more reversible, and thereby favor the formation of the thermodynamically preferred 2,6-cis tetrahydropyran ring. To test this hypothesis, α,β-unsaturated aldehyde 65 was prepared (Scheme 10) in two steps from ester 60 and subjected to cyclization using DBU. We found that the diastereoselectivity was improved to ~4–5 : 1. Recalling our previous experience, we suspected that the incorporation of a protecting group at C9 could further enhance distereoselectivity. Upon preparing the TIPS protected substrate, spontaneous cyclization was observed upon oxidizing allylic alcohol 67 with MnO2, and the desired tetrahydropyran (68) was isolated as a single diastereomer in an un-optimized 47% yield.
Scheme 10.
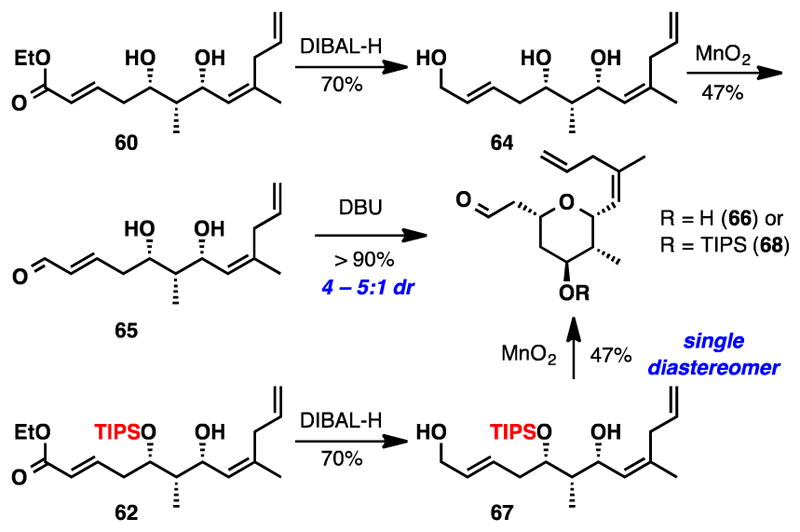
Further Studies Investigation THP Cyclizations
While encouraged by these results, access to allylic alcohol 67 was problematic due to several low yielding steps that were not amenable to scale-up. To circumvent these issues we decided to pursue an alternative reaction sequence that would feature a THP cyclization, bearing the necessary TIPS protection on the C9 hydroxyl, before installation of the C13 allyl segment (Scheme 11). However, in this alternate sequence, the alcohol required for the copper-mediated cross-coupling reaction would no longer be available. Therefore we decided to investigate a Hiyama cross coupling45 for the late stage installation of the allyl moiety. At the time this work was executed, the use of allyl acetate in the Hiyama cross coupling had not been described.
Scheme 11.
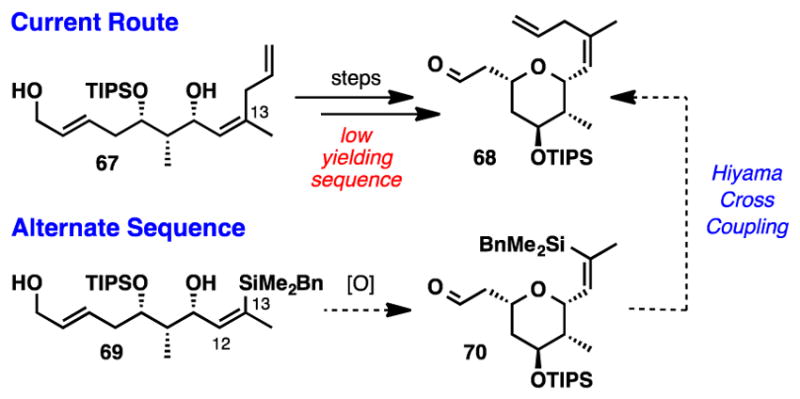
Revised Synthetic Approach to Access THP 68
The revised synthesis of fragment 11 began with the DIBAl-H reduction of ester 59 (Scheme 12). Allyic oxidation of alcohol 69 with MnO2 afforded tetrahydropyran 70 directly in nearly quantitative yield as a single diastereomer.46 Attempts to perform the Hiyama cross coupling with aldehyde 70 and allyl acetate were unsuccessful, presumably due to the insufficient stability of the starting aldehyde. To eliminate this problematic functionality, we first installed the dienoate moiety, before re-exploring the Hiyama coupling. Elaboration of the aldehyde into E,E-dienoate 71 was accomplished via Horner-Wadsworth-Emmons olefination using 4 Å MS and LiOH.
Scheme 12.
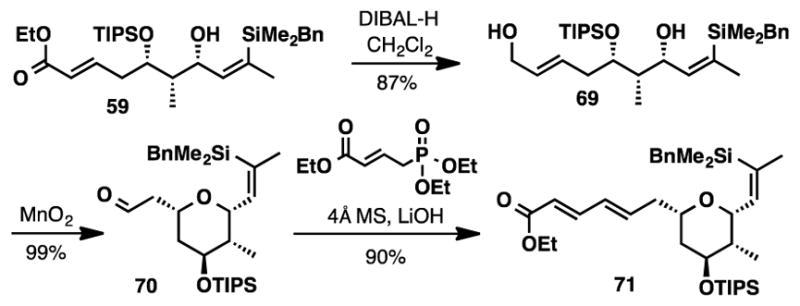
Synthesis of Vinyl Silane 71
Our investigation of the Hiyama coupling16 between allyl acetate47 and vinyl silane 71 began with utilizing 2 mol% Pd2dba3·CHCl3 and 4.2 equivalents of TBAF in a solution of THF (see Scheme 13 and SI Table S6). After 19 hours, a mixture of the desired product (72) along with an equimolar amount of silanol 73 (entry 1) was obtained in 75% combined yield. Extending the reaction time from 19 hours to 3 days also generated a 1:1 mixture of 72 : 73 but diminished the yield to 44% (entry 2). Increasing both the catalyst loading (10 mol%) and amount of TBAF (6.4 equiv.) was found to decrease the proportion of the remaining intermediate silanol 73, and alkene 72 could be isolated in 78% yield (entry 3). It is important to note that, when we repeated the experiment, we noticed that the highest yields for this reaction (85%) were obtained when fresh TBAF solutions were used (entry 4). This modest increase in yield may be related to the water concentration present in TBAF solutions.48 Alkene 75 was completed in two steps from 72; saponification using lithium hydroxide, followed by TBS protection and in situ hydrolysis of the resulting silyl ester (Scheme 13).
Scheme 13.
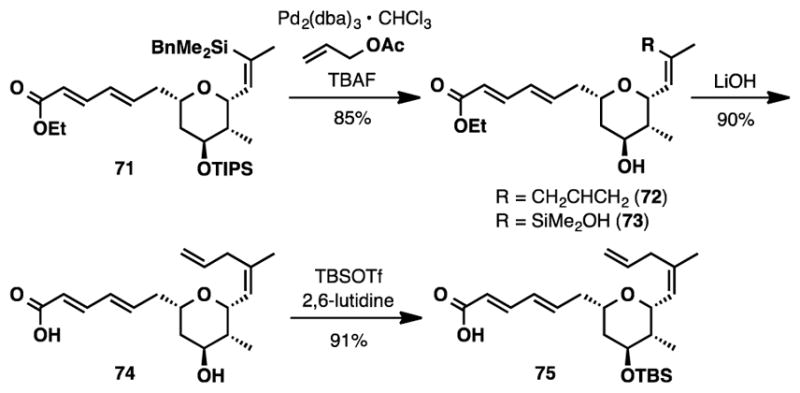
Synthesis of Alkene 75
Having achieved a synthesis of both fragments, we were poised to examine our key Ru-catalyzed alkene-alkyne coupling. Initial efforts were dedicated to exploring an intramolecular reaction to forge the macrocycle, since to date only three examples of intramolecular Ru-catalyzed alkene-alkyne couplings have been reported. These have appeared in the total syntheses of (+)-amphidinolide A, (+)-pinnatoxin A,49 and laulimalide. However, in each of these examples only branched products were generated. Figuring we possessed the necessary coordinating groups to favor the generation of a linear isomer, we were excited by the prospect of investigating a linear selective macrocyclization. Two substrates were examined in the macrocyclization reaction (76 and 78), each of which could easily be accessed using a Yamaguchi esterification.50 Unfortunately, after extensive investigation we were never able to form the desired macrocycle as rapid decomposition was observed in all experiments that were attempted (Scheme 14).
Scheme 14.
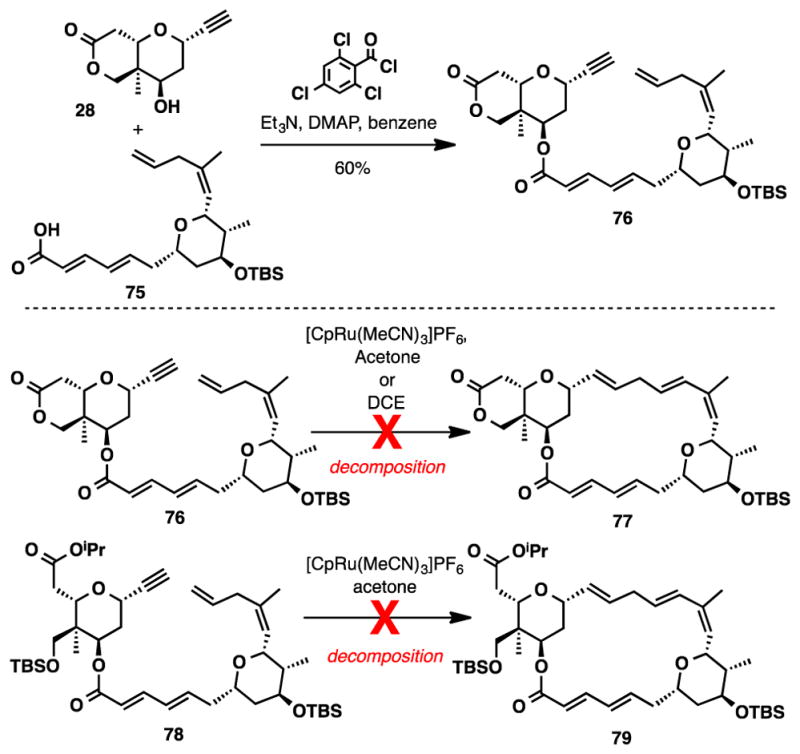
Attempted Macrocyclization via Ru-Catalyzed Alkene-Alkyne Coupling
We then decided to turn our attention to the exploration of an intermolecular alkene-alkyne coupling (Figure 5, eq 3).16 We were concerned that the π systems of electron deficient ester, present in alkene 11, could be coordinating to the ruthenium catalyst and inhibiting the coupling. As a result, we chose to use alkyne 28 in excess in an attempt to diminish this potential interaction (Table S7, entries 1–6). We observed 50% conversion of the alkene to the desired coupled products (80a and 80b) as an inseparable 2:1 mixture of linear : branched isomers (entry 1). Unfortunately, the excess alkyne that was used could not be recovered as it seems to decompose over the course of the reaction, likely to the hydrated dimer.9 Heating the reaction to 50 °C had little effect on the overall conversion (entry 2). However, when the alkyne was added to the reaction drop-wise over 15 minutes and then heated at 50°C we noticed an increase in conversion to 70% (entry 3). Increasing the concentration from 0.02 to 0.04 M (entry 4) also had no effect on conversion.
Figure 5.

Intermolecular Ru-Catalyzed Alkene-Alkyne Coupling
Content with our reaction conditions (entry 3), we increased the scale of the reaction from 5 μmol to 60 μmol in an attempt to obtain an isolated yield for the coupling. To our surprise, this seemingly minor modification was not tolerated and the conversion diminished significantly to ~20%. In this case, increasing the concentration to 0.12 M (entry 5) rescued the conversion slightly (35%) but not to the levels that were seen previously. Increasing both the catalyst loading and concentration had a deleterious effect on the overall conversion (entry 6).
Based on the experimental evidence presented in Table S7, entries 1–5 (see SI), we realized that our initial concerns related to catalyst deactivation via coordination to alkene 11 were largely attenuated due to the fact that in each of these entries, which successfully generated product, a 5-fold excess of alkene 11 to catalyst was present. Therefore we decided to screen reactions that used the alkene in excess. This change resulted in the complete consumption of alkyne 28 on a 68 μmol scale (Table S7, entry 7). Furthermore, after brief optimization, we determined that only 3 equiv. of alkene 11 were needed (using 10 mol% catalyst, in a 0.094 M solution of acetone) to achieve a 66% yield (entry 8).
With a reliable synthetic route, we advanced our synthesis adhering to the current synthesis plan (Scheme 15). Saponification of the ester and lactone, followed by re-lactonization delivered seco acid 81. Macrolactonization was attempted with a variety of reagents including 2,4,6-trichlorobenzoyl chloride, 2-methyl-6-nitrobenzoic anhydride,51 and dibutyltin oxide.52 Unfortunately, in all cases the macrocycle was never observed and the substrate decomposed under each of the reaction conditions. We speculated that the restricted conformational mobility of the C21 alcohol could be unable to access its optimal conformation for macrolactonization. Therefore, opening the lactone could enable further flexibility of the C21 alcohol and could allow for the desired cyclization to occur. As a result we decided to incorporate this idea into two modified substrates, 83 and 84.
Scheme 15.

Attempted Macrolactonization and Revised Synthetic Strategy
We had hoped to access the elaborated seco acid (83) from lactone 80a via reduction to the lactol, subsequent Wittig olefination, and saponification. Disappointingly, after completing the synthesis of Wittig salt 90, the proposed reaction sequence was unsuccessful in our model system (Scheme 16). Warming the reaction above −15°C decomposed the unstabilized Wittig reagent rapidly.
Scheme 16.
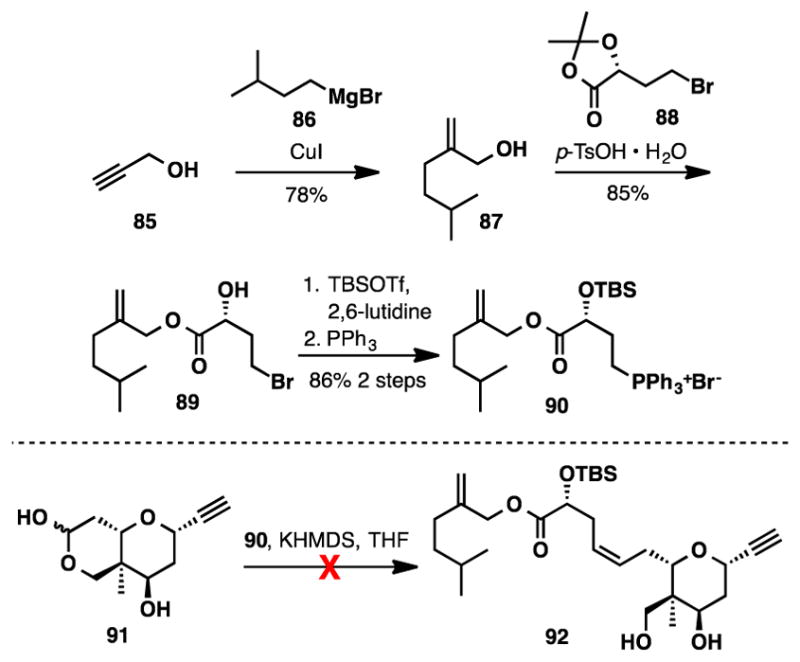
Preparation of Wittig Salt 90 and Attempted Olefination with Lactol 91
At this point, we decided to take a step back and, in turn, we began investigating a synthesis of seco acid 84. Ideally, an alkene-alkyne coupling between triol 93 and alkene 94 could provide rapid access to seco acid 84. Accordingly, we began screening conditions similar to those found to be optimal previously (Table S7). When acetone was used as solvent, we were able to obtain the desired products as their acetonides in 32% yield, as a 3.2:1 mixture of linear:branched isomers (Table 2, entry 1). Upon switching the solvent to cyclopentanone, the product ratio was enhanced to 3.8:1 (entry 2) – this was the highest ratio we had ever observed in our studies. We speculate that the enhanced selectivity could be related to several factors. Because the ruthenium catalyst has one open coordination site, the size of the solvent could have an effect on the product ratios. Alternatively, triol 93 might have been protected as its acetonide or cyclopentanone ketal before coupling occurs and the linear to branched ratios may be substrate specific. Finally, it is known that coordinating substituents in the substrates have dramatic effects on product distributions,53 and the presence of the C25 primary alcohol may be contributing to selectivity.
Table 2.
Probing at Structure and Solvent Effects in Ru-Catalyzed Alkene-Alkyne Coupling
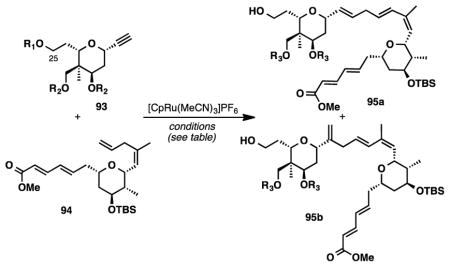
| ||||||
|---|---|---|---|---|---|---|
| Entrya | R1 | R2 | R3 | Solvent | Yieldb | 95a:95bc |
| 1 | H | H |
|
acetone | 32% | 3.2 : 1 |
| 2 | H | H |
|
cyclo-pentanone | 39% | 3.8 : 1 |
| 3 | H |
|
|
acetone | 41% | 3.7 : 1 |
| 4 | H |
|
|
cyclo-pentanone | 62% | 3 : 1 |
| 5 | TBS |
|
|
cyclo-pentanone | 36% | 3.7 : 1 |
All reactions were run using 3.0 equiv. 94, 15 mol% [CpRu(MeCN)3]PF6, in 0.047 M solvent, at 55°C for 2 h 10 min.
Mixture of 95a:95b.
Determined by 1H NMR.
We designed a series of experiments to probe these hypotheses. To determine the extent of contribution from solvent, we tested the cyclopentanone-protected substrate (Table 2, entry 3) in the coupling reaction using acetone as solvent. Only a slight decrease in the linear:branched ratio from 3.8:1 to 3.7:1 was observed. When the acetonide-protected substrate (entry 4) was tested, this time using cyclopentanone as solvent, we saw a decrease in linear:branched ratio to from 3.8:1 to 3:1. Finally, to probe the role of the C25 alcohol, we incorporated a TBS protecting group onto the cyclopentanone protected substrate (entry 5), expecting the linear:branched ratios to decrease if coordination was no longer possible. An identical 3.7:1 ratio of linear:branched isomers was obtained. The fact that the TBS protecting group was cleaved during the reaction invalidates any conclusions about the role of the hydroxyl group since the desilylation could have occurred prior to or after the alkene-alkyne coupling.
Several proposed ruthenacyclopentene intermediates for both the linear and branched products are depicted in Figure 5. After oxidative coupling, ruthenium has one open coordination site and can be occupied by solvent (S) or by a coordinating atom present in the substrate. For the linear isomer, the oxygen in the tetrahydropyran ring or the primary alcohol could occupy this site. For the branched isomer only coordination of the primary alcohol may be possible but, as depicted in Figure 6, seems to be rather unfavorable. The facile desilylation that was observed in the reaction (Table 2, entry 5) suggests that the alcohol may be coordinated to the Lewis acidic ruthenium rendering it more susceptible towards desilylation. Taken together, these results begin to provide evidence that the product ratio is highly dependent on the identity of the substrate and is less influenced by the solvent (i.e. cyclopentanone vs. acetone).
Figure 6.

Possible Ruthenacyclopentene Intermediates for Linear and Branched Products
From diene 95a, protection of the primary alcohol as its TBDPS ether and hydrolysis of the cyclopentanone ketal provided diol 96, an intermediate that was used in Shishido’s total synthesis of (+)-lasonolide A (Scheme 17).7e The physical data for diol 96 was in complete agreement with the reported data. The optical rotation of 96 was +11.4 (c 0.75, CHCl3), opposite to that reported in the Shishido synthesis ( (c 1.04, CHCl3), and consistent with our absolute stereochemical assignment for (−)-lasonolide A.
Scheme 17.
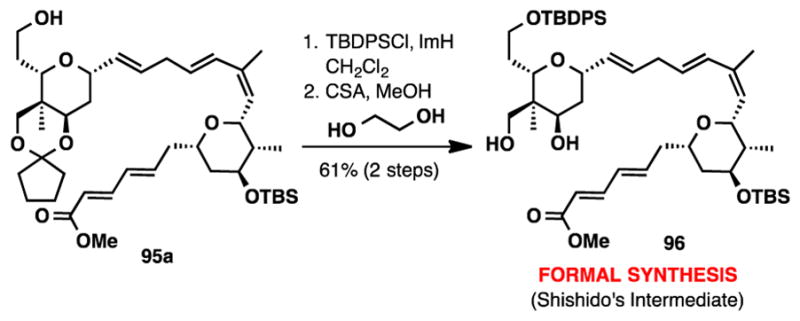
Formal Synthesis of (−)-Lasonolide A
At this point, we decided to further streamline our synthesis of lasonolide A to help fulfill the need for material that will help advance biological investigations. We turned our attention back to the Ru-catalyzed coupling reaction and questioned if a more elaborated coupling partner could be tolerated under the reaction conditions. Alkyne 92, which contains the fully elaborated side chain could be obtained in 4-steps from lactol 38 (Scheme 18). Although alkyne 92 contains two alkenes, disubstituted olefins are typically unreactive in the Ru-catalyzed coupling.53
Scheme 18.
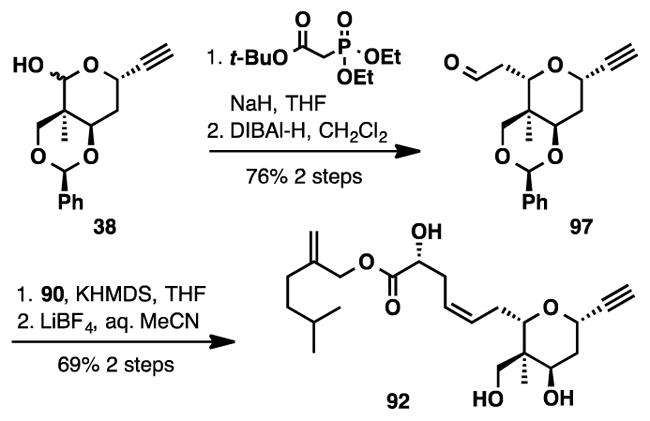
Synthesis of Alkyne 92
The alkene-alkyne coupling between 92 and 75 (Scheme 19), run under our standard set of conditions, took place in 43% yield with a linear:branched ratio of 3:1. The coupling between 92 and 74 also successfully occurred albeit with diminished linear:branched ratios (2:1) but in 69% yield. Reactions run in cyclopentanone led to poor conversion and slightly diminished linear:branched ratios in each case. Additionally, for the coupling between 92 and 74, decreasing the amount of catalyst (from 15 mol% to 5 mol%) while also increasing the concentration (from 0.047 M to 0.14M) decreased the yield to 28% (51% of 92 as its acetonide was recovered) without affecting the linear:branched ratio. It is also important to note, that in each of the examples described above, >80% of the alkene 74/75 was recovered.
Scheme 19.

Final Reaction Sequence Completing the Synthesis of (−)-Lasonolide A and Lasonolide Analogs
Completion of the synthesis was accomplished from both intermediates 98a and 99a.54 Removal of the acetonide with CSA followed by protection of the most accessible alcohols in both 100 and 101 provided seco acid 102, a common intermediate in both routes. Macrolactonization using the Yamaguchi reagent occurred without incident to provide the TBS protected lasonolide (103) in yields ranging from 40–62%. At this point, the undesired branched isomer that was generated during the coupling could be separated from the linear isomer. A final desilylation, using HF·Pyr, provided the target molecule, (−)-lasonolide A in 75% yield.
In addition to synthesizing the natural product we have also generated three analogs (Scheme 19). Compound 104 came from macrolactonization of the fully deprotected precursor hydroxyl acid. Compounds 105 and 106 were straightforwardly derived from the branched by-products of the alkene-alkyne couplings. The synthetic lasonolide A and the 3 analogs were submitted to in vitro testing in an attempt to explore their activity against various cell lines.16 Each analog tested was essentially inactive compared to the synthetic (−)-lasonolide A in all assays except for the HCT116.
SUMMARY
In conclusion, a synthesis of (−)-lasonolide A has been described. The synthesis has been accomplished in 16 linear steps and 34 total steps from commercially available starting materials. A formal intermediate was also prepared and matches the physical data that was reported in Shishido’s synthesis of (+)-lasonolide A. Biological studies, utilizing the synthetic material generated from this work, are currently underway in an effort to further understand the mechanism of action. Importantly also, these studies verify that the Ru catalyzed alkene-alkyne coupling is amenable for making linear as well as branched 1,4-diene motifs en route to natural products, notably biologically active macrocycles. This success of the intermolecular coupling between fully unprotected polyhydroxy dieneyne 92 and tetraene 74/75 demonstrates a remarkable chemoselectivity.
Supplementary Material
Figure 4.
THP Cyclizations of 60 and 62
Acknowledgments
Funding Sources
We thank the NIH (GM33049) for generous support to our programs. C.E.S., K.L.H., and A.H. are the recipients of NIH Ruth L. Kirschstein National Research Service Awards (GM101747, GM086093, and GM077840, respectively). C.P. is the recipient of a DAAD ISAP fellowship.
We thank Johnson-Matthey for generous gifts of palladium and ruthenium salts and Codexis, Inc. for the generous donation of enzymes. Finally we would like to thank Genentech (Gail Phillips, Vishal Verma and John Flygare) for screening the synthetic (−)-lasonolide A and lasonolide analogs.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, analytical data (1H NMR, 13C NMR, MS, IR, and [α]D) for all new compounds as well as additional reaction optimization tables.
References
- 1.Horton PA, Koehn FE, Longley RE, McConnell OJ. J Am Chem Soc. 1994;116:6015–6016. [Google Scholar]
- 2.(a) Lee E, Song HY, Kang JW, Kim DS, Jung CK, Joo JM. J Am Chem Soc. 2002;124:384–385. doi: 10.1021/ja017265d. [DOI] [PubMed] [Google Scholar]; (b) Song HY, Joo JM, Kang JW, Kim DS, Jung CK, Kwak HS, Park JH, Lee E, Hong CY, Jeong SW, Jeon K, Park JH. J Org Chem. 2003;68:8080–8087. doi: 10.1021/jo034930n. [DOI] [PubMed] [Google Scholar]
- 3.(a) Alley MC, Scudiero DA, Monks A, Hursey ML, Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH, Boyd MR. Cancer Res. 1988;48:589–601. [PubMed] [Google Scholar]; (b) Zhang YW, Ghosh AK, Pommier Y. Cell Cycle. 2012;11:4424–4435. doi: 10.4161/cc.22768. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jossé R, Zhang YW, Giroux V, Ghosh AK, Luo J, Pommier Y. Mar Drugs. 2015;13:3625–3639. doi: 10.3390/md13063625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Lee E, Song HY, Joo JM, Kang JW, Kim DS, Jung CK, Hong CY, Jeong SW, Jeon K. Bioorg Med Chem Lett. 2002;12:3519–3520. doi: 10.1016/s0960-894x(02)00821-1. [DOI] [PubMed] [Google Scholar]; (b) Joo JM, Kwak HS, Park JH, Song HY, Lee E. Bioorg Med Chem Lett. 2004;14:1905–1908. doi: 10.1016/j.bmcl.2004.01.088. [DOI] [PubMed] [Google Scholar]
- 5.Additional lasonolides isolated from their natural source: Wright AE, Chen Y, Winder PL, Pitts TP, Pomponi SA, Longley RE. J Nat Prod. 2004;67:1351–1355. doi: 10.1021/np040028e.
- 6.For synthetic studies towards the lasonolides see: Ghosh AK, Ren GB. J Org Chem. 2012;77:2559–2565. doi: 10.1021/jo202631e.Deba T, Yakushiji F, Mitsuru S, Shishido K. Synlett. 2003;10:1500–1502.Yoshimura T, Bando T, Shindo M, Shishido K. Tetrahedron Lett. 2004;45:9241–9244.Gurjar MK, Kumar P, Rao BV. Tetrahedron Lett. 1996;37:8617–8620.Gurjar MK, Chakrabarti A, Rao BV, Kumar P. Tetrahedron Lett. 1997;38:6885–6888.Nowakowski M, Hoffmann HMR. Tetrahedron Lett. 1997;38:1001–1004.Beck H, Hoffmann HMR. Eur J Org Chem. 1999;11:2991–2995.Hart DJ, Patterson S, Unch JP. Synlett. 2003;9:1334–1338.Dalgrad JE, Rychnovsky SD. Org Lett. 2005;7:1589–1591. doi: 10.1021/ol050270s.Sawant KB, Ding F, Jenings MP. Tetrahedron Lett. 2006;47:939–942.
- 7.For total syntheses of lasonolide A see: Trost BM, Stivala CE, Hull KL, Huang A, Fandrick DR. J Am Chem Soc. 2014;136:88–91. doi: 10.1021/ja411270d.Ghosh AK, Gong G. Org Lett. 2007;9:1437–1440. doi: 10.1021/ol0701013.Ghosh AK, Gong G. Chem Asian J. 2008;3:1811–1823. doi: 10.1002/asia.200800164.Kang SH, Kang SY, Kim CM, Choi H-w, Jun HS, Lee BM, Park CM, Jeong JW. Angew Chem Int Ed. 2003;42:4779–4782. doi: 10.1002/anie.200352016.Yoshimura T, Yakushiji F, Kondo S, Wu X, Shindo M, Shishido K. Org Lett. 2006;8:475–478. doi: 10.1021/ol0527678.
- 8.(a) Trost BM, Toste FD, Pinkerton AB. Chem Rev. 2005;101:2067–2096. doi: 10.1021/cr000666b. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Frederiksen MU, Rudd MT. Angew Chem Int Ed. 2005;44:6630–6666. doi: 10.1002/anie.200500136. [DOI] [PubMed] [Google Scholar]
- 9.Trost BM, Probst G, Schoop A. J Am Chem Soc. 1998;120:9228–9236. [Google Scholar]
- 10.(a) Trost BM, Harrington PE, Chisholm JD, Wrobleski ST. J Am Chem Soc. 2005;127:13598–13610. doi: 10.1021/ja053365y. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Wrobleski ST, Chisholm JD, Harrington PE, Jung M. J Am Chem Soc. 2005;127:13589–13597. doi: 10.1021/ja0533646. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Chisholm JD, Wrobleski SJ, Jung M. J Am Chem Soc. 2002;124:12420–12421. doi: 10.1021/ja027883+. [DOI] [PubMed] [Google Scholar]
- 11.(a) Trost BM, Papillon JPN. J Am Chem Soc. 2004;126:13618–13619. doi: 10.1021/ja045449x. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Papillon JPN, Nussbaumer T. J Am Chem Soc. 2005;127:17921–17937. doi: 10.1021/ja055967n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Trost BM, Amans D, Seganish WM, Chung CK. J Am Chem Soc. 2009;131:17087–17089. doi: 10.1021/ja907924j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Amans D, Seganish WM, Chung CK. Chem Eur J. 2012;18:2961–2971. doi: 10.1002/chem.201102899. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trost BM, Seganish WM, Chung CK, Amans D. Chem Eur J. 2012;18:2948–2960. doi: 10.1002/chem.201102898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Trost BM, Yang H, Brindle CS, Dong G. Chem– Eur J. 2011;17:9777–9788. doi: 10.1002/chem.201002930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.It is possible that alkyne 20 can dimerize under the reaction conditions. For examples of Ru-catalyzed alkyne dimerizations see: Trost BM, Rudd MT. J Am Chem Soc. 2001;123:8862–8863. doi: 10.1021/ja0111636.Trost BM, Rudd MT. J Am Chem Soc. 2002;124:4178–4179. doi: 10.1021/ja012672a.
- 15.Trost BM, Fettes A, Shireman BT. J Am Chem Soc. 2004;126:2660–2661. doi: 10.1021/ja038666r. [DOI] [PubMed] [Google Scholar]
- 16.See Supporting Information for details.
- 17.Chen KM, Hardtmann GE, Pradad K, Repic O, Shapiro MJ. Tetrahedorn Lett. 1987;28:155–158. [Google Scholar]
- 18.(a) Kiyooka S-j, Kuroda H, Shimasaki Y. Tetrahedron Lett. 1986;27:3009–3012. [Google Scholar]; (b) Mohr P. Tetrahedron Lett. 1991;32:2219–2222. [Google Scholar]
- 19.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994;7:639–666. [Google Scholar]
- 20.Smith AB, Chen SSY, Nelson FC, Reichert JM, Salvatore BA. J Am Chem Soc. 1995;117:12013–12014. [Google Scholar]
- 21.HWE olefination and in situ cyclizations of lactols have been reported by: Allevi P, Ciuffreda P, Colombo D, Mont D, Speranza G, Manitto P. J Chem Soc Perkin Trans 1. 1989:1281–1283.
- 22.Figueroa-Perez S, Schmidt RR. Carbohydr Res. 2000;328:95–102. doi: 10.1016/s0008-6215(00)00092-6. [DOI] [PubMed] [Google Scholar]
- 23.Bennett SM, Nguyen-Ba N, Ogilvie KK. J Med Chem. 1990;33:2162–2173. doi: 10.1021/jm00170a019. [DOI] [PubMed] [Google Scholar]
- 24.Bonner TG, Bourne EJ, McNally S. J Chem Soc. 1960:2929–2934. [Google Scholar]
- 25.Takano S, Hatakeyama S, Ogasawara K. J Chem Soc, Perkin Trans 1. 1980;2:457–461. [Google Scholar]
- 26.Liu Z-Y, Ji J-X, Li B-G. J Chem Soc, Perkin Trans 1. 2000:3519–3521. [Google Scholar]
- 27.Chen LY, Zaks A, Chackalamannil S, Dugar S. J Org Chem. 1996;61:8341–8343. doi: 10.1021/jo961096b. [DOI] [PubMed] [Google Scholar]
- 28.Lipshutz BH, Pegram JJ, Morey MC. Synth Commun. 1982;12:267–277.For a recent example of benzylidene acetal hydrolysis using LiBF4 in a complex setting see: Araoz R, Servent D, Molgo J, Iorga BH, Fruchart-Gaillard C, Benoit E, Gu Z, Stivala CE, Zakarian A. J Am Chem Soc. 2011;133:10499–10511. doi: 10.1021/ja201254c.
- 29.Suarez A, Fu GC. Angew Chem Int Ed. 2004;43:3580–3582. doi: 10.1002/anie.200454070. [DOI] [PubMed] [Google Scholar]
- 30.Trost BM, Kazmaier U. J Am Chem Soc. 1992;114:7933–7935. [Google Scholar]; (b) Trost BM, Li CJ. J Am Chem Soc. 1994;116:10819–10820. [Google Scholar]; (c) Rychnovsky SD, Kim J. J Org Chem. 1994;59:2959–2660. [Google Scholar]
- 31.(a) Noyori R, Ohkuma T, Kitamura M. J Am Chem Soc. 1987;109:5856–5858. [Google Scholar]; (b) Mashima K, Kusano K-h, Sato N, Matsumura Y, Kyoko N, Kumobayashi H, Sayo N, Hori Y, Ishizaki T, Akutagawa S, Takaya H. J Org Chem. 1994;59:3064–3076. [Google Scholar]; (c) Everaere K, Carpentier JF, Mortreux A, Bulliard M. Tetrahedron Asym. 1999;10:4083–4086. [Google Scholar]
- 32.Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 33.For examples of attempted diastereoselective asymmetric hydrogenation of α-methyl-β-keto-esters: Kitamura M, Ohkuma T, Tokunaga M, Noyori R. Tetrahedron Asym. 1990;1:1–4.Rychnovsky SD, Hoye RC. J Am Chem Soc. 1994;116:1753–1765.Burk MJ, Harper GP, Kalberg CS. J Am Chem Soc. 1995;117:4423–4424.Yamano T, Taya N, Kawada M, Huang T, Imamoto T. Tetrahedron Lett. 1999;40:2577–2580.Zhang Z, Qian H, Longmire J, Zhang X. J Org Chem. 2000;65:6223–6226. doi: 10.1021/jo000462v.Ireland T, Tappe K, Grossheimann G, Knochel P. Chem Eur J. 2002;8:843–852. doi: 10.1002/1521-3765(20020215)8:4<843::aid-chem843>3.0.co;2-9.Evans DA, Michael FE, Tedrow JS, Campos KR. J Am Chem Soc. 2003;125:3539–3543. doi: 10.1021/ja012639o.
- 34.(a) Nakamura K, Kawai Y, Miyai T, Ohno A. Tetrahedron Lett. 1990;31:3631–3632. [Google Scholar]; (b) Nakamura K, Kawai Y, Ohno A. Tetrahedron Lett. 1991;32:2927–2928. [Google Scholar]; (c) Hamdani M, De Jeso B, Deleuze H, Maillard B. Tetrahedron Asym. 1993;4:1229–1232. [Google Scholar]; (d) Shieh WR, Sih CJ. Tetrahedron Asym. 1993;4:1259–1269. [Google Scholar]; (e) North M. Tetrahedron Lett. 1996;37:1699–1702. [Google Scholar]; (f) Ji A, Wolberg M, Hummel W, Wandrey C, Muller M. Chem Commun. 2001:57–58. [Google Scholar]
- 35.Pedersen OS, Peterson L, Brandt M, Nielsen C, Pedersen EB. Monatsh Chem. 1999;130:1499–1512. [Google Scholar]
- 36.Stereochemistry also confirmed by comparing physical data to known β-hydroxyester: Hareau GPJ, Koiwa M, Hikichi S, Sato F. J Am Chem Soc. 1999;121:3640–3650.
- 37.(a) Kalaitzakis D, Rozzell JD, Kambourakis S, Smonou I. Org Lett. 2005;7:4799–4801. doi: 10.1021/ol051166d. [DOI] [PubMed] [Google Scholar]; (b) Kalaitzakis D, Rozzell JD, Kambourakis S, Smonou I. Eur J Org Chem. 2006:2309–2313. [Google Scholar]
- 38.The absolute stereochemistry was established by conversion of a 4 : 1 (syn:anti) mixture of diastereomers to known saturated hydroxyl esters. See Supporting Information for details Oppolzer W, Blagg J, Walther E. J Am Chem Soc. 1990;112:2767–2772.Ghosh AK, Onishi M. J Am Chem Soc. 1996;118:2527–2528. doi: 10.1021/ja9539148.
- 39.Williams JM, Jobson RB, Yasuda N, Marchesini G, Dolling UH, Grabowski EJJ. Tetrahedron Lett. 1995;36:5461–5464. [Google Scholar]
- 40.Yu CM, Kim C, Kweon JH. Chem Commun. 2004;21:2494–2495. doi: 10.1039/b407387h. [DOI] [PubMed] [Google Scholar]
- 41.Trost BM, Ball ZT, Joge T. Angew Chem Int Ed. 2003;42:3415–1418. doi: 10.1002/anie.200351587. [DOI] [PubMed] [Google Scholar]
- 42.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Soc Chem. 2000;122:8168. [Google Scholar]
- 43.Taguchi H, Ghoroku K, Tadaki M, Tsubouchi A, Takeda T. Org Lett. 2001;3:3811. doi: 10.1021/ol016837w. [DOI] [PubMed] [Google Scholar]; b) Taguchi H, Ghoroku K, Tadaki M, Tsubouchi A, Takeda T. J Org Chem. 2002;67:8450. doi: 10.1021/jo025973r. [DOI] [PubMed] [Google Scholar]; c) Tsubouchi A, Itoh M, Onishi K, Takeda T. Synthesis. 2004:1504. [Google Scholar]; d) Taguchi H, Miyashita H, Tsubouchi A, Takeda T. Chem Commun. 2002:2218. doi: 10.1039/b207325k. [DOI] [PubMed] [Google Scholar]; e) Denmark S, Wehrli D, Choi J. Org Lett. 2000;2:2491. doi: 10.1021/ol006170y. [DOI] [PubMed] [Google Scholar]
- 44.Paterson I, Arnott EA. Tetrahedron Lett. 1998;39:7185–7188. [Google Scholar]
- 45.Hiyama T, Hatanaka Y. Pure Appl Chem. 1994;66:1471–1478.Trost BM, Machacek MR, Ball ZT. Org Lett. 2003;5:1895–1898. doi: 10.1021/ol034463w.Dey R, Chattopadhyay K, Ranu BC. J Org Chem. 2008;73:9461–9464. doi: 10.1021/jo802214m.For a review see: Denmark SE, Sweis RF. Chem Pharm Bull. 2002;50:1531–1541. doi: 10.1248/cpb.50.1531.
- 46.In our initial communication (Ref 7a) we described the formation of the substituted tetrahydropyran ring through a simultaneous Hoveyda-Grubbs 2nd generation catalyzed cross metathesis/oxa-Michael sequence: Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179.
- 47.Allyl chloride and allyl methyl carbonate could also be utilized in the Hiyama coupling.
- 48.Hogrefe RI, McCaffrey AP, Corozdina LU, McCampbell ES, Vaghefi MW. Nucleic Acids Res. 1993;21:4739–4741. doi: 10.1093/nar/21.20.4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura S, Kikuchi F, Hashimoto S. Angew Chem Int Ed. 2008;47:7091–7094. doi: 10.1002/anie.200802729. [DOI] [PubMed] [Google Scholar]
- 50.(a) Yoshimura T, Yakushiji F, Kondo S, Wu X, Shindo M, Shishido K. Org Lett. 2006;8:475–478. doi: 10.1021/ol0527678. [DOI] [PubMed] [Google Scholar]; (b) Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 51.Shiina I, Kubota M, Oshiumi H, Hashizume M. J Org Chem. 2004;69:1822–1830. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]
- 52.Steliou K, Szczygielska-Nowosielska A, Favre A, Poupart MA, Hanessian S. J Am Chem Soc. 1980;102:7579–7581. [Google Scholar]
- 53.Trost BM, Cregg JJ. J Am Chem Soc. 2015;137:620–623. doi: 10.1021/ja511911b. [DOI] [PubMed] [Google Scholar]
- 54.For clarity only the major (linear) isomer, from the alkene-alkyne coupling, is drawn. In the case where R=H a 2:1 ratio of products is advanced and in the case of R=TBS a 3:1 ratio of products is advanced. The linear and branched isomers become separable after macrolactonization.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.